
A Computational Approach to Investigate TDP-43 RNA-Recognition Motif 2 C-Terminal Fragments Aggregation in Amyotrophic Lateral Sclerosis

 , , ,
, , ,  ,
,  and
and
Abstract
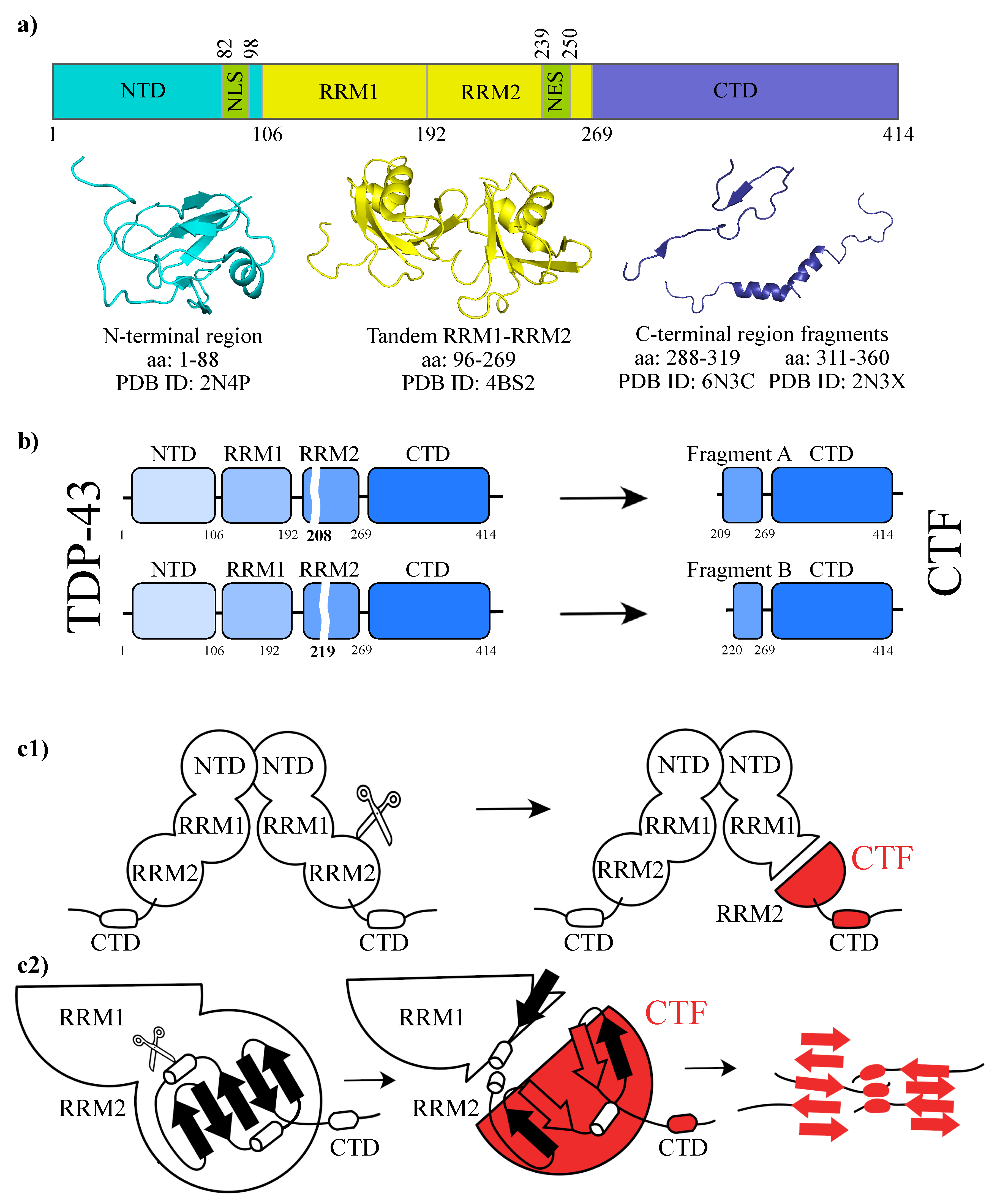
:1. Introduction
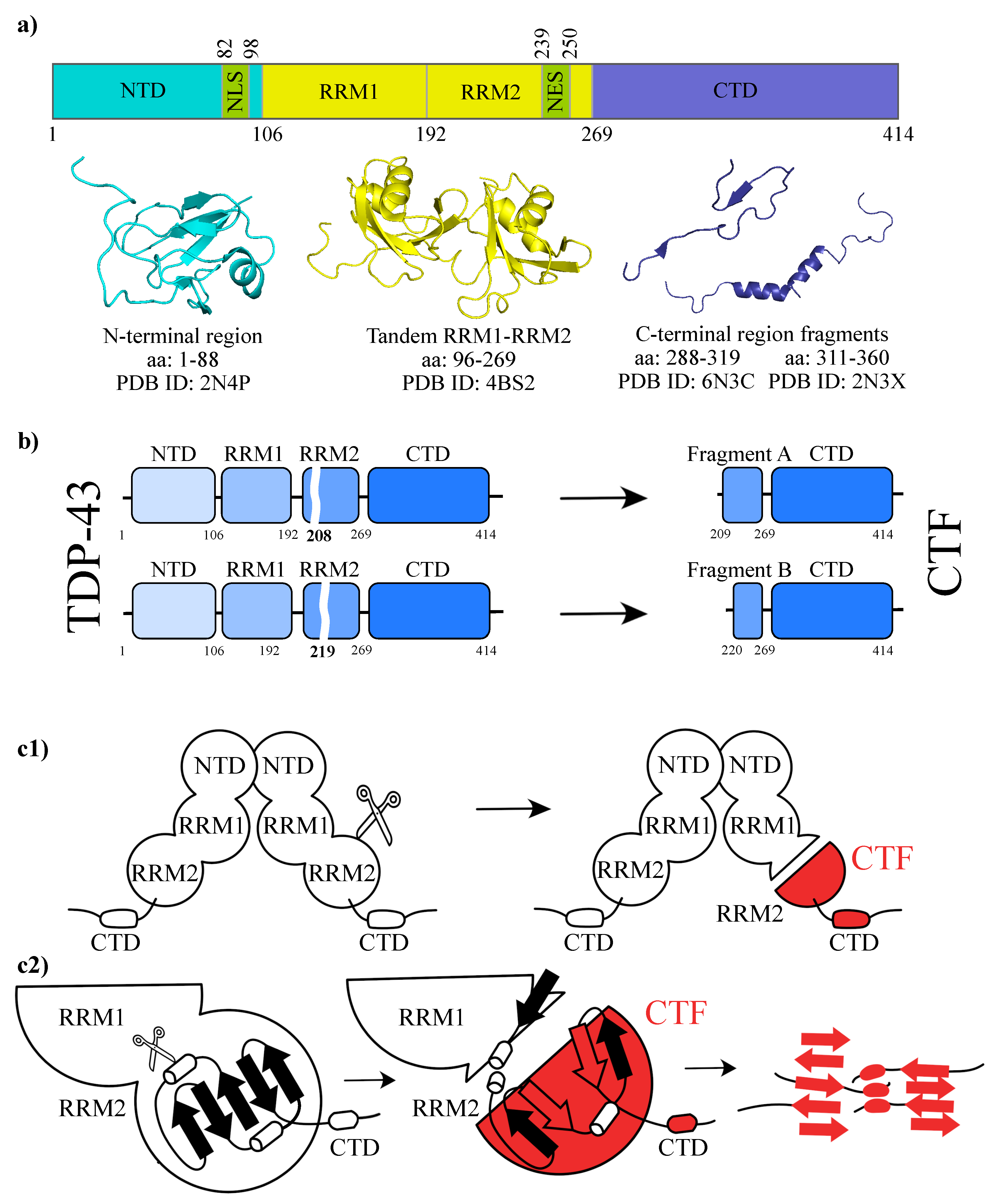
2. Results and Discussions
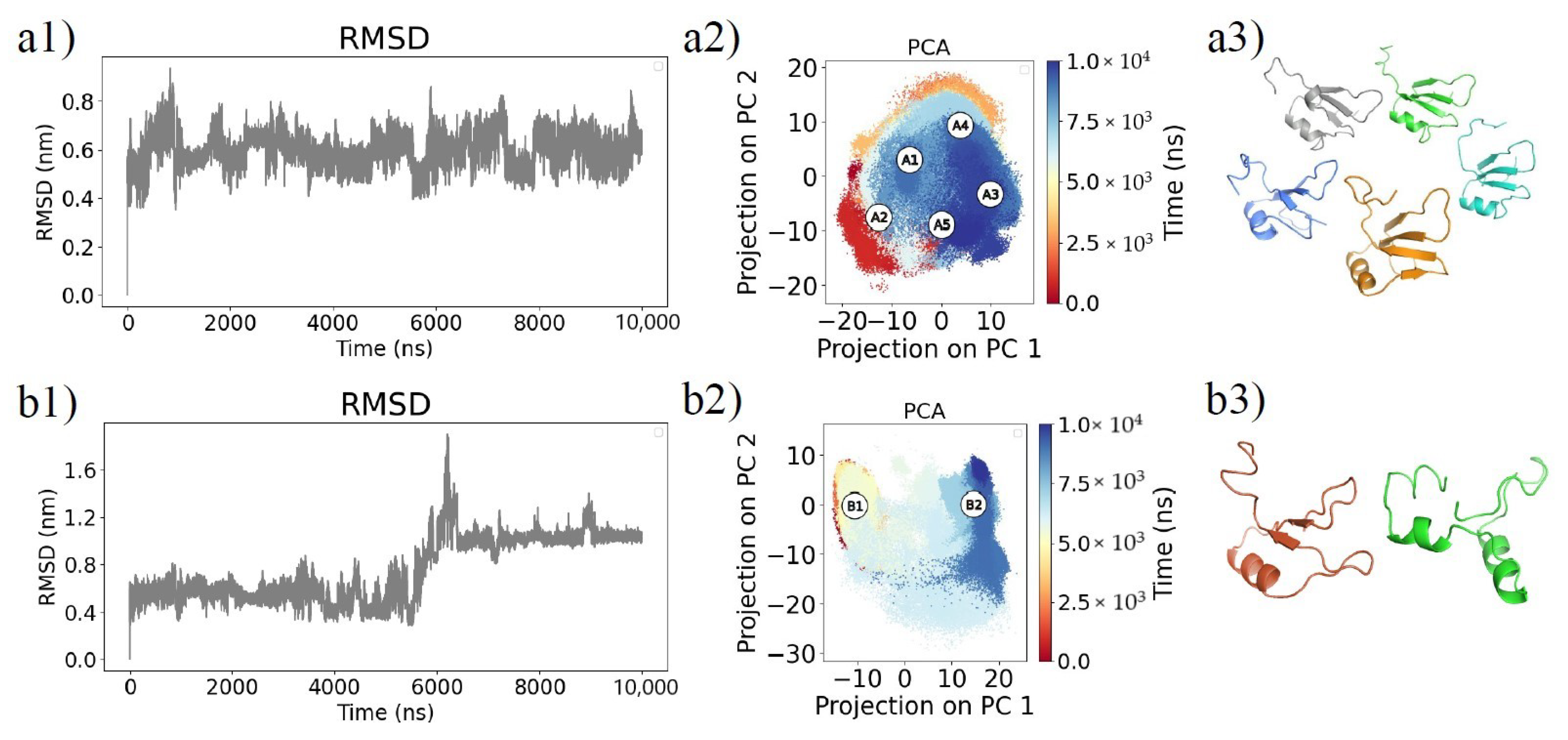
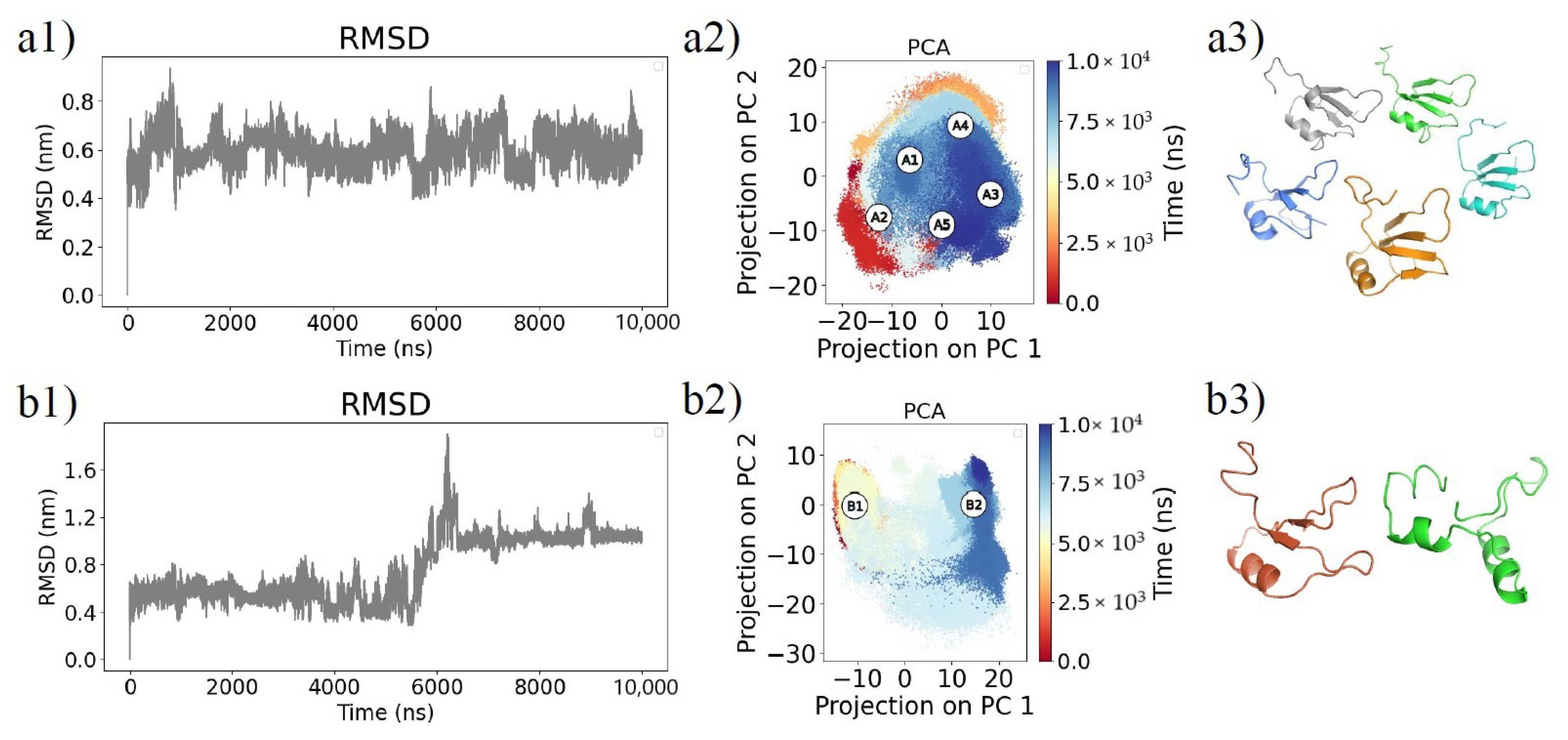
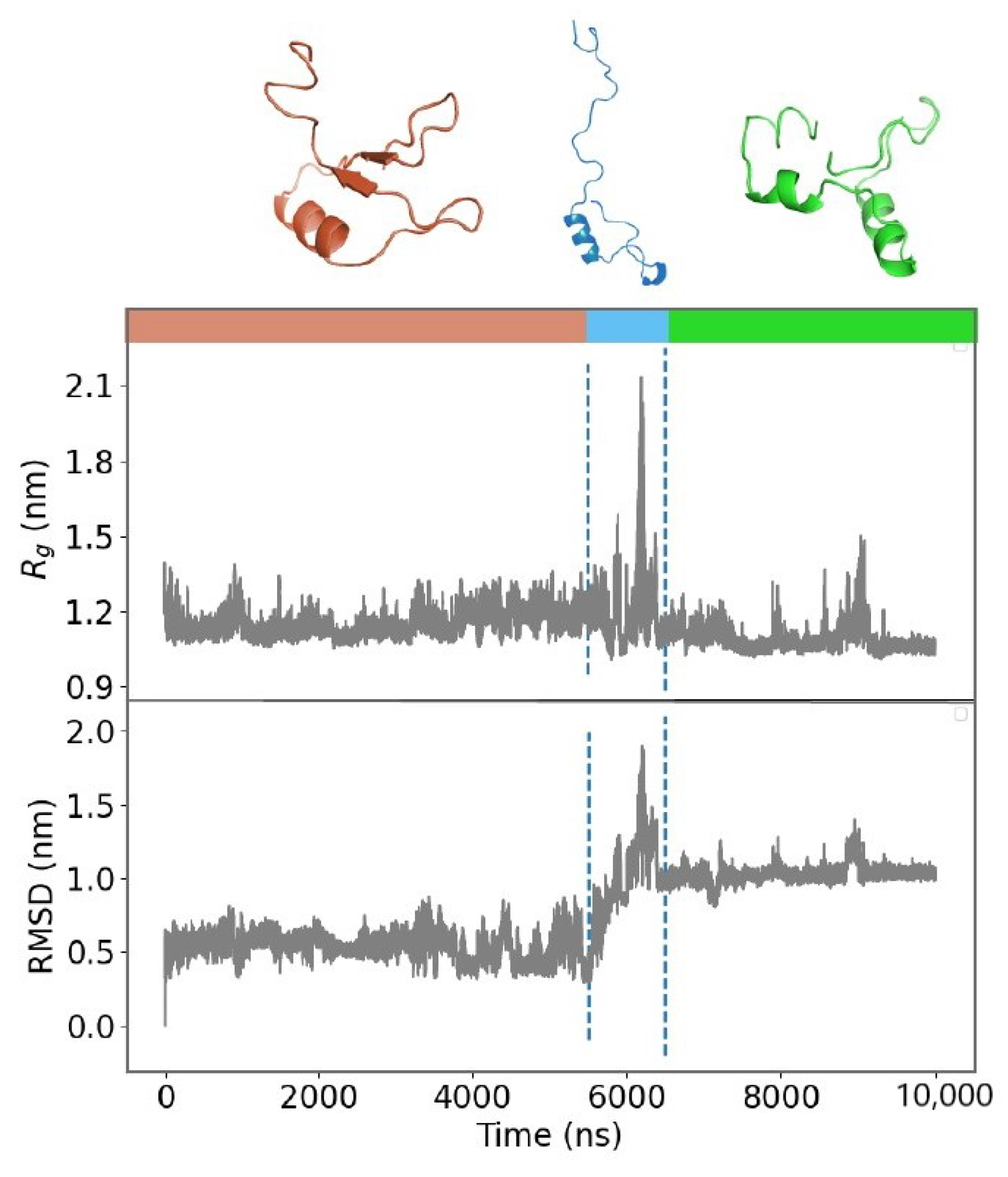
2.1. Molecular Dynamics Simulations and Equilibrium Conformations
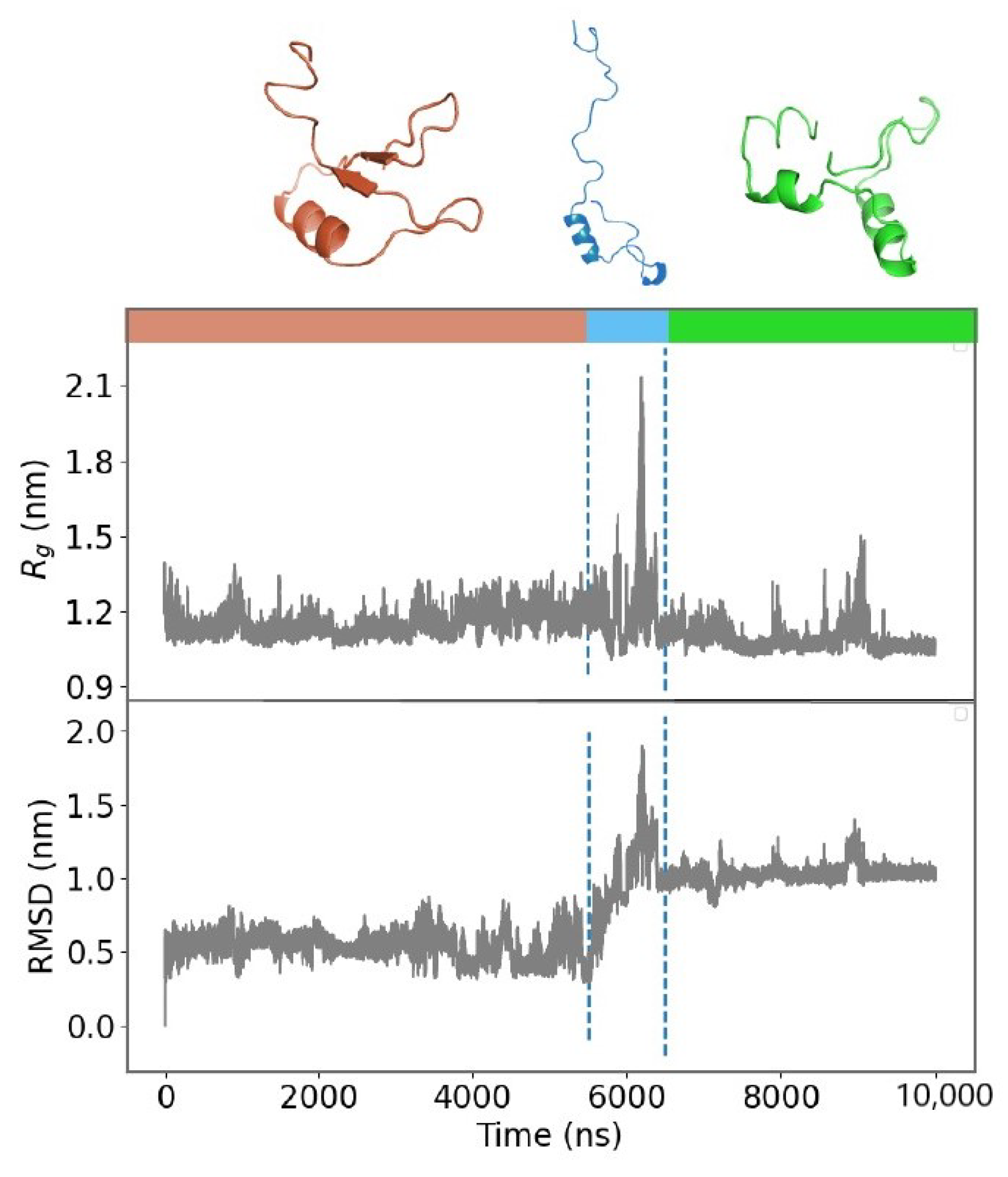
2.2. Unfolding Process of Fragment B
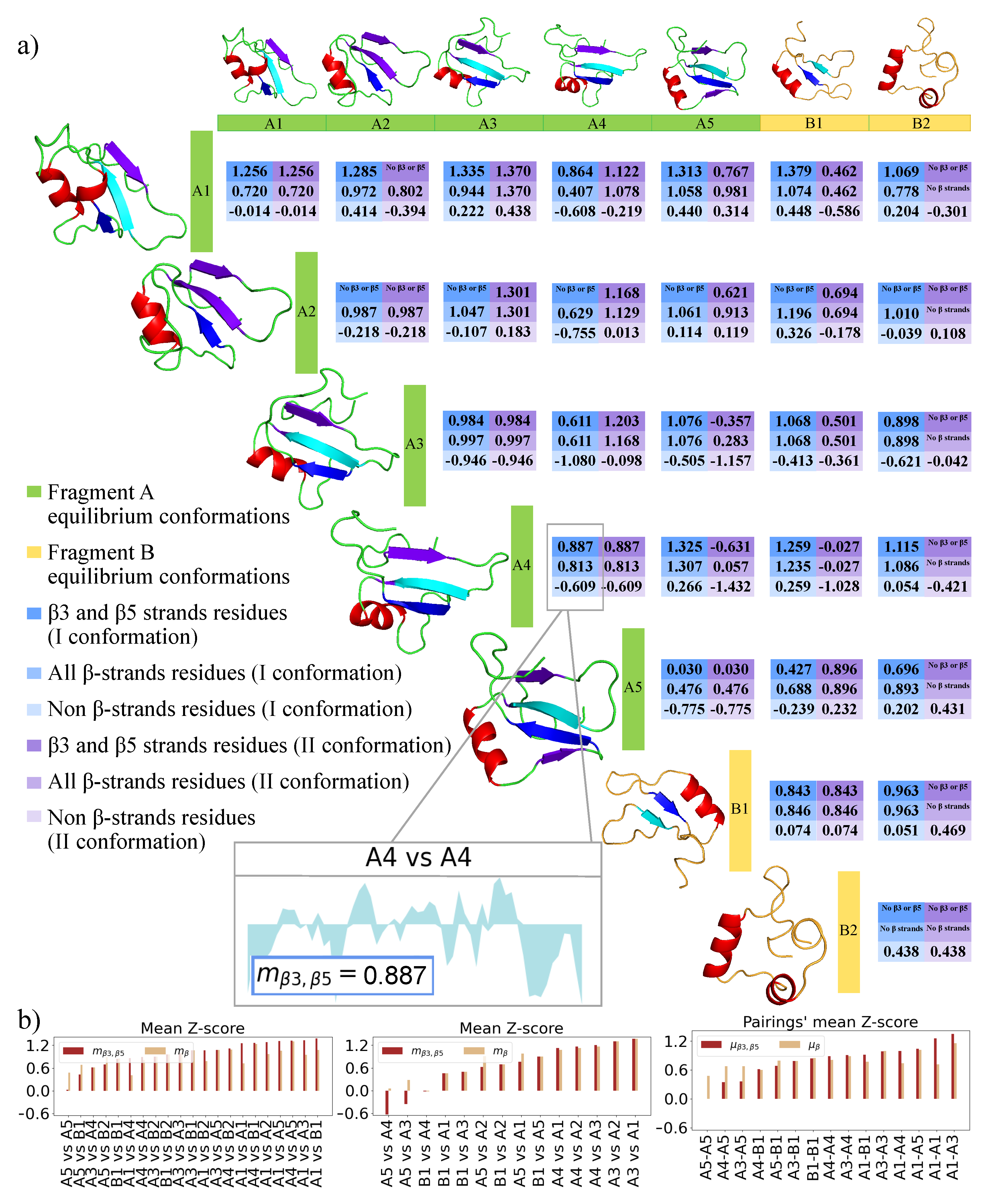
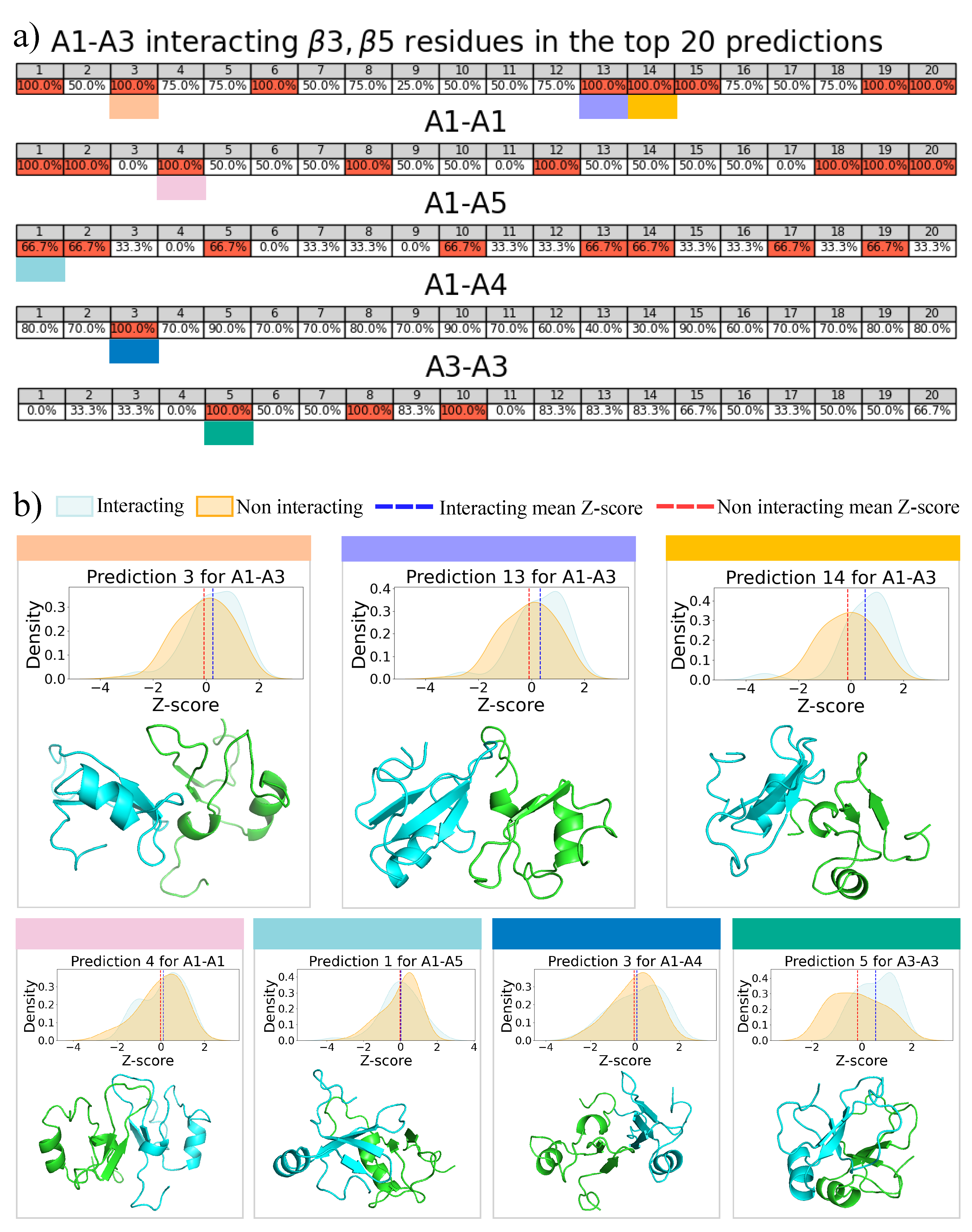
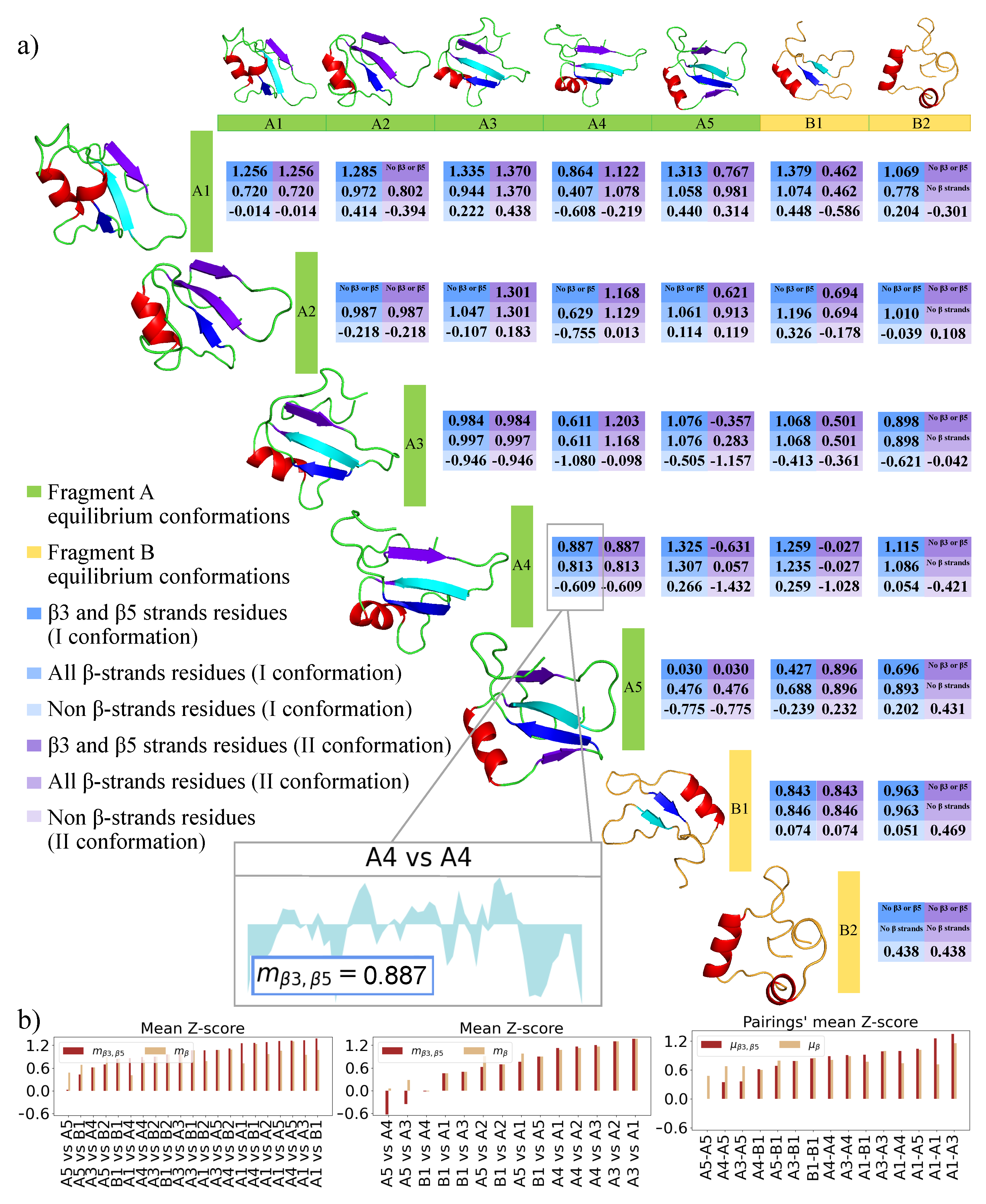
2.3. 2D Zernike Polynomial Expansion for Binding Regions Prediction
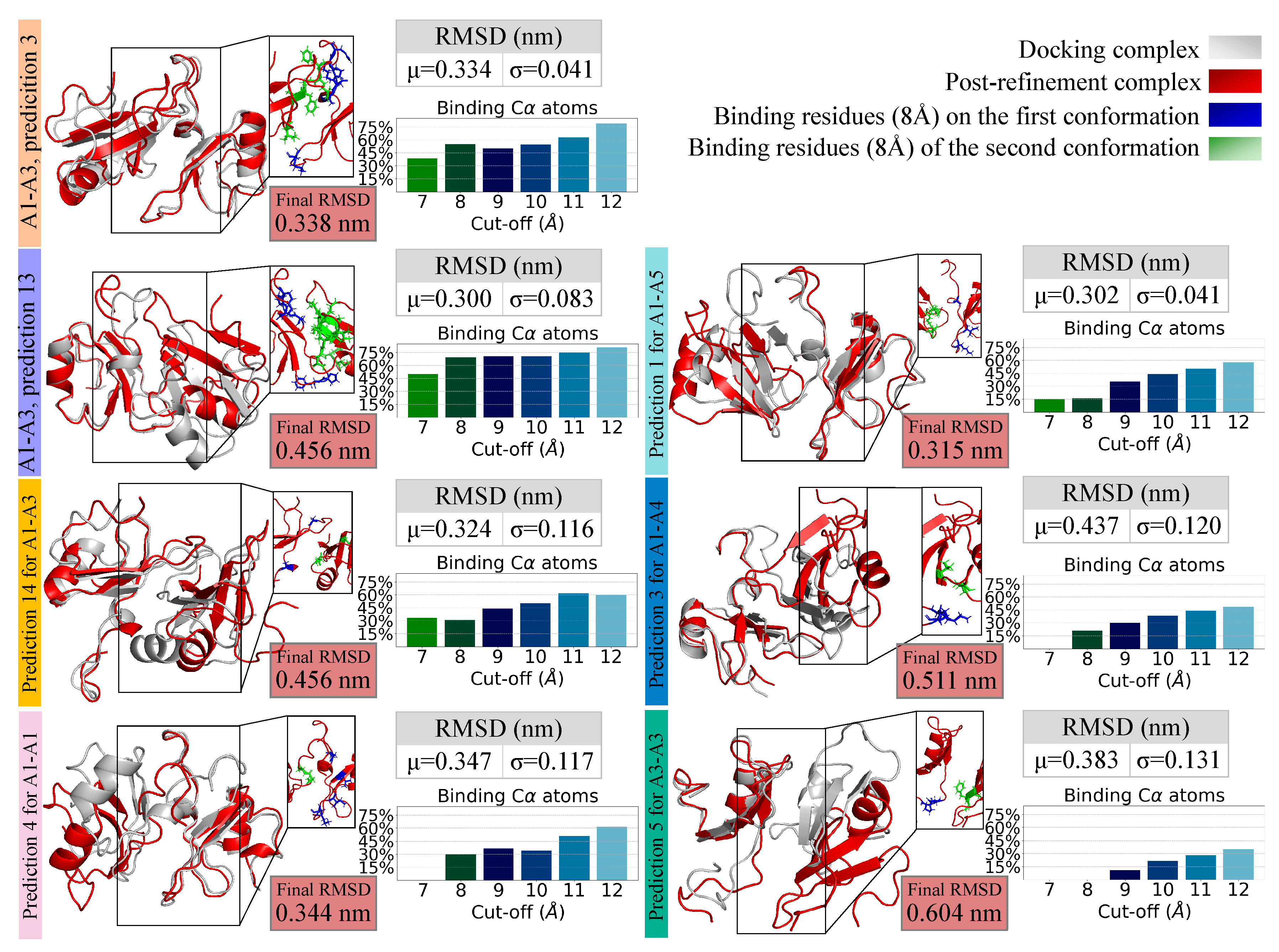
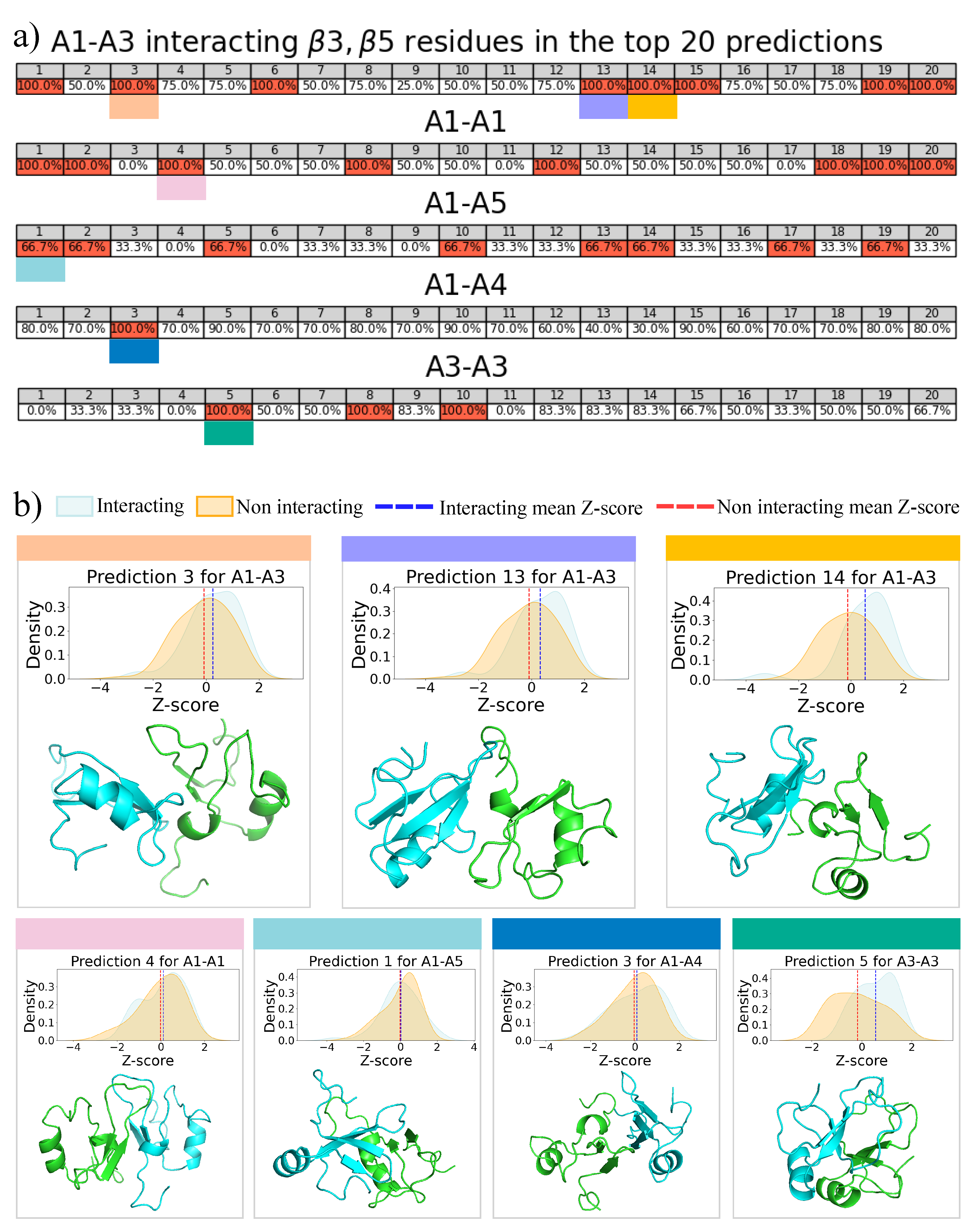
2.4. Molecular Docking for Complexes Binding Poses Prediction of Different Conformations
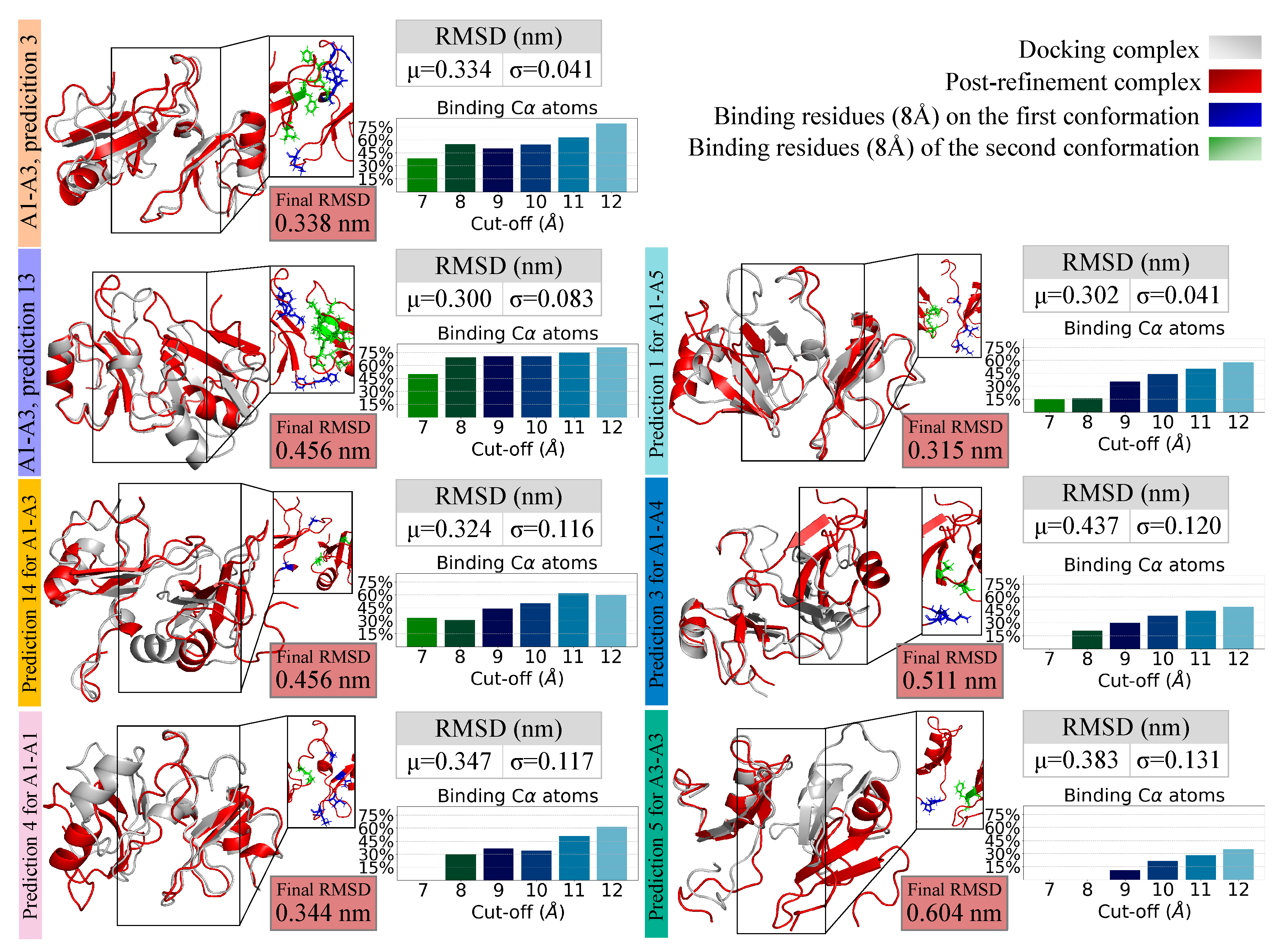
2.5. Refining and Stability Analysis of the Selected Docked Complexes through MD Simulations
3. Conclusions
4. Materials and Methods
4.1. Dataset
4.2. Molecular Dynamics Simulations
4.3. Principal Component Analysis and Clustering Analysis
4.4. Computation of Molecular Surfaces
4.5. Evaluation of Shape Complementarity
