
NAMPT Inhibitor and P73 Activator Represses P53 R175H Mutated HNSCC Cell Proliferation in a Synergistic Manner

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatment
2.2. CCK8 Assay
2.3. Thioflavin T Staining
2.4. Immunocytochemistry
2.5. Real Time RT-PCR
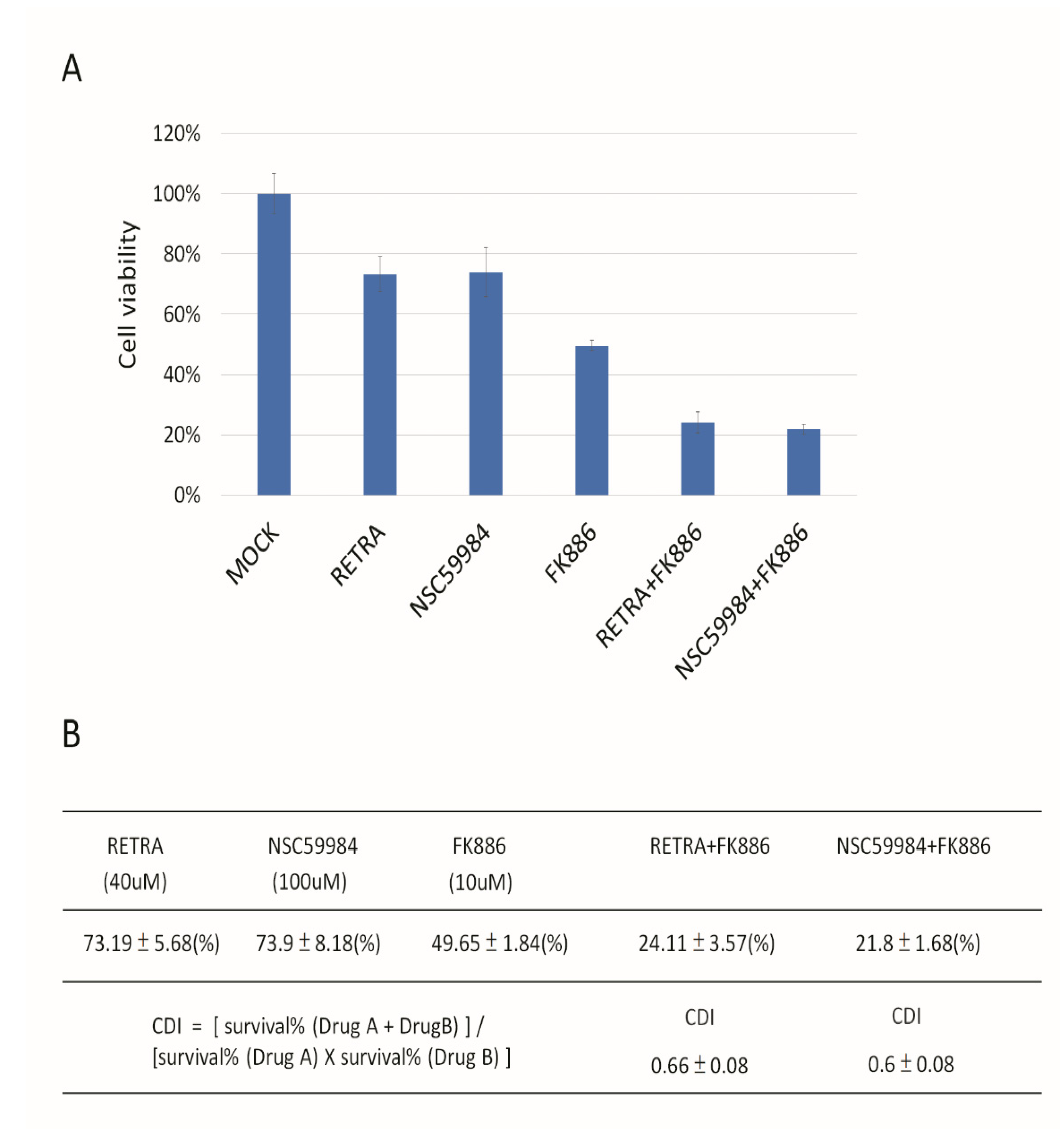
2.6. Drug Interaction Analysis
3. Results
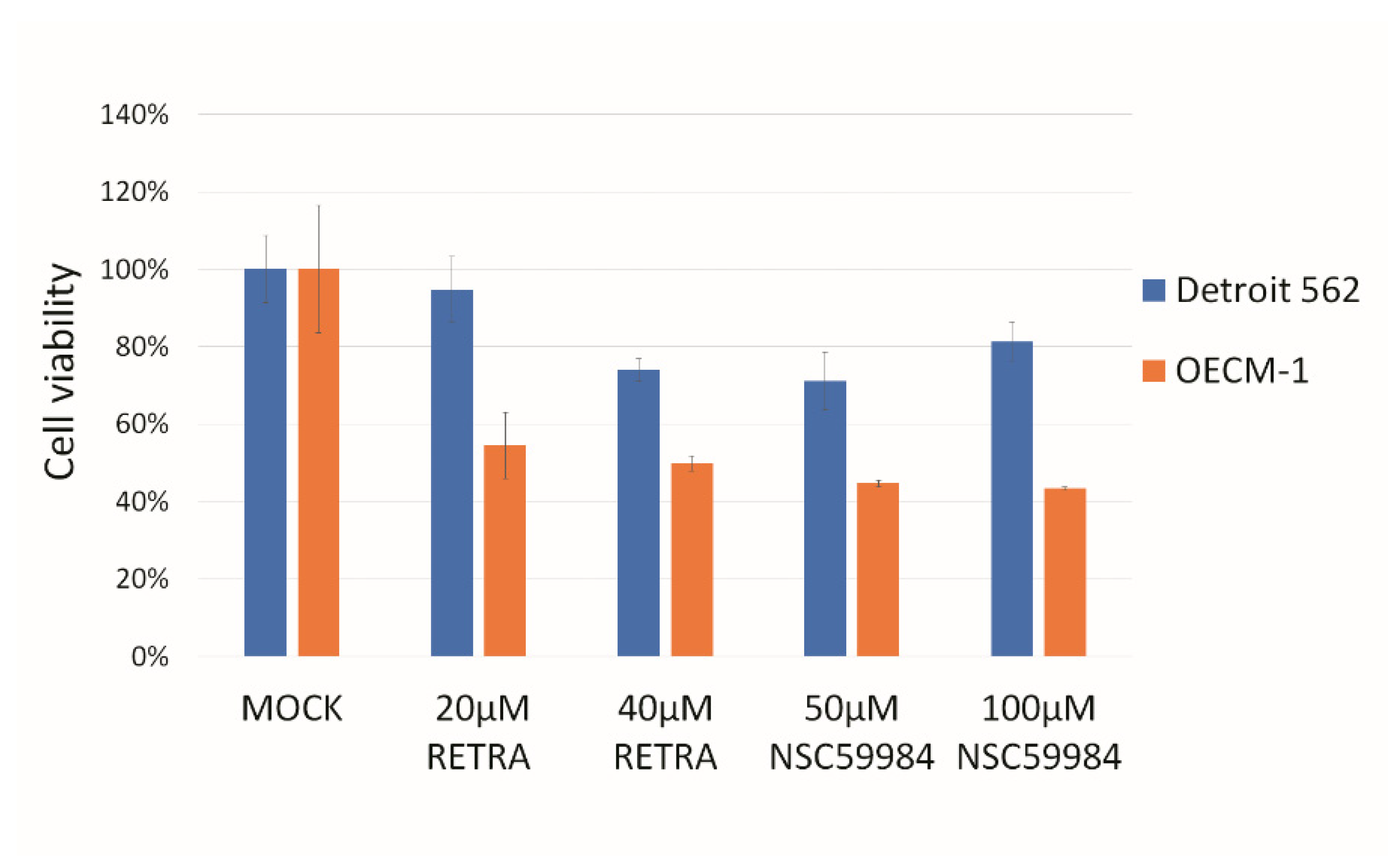
3.1. p73 Activators Have Different Responses in HNSCC Cell Lines with p53 V173L or p53 R175H
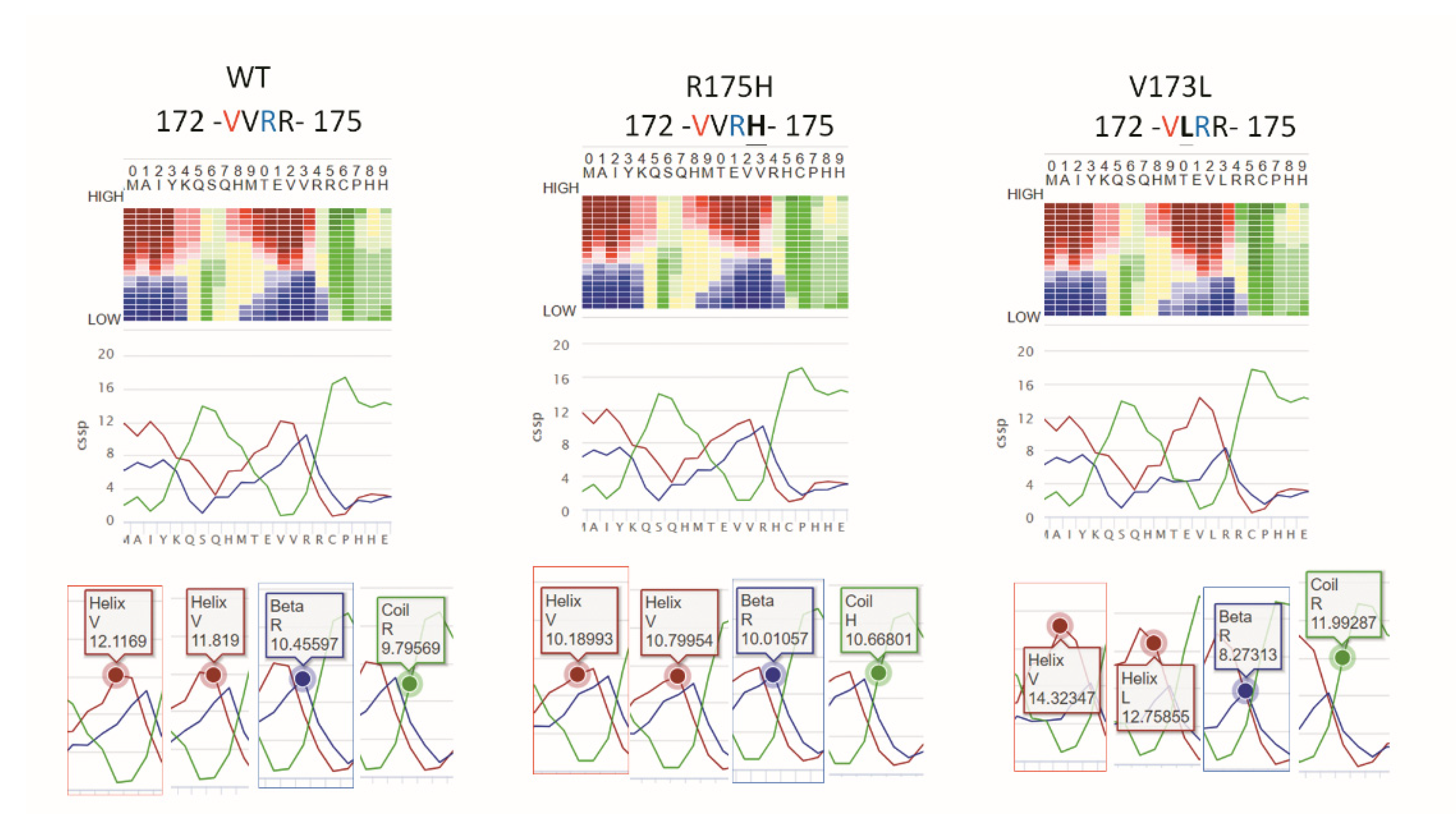
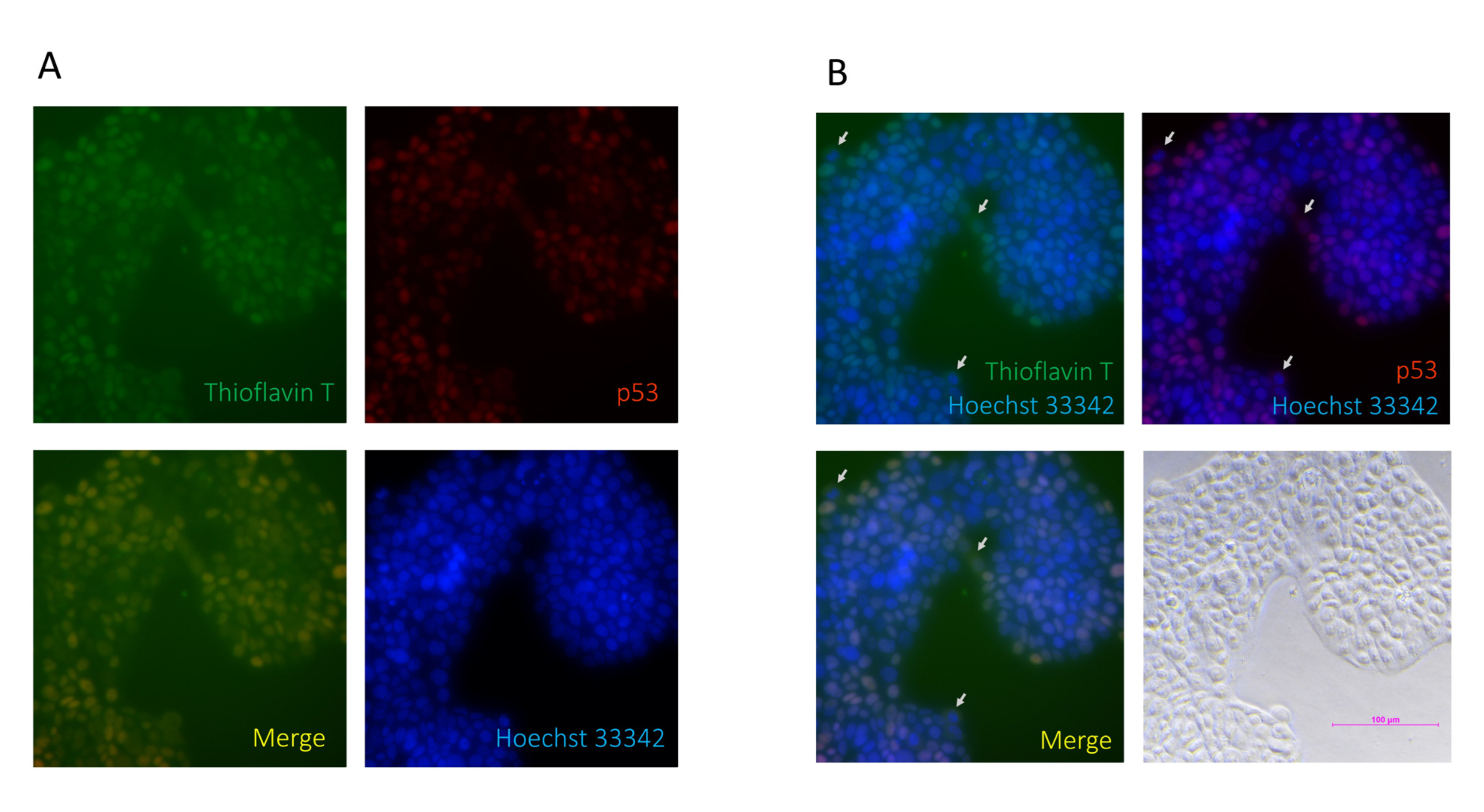
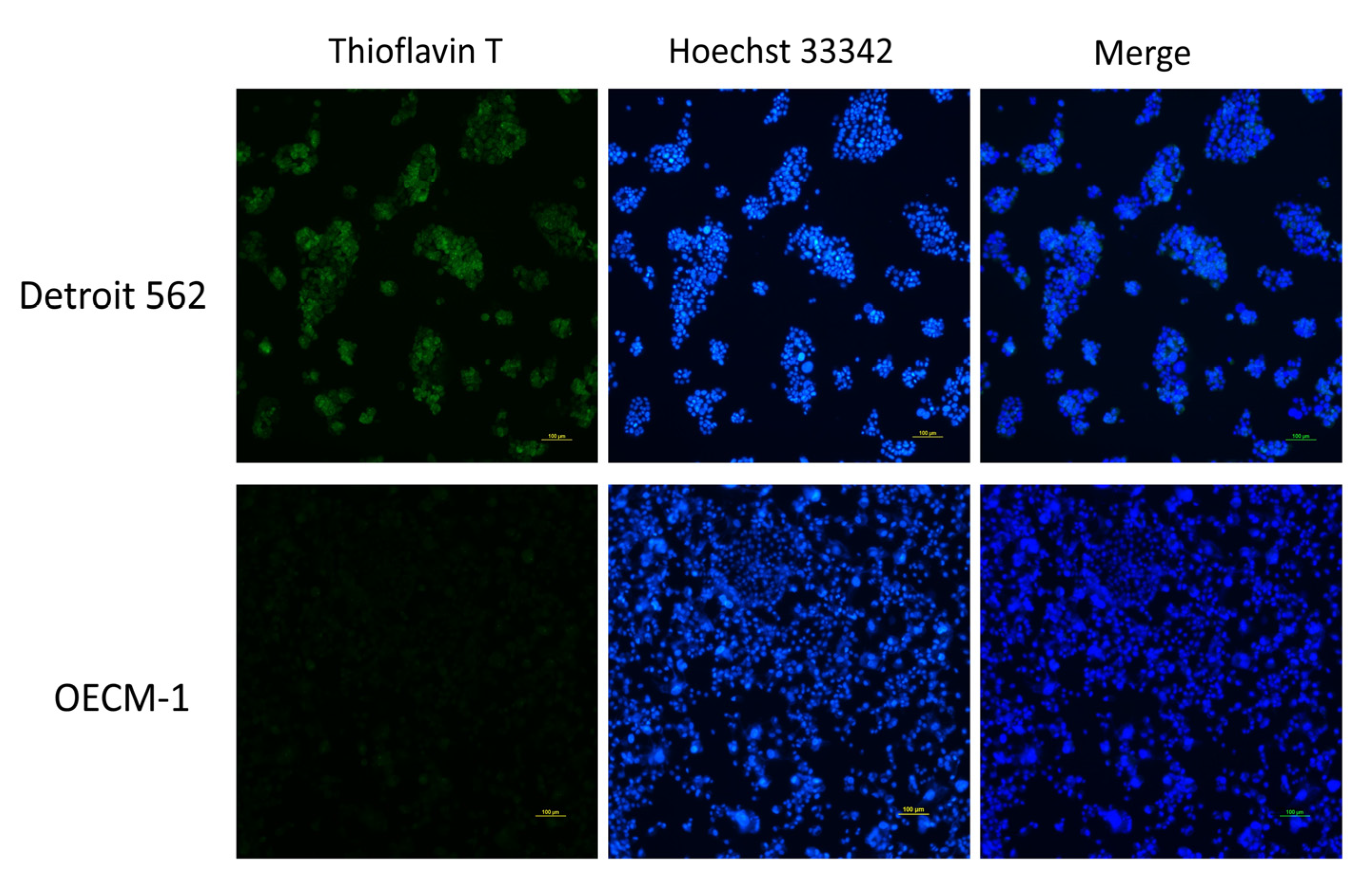
3.2. p53 V173L is A Non-Aggregative Type of p53 Mutant
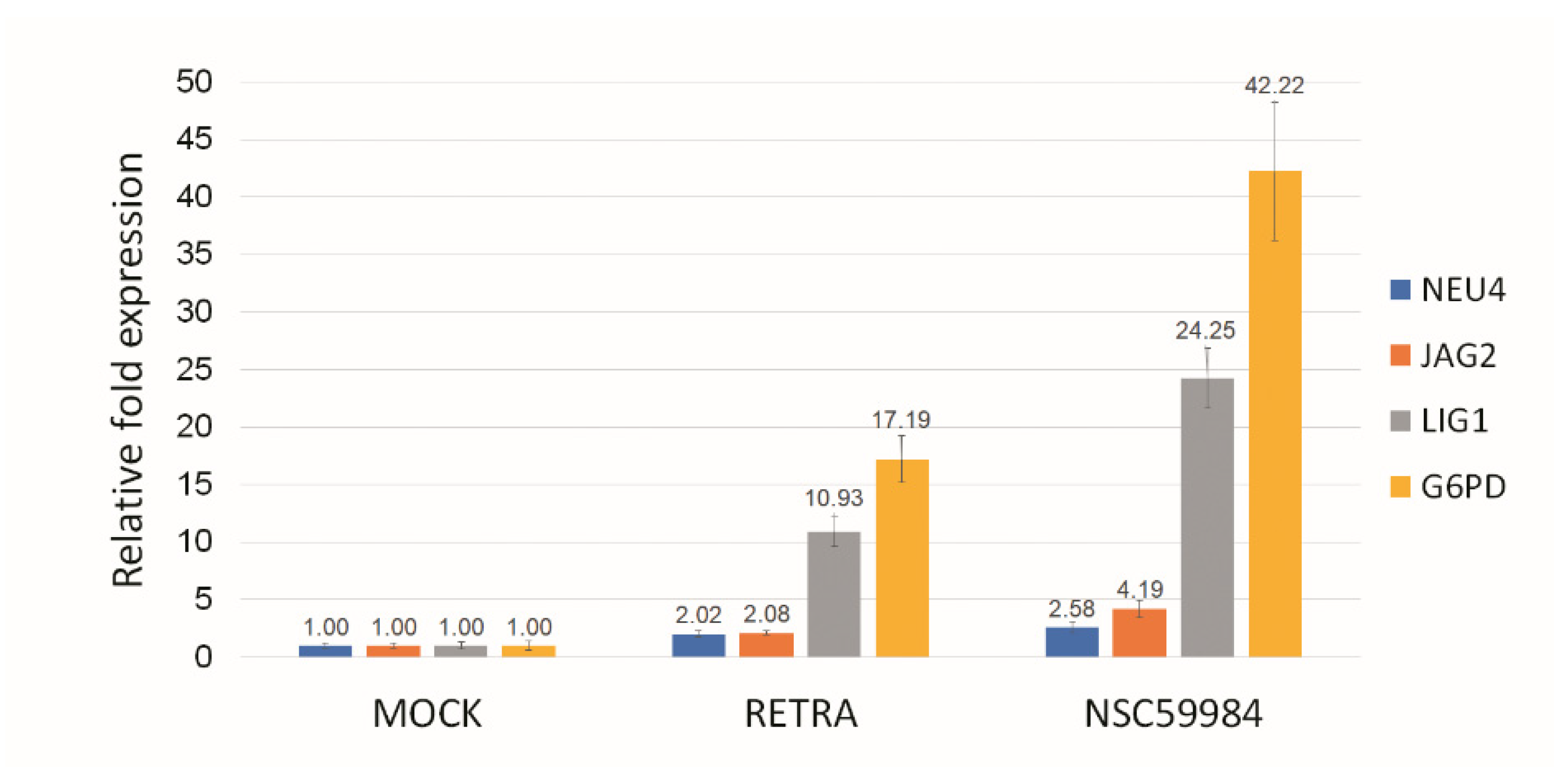
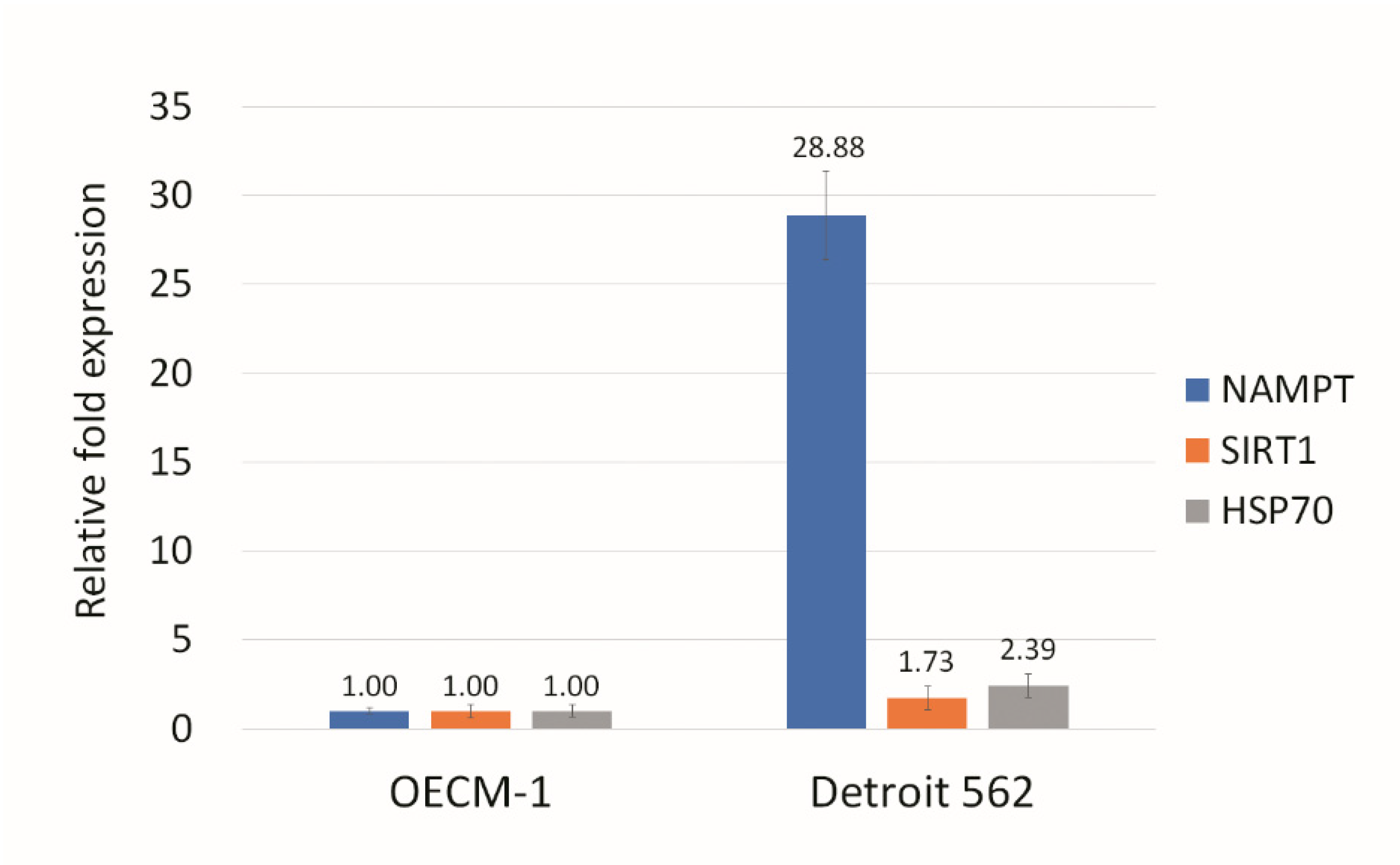
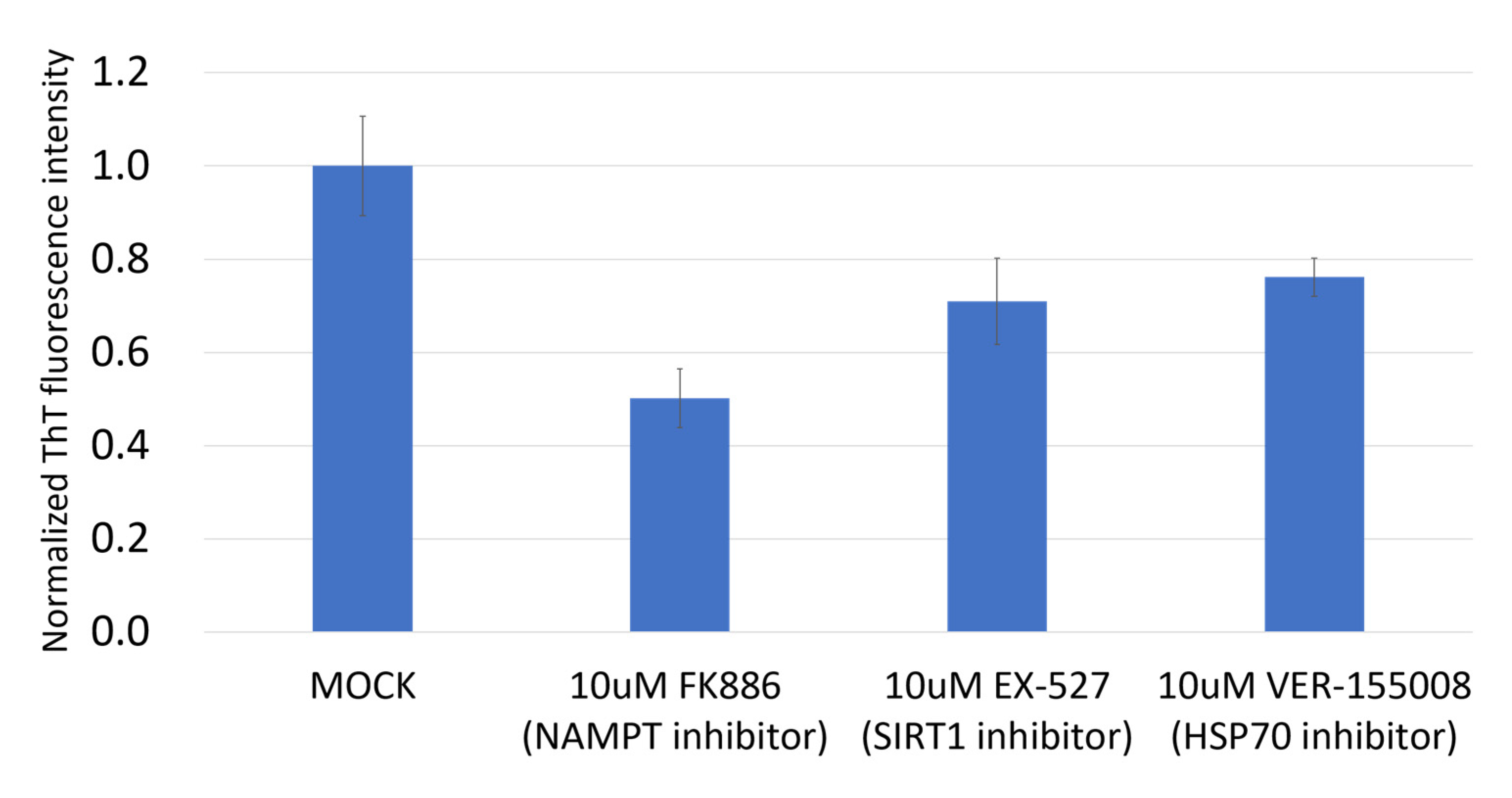
3.3. NAMPT Inhibitor and p73 Activator Have Synergic Effects That Repress the Growth of HNSCC Cells with p53 R175H Mutation
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levrero, M.; De Laurenzi, V.; Costanzo, A.; Gong, J.; Wang, J.Y.; Melino, G. The p53/p63/p73 family of transcription factors: Overlapping and distinct functions. J. Cell Sci. 2000, 113 Pt 10, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Liu, S.; Tian, X.; Wang, X.; Gao, P. A TP53 mutation model for the prediction of prognosis and therapeutic responses in head and neck squamous cell carcinoma. BMC Cancer 2021, 21, 1035. [Google Scholar] [CrossRef] [PubMed]
