
Acetylation, Phosphorylation, Ubiquitination (Oh My!): Following Post-Translational Modifications on the Ubiquitin Road


Abstract
1. Introduction
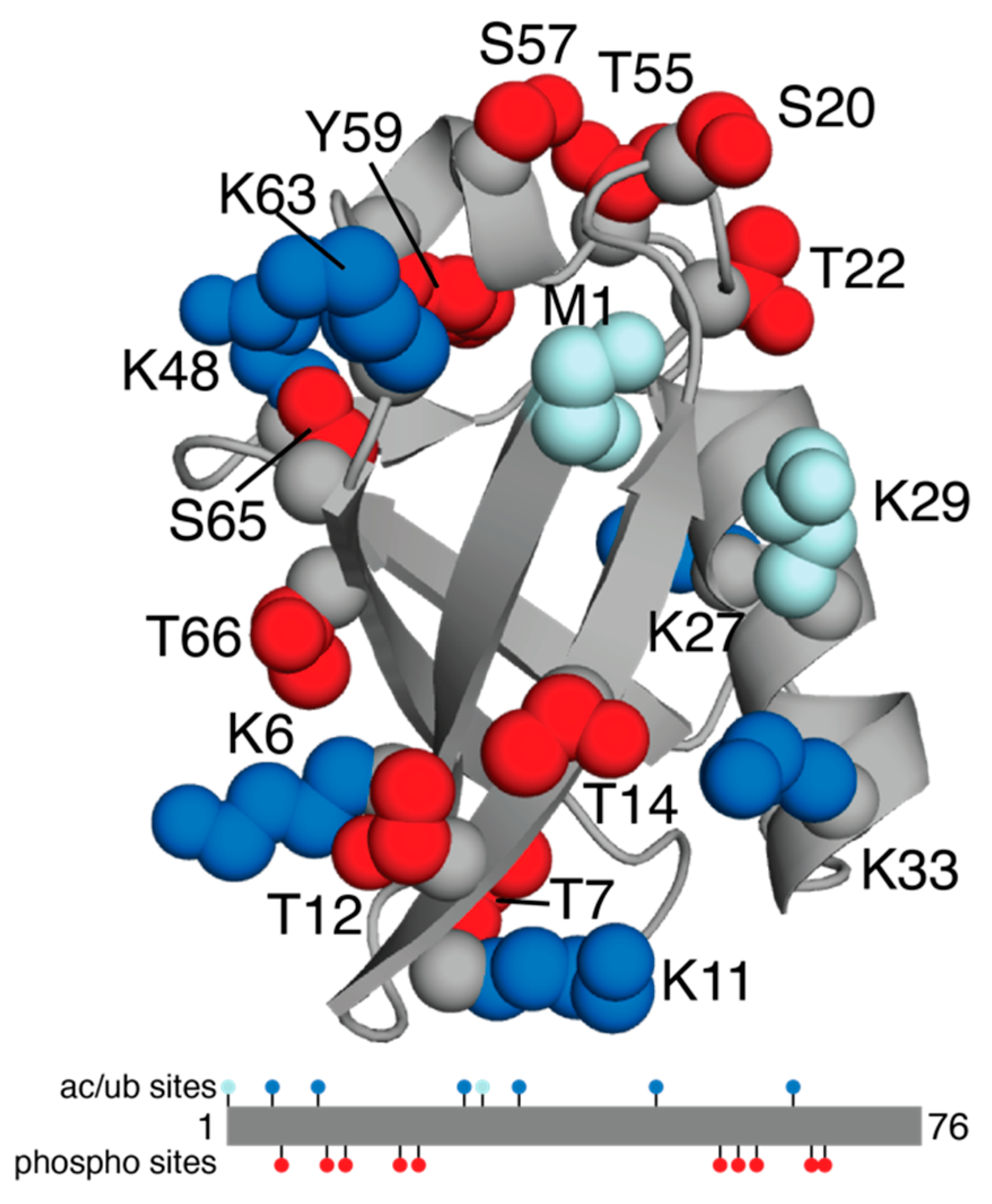
2. Ubiquitin
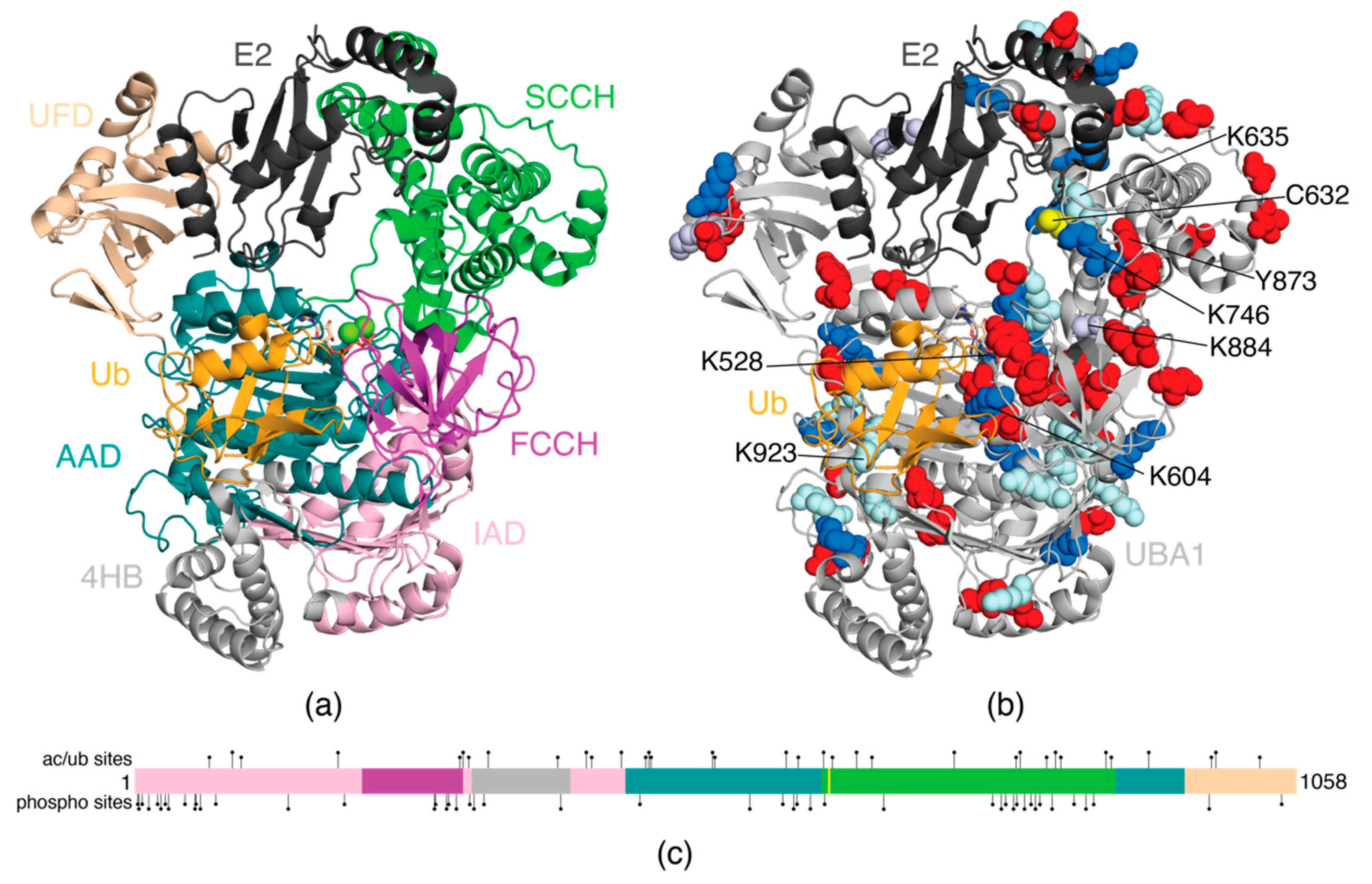
3. E1 Activating Enzyme (UBA1)
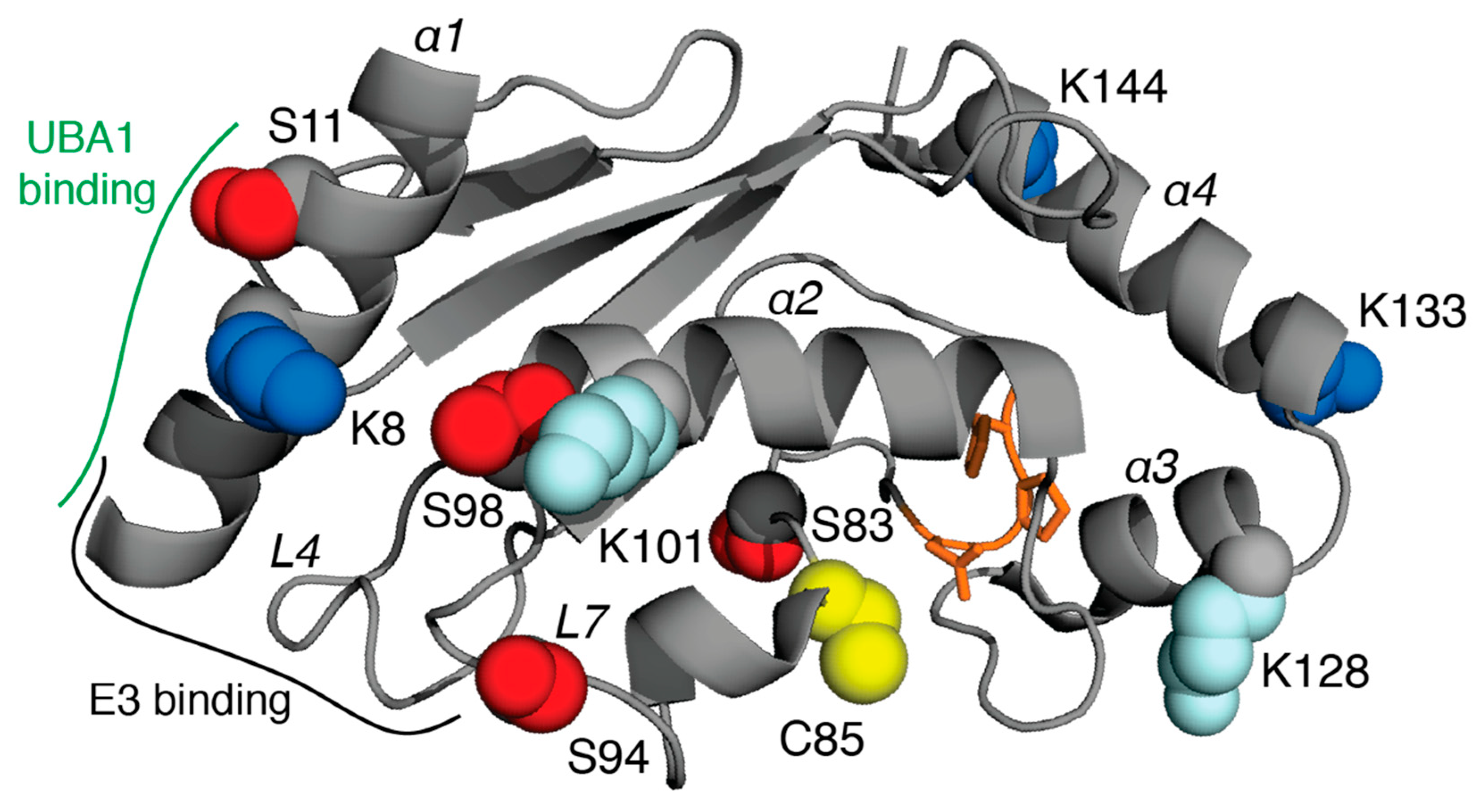
4. E2 Conjugating Enzymes
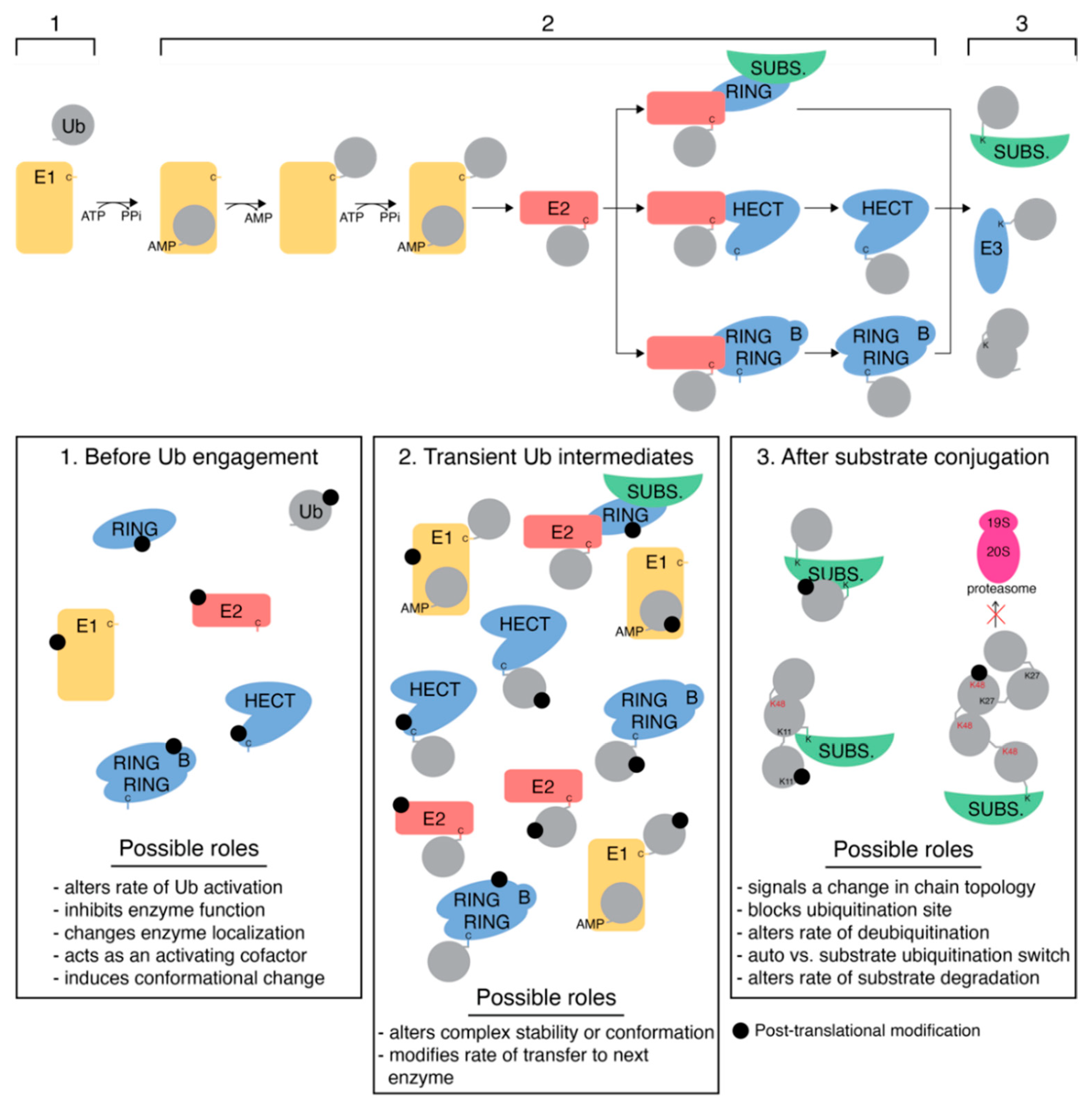
5. E3 Ligases
5.1. RING E3 Ligases
5.2. HECT Ligases
5.3. RBR Ligases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ye, Y.; Rape, M. Building Ubiquitin Chains: E2 Enzymes at Work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef] [PubMed]
- Buuh, Z.Y.; Lyu, Z.; Wang, R.E. Interrogating the Roles of Post-Translational Modifications of Non-Histone Proteins: Miniperspective. J. Med. Chem. 2018, 61, 3239–3252. [Google Scholar] [CrossRef] [PubMed]
- Rittinger, K.; Ikeda, F. Linear Ubiquitin Chains: Enzymes, Mechanisms and Biology. Open Biol. 2017, 7, 170026. [Google Scholar] [CrossRef] [PubMed]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and Its Interactions with the PI3K/AKT/MTOR Signalling Network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 36. [Google Scholar] [CrossRef]
- Kurtishi, A.; Rosen, B.; Patil, K.S.; Alves, G.W.; Møller, S.G. Cellular Proteostasis in Neurodegeneration. Mol. Neurobiol. 2019, 56, 3676–3689. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated Proteomic Analysis of Post-Translational Modifications by Serial Enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-F.; Wang, Y.-T.; Yen, H.-Y.; Tsou, C.-C.; Ku, W.-C.; Lin, P.-Y.; Chen, H.-Y.; Nesvizhskii, A.I.; Ishihama, Y.; Chen, Y.-J. Large-Scale Determination of Absolute Phosphorylation Stoichiometries in Human Cells by Motif-Targeting Quantitative Proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.N.; Jefferson, L.S.; Kimball, S.R. ERK and Akt Signaling Pathways Function through Parallel Mechanisms to Promote MTORC1 Signaling. Am. J. Physiol. Cell Physiol. 2011, 300, C1172–C1180. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of P53 Stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Mayat, R.; Kazazt, A.; Orent, M. Mdm2 Promotes the Rapid Degradation of P53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-W.; Lee, M.-S.; Camus, S.; Ghim, J.; Yang, M.-R.; Oh, W.; Ha, N.-C.; Lane, D.P.; Song, J. Differential Regulation of P53 and P21 by MKRN1 E3 Ligase Controls Cell Cycle Arrest and Apoptosis. EMBO J. 2009, 28, 2100–2113. [Google Scholar] [CrossRef] [PubMed]
- Allton, K.; Jain, A.K.; Herz, H.-M.; Tsai, W.-W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 Targets Endogenous P53 for Degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Bae, S.; Lee, J.Y.; Woo, S.R.; Cha, H.J.; Yoon, Y.; Suh, K.-S.; Lee, S.-J.; Park, I.-C.; Jin, Y.-W.; et al. E3 Ubiquitin Ligase Hades Negatively Regulates the Exonuclear Function of P53. Cell Death Differ. 2011, 18, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L.; et al. Cytoplasmic Destruction of P53 by the Endoplasmic Reticulum-Resident Ubiquitin Ligase ‘Synoviolin’. EMBO J. 2007, 26, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Dharmarajan, A.; Warrier, S.; Bishayee, A.; Kumar, A.P.; Sethi, G.; Ahn, K.S. Role of Histone Acetyltransferase Inhibitors in Cancer Therapy. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 125, pp. 149–191. ISBN 978-0-323-85315-6. [Google Scholar]
- José-Enériz, S.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Frogne, T.; Laenkholm, A.-V.; Lyng, M.B.; Henriksen, K.L.; Lykkesfeldt, A.E. Determination of HER2 Phosphorylation at Tyrosine 1221/1222 Improves Prediction of Poor Survival for Breast Cancer Patients with Hormone Receptor-Positive Tumors. Breast Cancer Res. 2009, 11, R11. [Google Scholar] [CrossRef]
- Gijsen, M.; King, P.; Perera, T.; Parker, P.J.; Harris, A.L.; Larijani, B.; Kong, A. HER2 Phosphorylation Is Maintained by a PKB Negative Feedback Loop in Response to Anti-HER2 Herceptin in Breast Cancer. PLoS Biol. 2010, 8, e1000563. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Murase, K.; Saito, M.; Imamura, M.; Oh, K. Mechanisms of Estrogen Receptor-α Upregulation in Breast Cancers. Med. Mol. Morphol. 