4.6. Contact Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baloh, R.H. TDP-43: The relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FEBS J. 2011, 278, 3539–3549. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Lee, S.; Jeon, Y.M.; Kim, S.; Kwon, Y.; Kim, H.J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Baralle, M.; Buratti, E.; Baralle, F.E. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem. Soc. Trans. 2013, 41, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.L.; Xue, W.; Hong, J.Y.; Zhang, J.T.; Li, M.J.; Yu, S.N.; He, J.H.; Hu, H.Y. The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep. 2017, 7, 6196. [Google Scholar] [CrossRef]
- Lee, D.Y.; McMurray, C.T. Trinucleotide expansion in disease: Why is there a length threshold? Curr. Opin. Genet. Dev. 2014, 26, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.T.; Kuo, P.H.; Chiang, C.H.; Liang, J.R.; Chen, Y.R.; Wang, S.; Shen, J.C.; Yuan, H.S. The Truncated C-terminal RNA Recognition Motif of TDP-43 Protein Plays a Key Role in Forming Proteinaceous Aggregates. J. Biol. Chem. 2013, 288, 9049–9057. [Google Scholar] [CrossRef] [Green Version]
- Tavella, D.; Zitzewitz, J.A.; Massi, F. Characterization of TDP-43 RRM2 Partially Folded States and Their Significance to ALS Pathogenesis. Biophys. J. 2018, 115, 1673–1680. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- François-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Guenther, E.L.; Ge, P.; Trinh, H.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Gonen, T.; Zhou, Z.H.; Eisenberg, D.S. Atomic-level evidence for packing and positional amyloid polymorphism by segment from TDP-43 RRM2. Nat. Struct. Mol. Biol. 2018, 25, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackness, B.C.; Tran, M.T.; McClain, S.P.; Matthews, C.R.; Zitzewitz, J.A. Folding of the RNA Recognition Motif (RRM) Domains of the Amyotrophic Lateral Sclerosis (ALS)-linked Protein TDP-43 Reveals an Intermediate State. J. Biol. Chem. 2014, 289, 8264–8276. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Wahiduzzaman.; Prakash, A.; Tomar, A.K.; Srivastava, A.; Kundu, B.; Lynn, A.M.; Hassan, M.I. Exploring the aggregation-prone regions from structural domains of human TDP-43. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef]
- Zacco, E.; Martin, S.R.; Thorogate, R.; Pastore, A. The RNA-recognition motifs of TAR DNA-binding protein 43 may play a role in the aberrant self-assembly of the protein. Front. Mol. Neurosci. 2018, 11, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacco, E.; Graña-Montes, R.; Martin, S.R.; de Groot, N.S.; Alfano, C.; Tartaglia, G.G.; Pastore, A. RNA as a key factor in driving or preventing self-assembly of the TAR DNA-binding protein 43. J. Mol. Biol. 2019, 431, 1671–1688. [Google Scholar] [CrossRef]