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donzelli, S.; Biagioni, F.; Fausti, F.; Strano, S.; Fontemaggi, G.; Blandino, G. Oncogenomic Approaches in Exploring Gain of Function of Mutant p53. Curr. Genom. 2008, 9, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Liebl, M.C.; Hofmann, T.G. The Role of p53 Signaling in Colorectal Cancer. Cancers 2021, 13, 2125. [Google Scholar] [CrossRef]
- Koga, H.; Deppert, W. Identification of genomic DNA sequences bound by mutant p53 protein (Gly245-->Ser) in vivo. Oncogene 2000, 19, 4178–4183. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol. Cell 2017, 68, 1134–1146.e1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stindt, M.H.; Muller, P.A.; Ludwig, R.L.; Kehrloesser, S.; Dötsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015, 34, 4300–4310. [Google Scholar] [CrossRef]
- De Oliveira, G.A.P.; Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Vieira, T.C.R.G.; Cino, E.A.; Silva, J.L. The Status of p53 Oligomeric and Aggregation States in Cancer. Biomolecules 2020, 10, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrote, M.M.; Motta, M.F.; Ferretti, G.D.S.; Norberto, D.R.; Spohr, T.C.L.S.; Lima, F.R.S.; Gratton, E.; Silva, J.L.; de Oliveira, G.A.P. Oncogenic Gain of Function in Glioblastoma Is Linked to Mutant p53 Amyloid Oligomers. iScience 2020, 23, 100820. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhou, L.; Hong, B.; van den Heuvel, A.P.