2010, 43, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Pegueroles, J.; Herrmann, A.G.; Henstridge, C.M.; Muñoz, L.; Querol-Vilaseca, M.; Martín-Paniello, C.S.; Luque-Cabecerans, J.; Clarimon, J.; Belbin, O.; et al. Synaptic Phosphorylated α-Synuclein in Dementia with Lewy Bodies. Brain 2017, 140, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Cork, L.C.; Sternberger, N.H.; Sternberger, L.A.; Casanova, M.F.; Struble, R.G.; Price, D.L. Phosphorylated Neurofilament Antigens in Neurofibrillary Tangles in Alzheimer’s Disease. J. Neuropathol. Exp. Neurol. 1986, 45, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Sternberger, N.H.; Sternberger, L.A.; ULRICHt, J. Aberrant Neurofilament Phosphorylation in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1985, 82, 4274–4276. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Wisniewski, H.M.; Binder, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein X (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced Expression of Class II Histone Deacetylase Genes Is Associated with Poor Prognosis in Lung Cancer Patients. Int. J. Cancer 2004, 112, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The Acetylation of Tau Inhibits Its Function and Promotes Pathological Tau Aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.H.; Ko, J.; Donoghue, D.J. The Importance of Regulatory Ubiquitination in Cancer and Metastasis. Cell Cycle 2017, 16, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Pascovici, D.; Wu, J.X.; McKay, M.J.; Joseph, C.; Noor, Z.; Kamath, K.; Wu, Y.; Ranganathan, S.; Gupta, V.; Mirzaei, M. Clinically Relevant Post-Translational Modification Analyses—Maturing Workflows and Bioinformatics Tools. Int. J. Mol. Sci. 2018, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the Immune System. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Basar, M.A.; Beck, D.B.; Werner, A. Deubiquitylases in Developmental Ubiquitin Signaling and Congenital Diseases. Cell Death Differ. 2021, 28, 538–556. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-Mediated Phosphorylation of the Parkin Ubiquitin-like Domain Primes Mitochondrial Translocation of Parkin and Regulates Mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin Is Activated by PINK1-Dependent Phosphorylation of Ubiquitin at Ser65. Biochem. J. 2014, 460, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Altiok, S.; Batt, D.; Altiok, N.; Papautsky, A.; Downward, J.; Roberts, T.M.; Avraham, H. Heregulin Induces Phosphorylation of BRCA1 through Phosphatidylinositol 3-Kinase/AKT in Breast Cancer Cells. J. Biol. Chem. 1999, 274, 32274–32278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nelson, A.C.; Lyons, T.R.; Young, C.D.; Hansen, K.C.; Anderson, S.M.; Holt, J.T. AKT Regulates BRCA1 Stability in Response to Hormone Signaling. Mol. Cell Endocrinol. 2010, 319, 129–142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1–BARD1 Promotes Replication Fork Protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhou, W.; Jeon, M.; Demydenko, D.; Harada, Y.; Zhou, H.; Liu, Y.-C. Negative Regulation of the E3 Ubiquitin Ligase Itch via Fyn-Mediated Tyrosine Phosphorylation. Mol. Cell 2006, 21, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.; Gao, M.; Liu, Y.-C.; Karin, M. Activation of the E3 Ubiquitin Ligase Itch through a Phosphorylation-Induced Conformational Change. Proc. Natl. Acad. Sci. USA 2006, 103, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Shen, L.; Zhou, H.; Gao, J.; Pan, H.; Zheng, L.; Armstrong, B.; Peng, Y.; Peng, G.; Zhou, B.P.; et al. ITCH Nuclear Translocation and H1.2 Polyubiquitination Negatively Regulate the DNA Damage Response. Nucleic Acids Res. 2019, 47, 824–842. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin Chain Diversity at a Glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- French, M.E.; Koehler, C.F.; Hunter, T. Emerging Functions of Branched Ubiquitin Chains. Cell Discov. 2021, 7, 10. [Google Scholar] [CrossRef]