- Paul, F.; Weikl, T.R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical Relaxation Rates. PLoS Comput. Biol. 2016, 12, e1005067. [Google Scholar] [CrossRef] [Green Version]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Nat. Preced. 2010, 35, 539–546. [Google Scholar]
- Milanetti, E.; Miotto, M.; Di Rienzo, L.; Monti, M.; Gosti, G.; Ruocco, G. 2D Zernike polynomial expansion: Finding the protein-protein binding regions. Comput. Struct. Biotechnol. J. 2021, 19, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Milanetti, E.; Miotto, M.; Di Rienzo, L.; Nagaraj, M.; Monti, M.; Golbek, T.W.; Gosti, G.; Roeters, S.J.; Weidner, T.; Otzen, D.E.; et al. In-Silico Evidence for a Two Receptor Based Strategy of SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 690655. [Google Scholar] [CrossRef] [PubMed]
- Miotto, M.; Di Rienzo, L.; Bò, L.; Boffi, A.; Ruocco, G.; Milanetti, E. Molecular Mechanisms Behind Anti SARS-CoV-2 Action of Lactoferrin. Front. Mol. Biosci. 2021, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Bò, L.; Miotto, M.; Di Rienzo, L.; Milanetti, E.; Ruocco, G. Exploring the association between sialic acid and SARS-CoV-2 spike protein through a molecular dynamics-based approach. Front. Med. Technol. 