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrloesser, S.; Osterburg, C.; Tuppi, M.; Schäfer, B.; Vousden, K.H.; Dötsch, V. Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ. 2016, 23, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Kawasumi, M.; Lan, T.Y.; Poon, C.L.; Lin, Y.S.; Wu, P.J.; Chen, Y.C.; Chen, B.H.; Wu, C.H.; Lo, J.F.; et al. Adaptation to Endoplasmic Reticulum Stress Enhances Resistance of Oral Cancer Cells to Cisplatin by Up-Regulating Polymerase η and Increasing DNA Repair Efficiency. Int. J. Mol. Sci. 2020, 22, 355. [Google Scholar] [CrossRef]
- Kim, C.; Choi, J.; Lee, S.J.; Welsh, W.J.; Yoon, S. NetCSSP: Web application for predicting chameleon sequences and amyloid fibril formation. Nucleic Acids Res. 2009, 37, W469–W473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Cai, B.H.; Wu, P.H.; Chou, C.K.; Huang, H.C.; Chao, C.C.; Chung, H.Y.; Lee, H.Y.; Chen, J.Y.; Kannagi, R. Synergistic activation of the NEU4 promoter by p73 and AP2 in colon cancer cells. Sci. Rep. 2019, 9, 950. [Google Scholar] [CrossRef]
- Fontemaggi, G.; Kela, I.; Amariglio, N.; Rechavi, G.; Krishnamurthy, J.; Strano, S.; Sacchi, A.; Givol, D.; Blandino, G. Identification of direct p73 target genes combining DNA microarray and chromatin immunoprecipitation analyses. J. Biol. Chem. 2002, 277, 43359–43368. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013, 15, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Chao, C.F.; Huang, H.C.; Lee, H.Y.; Kannagi, R.; Chen, J.Y. Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation. Int. J. Mol. Sci. 2019, 20, 3681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smet, F.; Rubio, M.S.; Hompes, D.; Naus, E.; De Baets, G.; Langenberg, T.; Hipp, M.S.; Houben, B.; Claes, F.; Charbonneau, S.; et al. Nuclear inclusion bodies of mutant and wild-type p53 in cancer: A hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J. Pathol. 2017, 242, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.T.; Chien, Y.C.; Lin, Y.H.; Wu, H.H.; Lee, D.F.; Yu, Y.L. The Function of the Mutant p53-R175H in Cancer. Cancers 2021, 13, 4088. [Google Scholar] [CrossRef] [PubMed]
- Shivu, B.; Seshadri, S.; Li, J.; Oberg, K.A.; Uversky, V.N.; Fink, A.L. Distinct β-sheet structure in protein aggregates determined by ATR-FTIR spectroscopy. Biochemistry 2013, 52, 5176–5183. [Google Scholar] [CrossRef]
- Kozyrska, K.; Pilia, G.; Vishwakarma, M.; Wagstaff, L.; Goschorska, M.; Cirillo, S.; Mohamad, S.; Gallacher, K.; Carazo Salas, R.E.; Piddini, E. p53 directs leader cell behavior, migration, and clearance during epithelial repair. Science 2022, 375, eabl8876. [Google Scholar] [CrossRef]
- Tong, X.; Xu, D.; Mishra, R.K.; Jones, R.D.; Sun, L.; Schiltz, G.E.; Liao, J.; Yang, G.Y. Identification of a druggable protein-protein interaction site between mutant p53 and its stabilizing chaperone DNAJA1. J. Biol. Chem. 2021, 296, 100098. [Google Scholar] [CrossRef] [PubMed]