- Ohtake, F.; Saeki, Y.; Sakamoto, K.; Ohtake, K.; Nishikawa, H.; Tsuchiya, H.; Ohta, T.; Tanaka, K.; Kanno, J. Ubiquitin Acetylation Inhibits Polyubiquitin Chain Elongation. EMBO Rep. 2015, 16, 192–201. [Google Scholar] [CrossRef]
- Lacoursiere, R.E.; Shaw, G.S. Acetylated Ubiquitin Modulates the Catalytic Activity of the E1 Enzyme Uba1. Biochemistry 2021, 60, 1276–1285. [Google Scholar] [CrossRef]
- Wu, Q.; Cheng, Z.; Zhu, J.; Xu, W.; Peng, X.; Chen, C.; Li, W.; Wang, F.; Cao, L.; Yi, X.; et al. Suberoylanilide Hydroxamic Acid Treatment Reveals Crosstalks among Proteome, Ubiquitylome and Acetylome in Non-Small Cell Lung Cancer A549 Cell Line. Sci. Rep. 2015, 5, 9520. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.R.; Mohammed, S. Toward a Comprehensive Characterization of a Human Cancer Cell Phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics Connects Somatic Mutations to Signalling in Breast Cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative Phosphoproteomics Identifies Substrates and Functional Modules of Aurora and Polo-Like Kinase Activities in Mitotic Cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative Phosphoproteomics Reveals Widespread Full Phosphorylation Site Occupancy During Mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.J.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A Proteome-Wide, Quantitative Survey of In Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol. Cell Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villen, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-Scale Characterization of HeLa Cell Nuclear Phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic Investigations Reveal a Role for RNA Processing Factor THRAP3 in the DNA Damage Response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygün, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.P.; et al. Multiomic Analysis of the UV-Induced DNA Damage Response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Bordoli, M.R.; Yum, J.; Breitkopf, S.B.; Thon, J.N.; Italiano, J.E.; Xiao, J.; Worby, C.; Wong, S.