2020, 2, 24. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Jandova, Z.; Vargiu, A.V.; Bonvin, A.M.J.J. Native or Non-Native Protein–Protein Docking Models? Molecular Dynamics to the Rescue. J. Chem. Theory Comput. 2021, 17, 5944–5954. [Google Scholar] [CrossRef]
- Radom, F.; Plückthun, A.; Paci, E. Assessment of ab initio models of protein complexes by molecular dynamics. PLoS Comput. Biol. 2018, 14, e1006182. [Google Scholar] [CrossRef]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations: A cross-docking study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Guterres, H.; Im, W. Improving protein-ligand docking results with high-throughput molecular dynamics simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Pfeiffenberger, E.; Bates, P.A. Refinement of protein-protein complexes in contact map space with metadynamics simulations. Proteins Struct. Funct. Bioinform. 2019, 87, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank. A Computer-Based Archival File for Macromolecular Structures. Eur. J. Biochem. 1977, 80, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.; Daujotyte, D.; Tollervey, J.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.; Damberger, F.; Allain, F. NMR structure of human TDP-43 tandem RRMs in complex with UG-rich RNA. Nat. Struct. Biol. 2013, 10, 980. [Google Scholar] [CrossRef]
- Lindahl, A.; Hess, S.V.D.; van der Spoel, D. GROMACS 2020 Source Code. Zenodo 2020. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Cheatham, T.E.I.; Miller, J.L.; Fox, T.; Darden, T.A.; Kollman, P.A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar] [CrossRef]
- Hayward, S.; Go, N. Collective Variable Description of Native Protein Dynamics. Annu. Rev. Phys. Chem. 1995, 46, 223–250. [Google Scholar] [CrossRef]
- Wolf, A.; Kirschner, K.N. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11·23S subdomain. J. Mol. Model. 2012, 19, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, R.D.; Quevedo, C.V.; Ruiz, D.D.; de Souza, O.N.; Barros, R.C. Clustering Molecular Dynamics Trajectories for Optimizing Docking Experiments. Comput. Intell. Neurosci. 2015, 2015, 916240. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176. [Google Scholar] [CrossRef] [PubMed]
- Gadkari, R.A.; Varughese, D.; Srinivasan, N. Recognition of Interaction Interface Residues in Low-Resolution Structures of Protein Assemblies Solely from the Positions of C-alpha Atoms. PLoS ONE 2009, 4, e4476. [Google Scholar] [CrossRef]