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-Mediated Regulation of the Conformation of p53 DNA Binding Domain and p53 Cancer Variants. Mol. Cell. 2019, 74, 831–843.e4. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr. Opin. Gastroenterol. 2007, 23, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Berrocal, J.G.; Frizzell, K.M.; Gamble, M.J.; DuMond, M.E.; Krishnakumar, R.; Yang, T.; Sauve, A.A.; Kraus, W.L. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem. 2009, 284, 20408–20417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Nguyen, T.Q.A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; An, S.S. Role of p53 isoforms and aggregations in cancer. Medicine 2016, 95, e3993. [Google Scholar] [CrossRef] [PubMed]
- Kanapathipillai, M. Treating p53 Mutant Aggregation-Associated Cancer. Cancers (Basel) 2018, 10, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, C.; Schulze, L.; Weiske, J.; Meyer, H.; Braeuer, N.; Barak, N.; Eberspächer, U.; Werbeck, N.; Stresemann, C.; Lange, M.; et al. Identification of Small Molecules that Modulate Mutant p53 Condensation. iScience 2020, 23, 101517. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kempen, P.M.; Verdam, F.J.; de Poel, E.; Braunius, W.W.; de Weger, R.A.; van Es, R.J.; Grolman, W.; Willems, S.M. TP53 Y220C Is a Hotspot Mutation in Oropharyngeal Squamous Cell Carcinoma. Pathobiology 2015, 82, 21–27. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, L.; Nicolau, M.; Giannoccaro, M.; Daidone, M.G.; Bossi, P.; Locati, L.; Licitra, L.; Canevari, S. Head and neck cancer subtypes with biological and clinical relevance: Meta-analysis of gene-expression data. Oncotarget 2015, 6, 9627–9642. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.J.; Eun, Y.G.; Rho, Y.S.; Kim, E.H.; Yim, S.Y.; Kang, S.H.; Sohn, B.H.; Kwon, G.H.; Lee, J.S. Three distinct genomic subtypes of head and neck squamous cell carcinoma associated with clinical outcomes. Oral Oncol. 2018, 85, 44–51. [Google Scholar] [CrossRef]
- Chan, W.M.; Siu, W.Y.; Lau, A.; Poon, R.Y. How many mutant p53 molecules are needed to inactivate a tetramer? Mol. Cell. Biol. 2004, 24, 3536–3551. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.B.; Beeler, J.S.; Lehmann, B.D.; Gonzalez-Ericsson, P.; Sanchez, V.; Sanders, M.E.; Boyd, K.L.; Pietenpol, J.A. Tissue-specific expression of p73 and p63 isoforms in human tissues. Cell Death Dis. 2021, 12, 745. [Google Scholar] [CrossRef]
- Conforti, F.; Yang, A.L.; Agostini, M.; Rufini, A.; Tucci, P.; Nicklison-Chirou, M.V.; Grespi, F.; Velletri, T.; Knight, R.A.; Melino, G.; et al. Relative expression of TAp73 and ΔNp73 isoforms. Aging (Albany NY) 2012, 4, 202–205. [Google Scholar] [CrossRef]
- Russo, C.; Osterburg, C.; Sirico, A.; Antonini, D.; Ambrosio, R.; Würz, J.M.; Rinnenthal, J.; Ferniani, M.; Kehrloesser, S.; Schäfer, B.; et al. Protein aggregation of the p63 transcription factor underlies severe skin fragility in AEC syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E906–E915. [Google Scholar] [CrossRef] [Green Version]