-K.; Lin, G.; Edenius, M.; et al. A Secreted Tyrosine Kinase Acts in the Extracellular Environment. Cell 2014, 158, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Christensen, G.L.; Bøgebo, R.; Sheikh, S.P.; Gammeltoft, S.; Olsen, J.V.; Hansen, J.L. Quantitative Phosphoproteomics Dissection of Seven-Transmembrane Receptor Signaling Using Full and Biased Agonists. Mol. Cell Proteom. 2010, 9, 1540–1553. [Google Scholar] [CrossRef] [PubMed]
- Demirkan, G.; Yu, K.; Boylan, J.M.; Salomon, A.R.; Gruppuso, P.A. Phosphoproteomic Profiling of In Vivo Signaling in Liver by the Mammalian Target of Rapamycin Complex 1 (MTORC1). PLoS ONE 2011, 6, e21729. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A Quantitative Atlas of Mitotic Phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed]
- Heibeck, T.H.; Ding, S.-J.; Opresko, L.K.; Zhao, R.; Schepmoes, A.A.; Yang, F.; Tolmachev, A.V.; Monroe, M.E.; Camp, D.G.; Smith, R.D.; et al. An Extensive Survey of Tyrosine Phosphorylation Revealing New Sites in Human Mammary Epithelial Cells. J. Proteome Res. 2009, 8, 3852–3861. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klammer, M.; Kaminski, M.; Zedler, A.; Oppermann, F.; Blencke, S.; Marx, S.; Müller, S.; Tebbe, A.; Godl, K.; Schaab, C. Phosphosignature Predicts Dasatinib Response in Non-Small Cell Lung Cancer. Mol. Cell Proteom. 2012, 11, 651–668. [Google Scholar] [CrossRef]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in Tumors Induces Early and Sustained Phosphorylation Changes in Stress Kinase Pathways but Does Not Affect Global Protein Levels. Mol. Cell Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed]
- Rigbolt, K.T.G.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-Wide Temporal Characterization of the Proteome and Phosphoproteome of Human Embryonic Stem Cell Differentiation. Sci. Signal. 2011, 4, rs3. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, D.K.; Rigas, J.R.; Gerber, S.A. Quantitative Phosphoproteomic Profiling of Human Non-Small Cell Lung Cancer Tumors. J. Proteom. 2013, 91, 286–296. [Google Scholar] [CrossRef]