- Miotto, M.; Di Rienzo, L.; Gosti, G.; Bo’, L.; Parisi, G.; Piacentini, R.; Boffi, A.; Ruocco, G.; Milanetti, E. Inferring the stabilization effects of SARS-CoV-2 variants on the binding with ACE2 receptor. BioRxiv 2021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A1–A3, | I conformation | 223, 259, 260, 261, 262, 263 |
| prediction 3 | II conformation | 225, 226, 227, 228, 229, 256 |
| A1–A3, | I conformation | 224, 225, 259, 260, 261, 262 |
| prediction 13 | II conformation | 227, 228, 229, 247, 248, 249, 252, 253, 254, 267, 268, 269 |
| A1–A3, | I conformation | 227, 259 |
| prediction 14 | II conformation | 229, 246 |
| A1–A1, | I conformation | 221, 223, 231, 260 |
| prediction 4 | II conformation | 258, 259 |
| A3–A3, | I conformation | 260, 261 |
| prediction 5 | II conformation | 227, 228, 229 |
| A1–A5, | I conformation | 221, 223, 259 |
| prediction 1 | II conformation | 221, 222, 225, 226 |
| A1–A4, | I conformation | 223, 224 |
| prediction 3 | II conformation | 247, 254 |
| Number of Clusters | Fragment A | Fragment B |
|---|---|---|
| k = 2 | 0.4489 | 0.7095 |
| k = 3 | 0.4568 | 0.7066 |
| k = 4 | 0.5023 | 0.6795 |
| k = 5 | 0.5148 | 0.6393 |
| k = 6 | 0.4833 | 0.6735 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grassmann, G.; Miotto, M.; Di Rienzo, L.; Salaris, F.; Silvestri, B.; Zacco, E.; Rosa, A.; Tartaglia, G.G.; Ruocco, G.; Milanetti, E. A