- Arsic, N.; Slatter, T.; Gadea, G.; Villain, E.; Fournet, A.; Kazantseva, M.; Allemand, F.; Sibille, N.; Seveno, M.; de Rossi, S.; et al. Δ133p53β isoform pro-invasive activity is regulated through an aggregation-dependent mechanism in cancer cells. Nat. Commun. 2021, 12, 5463. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Candelaria, N.; Wong, K.K.; Nikolai, B.C.; Lonard, D.M.; O’Malley, B.W.; Richards, J.S. USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat. Commun. 2018, 9, 1270. [Google Scholar] [CrossRef] [Green Version]
- Davison, T.S.; Vagner, C.; Kaghad, M.; Ayed, A.; Caput, D.; Arrowsmith, C.H. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J. Biol. Chem. 1999, 274, 18709–18714. [Google Scholar] [CrossRef] [Green Version]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [Green Version]
- Saha, M.N.; Jiang, H.; Yang, Y.; Reece, D.; Chang, H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol. Cancer Ther. 2013, 12, 2331–2341. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.; Prabhu, V.V.; Zhang, S.; van den Heuvel, A.P.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin rescues deficient p53 signaling and antitumor effects via upregulating p73 and disrupting its interaction with mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Abrams, S.L.; Steelman, L.S.; Cocco, L.; Ratti, S.; Martelli, A.M.; Lombardi, P.; Gizak, A.; Duda, P. APR-246—The