Computational Approach to Investigate TDP-43 RNA-Recognition Motif 2 C-Terminal Fragments Aggregation in Amyotrophic Lateral Sclerosis. Biomolecules 2021, 11, 1905. https://doi.org/10.3390/biom11121905
Grassmann G, Miotto M, Di Rienzo L, Salaris F, Silvestri B, Zacco E, Rosa A, Tartaglia GG, Ruocco G, Milanetti E. A Computational Approach to Investigate TDP-43 RNA-Recognition Motif 2 C-Terminal Fragments Aggregation in Amyotrophic Lateral Sclerosis. Biomolecules. 2021; 11(12):1905. https://doi.org/10.3390/biom11121905
Chicago/Turabian StyleGrassmann, Greta, Mattia Miotto, Lorenzo Di Rienzo, Federico Salaris, Beatrice Silvestri, Elsa Zacco, Alessandro Rosa, Gian Gaetano Tartaglia, Giancarlo Ruocco, and Edoardo Milanetti. 2021. "A Computational Approach to Investigate TDP-43 RNA-Recognition Motif 2 C-Terminal Fragments Aggregation in Amyotrophic Lateral Sclerosis" Biomolecules 11, no. 12: 1905. https://doi.org/10.3390/biom11121905
APA StyleGrassmann, G., Miotto, M., Di Rienzo, L., Salaris, F., Silvestri, B., Zacco, E., Rosa, A., Tartaglia, G. G., Ruocco, G., & Milanetti, E. (2021). A Computational Approach to Investigate TDP-43 RNA-Recognition Motif 2 C-Terminal Fragments Aggregation in Amyotrophic Lateral Sclerosis. Biomolecules, 11(12), 1905. https://doi.org/10.3390/biom11121905