Mutant TP53 Reactivator—Increases the Effectiveness of Berberine and Modified Berberines to Inhibit the Proliferation of Pancreatic Cancer Cells. Biomolecules 2022, 12, 276. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, B.-H.; Bai, Z.-Y.; Lien, C.-F.; Yu, S.-J.; Lu, R.-Y.; Wu, M.-H.; Wu, W.-C.; Chen, C.-C.; Hsu, Y.-C. NAMPT Inhibitor and P73 Activator Represses P53 R175H Mutated HNSCC Cell Proliferation in a Synergistic Manner. Biomolecules 2022, 12, 438. https://doi.org/10.3390/biom12030438
Cai B-H, Bai Z-Y, Lien C-F, Yu S-J, Lu R-Y, Wu M-H, Wu W-C, Chen C-C, Hsu Y-C. NAMPT Inhibitor and P73 Activator Represses P53 R175H Mutated HNSCC Cell Proliferation in a Synergistic Manner. Biomolecules. 2022; 12(3):438. https://doi.org/10.3390/biom12030438
Chicago/Turabian StyleCai, Bi-He, Zhi-Yu Bai, Ching-Feng Lien, Si-Jie Yu, Rui-Yu Lu, Ming-Han Wu, Wei-Chen Wu, Chia-Chi Chen, and Yi-Chiang Hsu. 2022. "NAMPT Inhibitor and P73 Activator Represses P53 R175H Mutated HNSCC Cell Proliferation in a Synergistic Manner" Biomolecules 12, no. 3: 438. https://doi.org/10.3390/biom12030438
APA StyleCai, B.-H., Bai, Z.-Y., Lien, C.-F., Yu, S.-J., Lu, R.-Y., Wu, M.-H., Wu, W.-C., Chen, C.-C., & Hsu, Y.-C. (2022). NAMPT Inhibitor and P73 Activator Represses P53 R175H Mutated HNSCC Cell Proliferation in a Synergistic Manner. Biomolecules, 12(3), 438. https://doi.org/10.3390/biom12030438