- Weinert, B.T.; Schölz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine Succinylation Is a Frequently Occurring Modification in Prokaryotes and Eukaryotes and Extensively Overlaps with Acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Cheng, D.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Peng, J. Phosphoproteomic Analysis of Human Brain by Calcium Phosphate Precipitation and Mass Spectrometry. J. Proteome Res. 2008, 7, 2845–2851. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.H.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Schölz, C.; Weinert, B.T.; Wagner, S.A.; Beli, P.; Miyake, Y.; Qi, J.; Jensen, L.J.; Streicher, W.; McCarthy, A.R.; Westwood, N.J.; et al. Acetylation Site Specificities of Lysine Deacetylase Inhibitors in Human Cells. Nat. Biotechnol. 2015, 33, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.K.; Gupta, R.; Baldus, L.; Lyon, D.; Narita, T.; Lammers, M.; Choudhary, C.; Weinert, B.T. Analysis of Human Acetylation Stoichiometry Defines Mechanistic Constraints on Protein Regulation. Nat. Commun. 2019, 10, 1055. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, Y.; Jin, M.; He, J.; Guli, A.; Yan, C.; Ding, S. Comprehensive Analysis of Lysine Acetylome Reveals a Site-Specific Pattern in Rapamycin-Induced Autophagy. J. Proteome Res. 2019, 18, 865–877. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, W.; Yang, J.S.; Cheng, Z.; Luo, H.; Lu, Z.; Tan, M.; Gu, W.; Zhao, Y. Quantitative Acetylome Analysis Reveals the Roles of SIRT1 in Regulating Diverse Substrates and Cellular Pathways. Mol. Cell Proteom. 2012, 11, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Ramírez-Torres, A.; Chiappe, D.; Luna-Peñaloza, J.; Fernandez-Reyes, F.C.; Arcos-Encarnación, B.; Contreras, S.; Encarnación-Guevara, S. Lysine Acetylation Stoichiometry and Proteomics Analyses Reveal Pathways Regulated by Sirtuin 1 in Human Cells. J. Biol. Chem. 2017, 292, 18129–18144. [Google Scholar] [CrossRef]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Schölz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/P300 Acetylome. Cell 2018, 174, 231–244.e12. [Google Scholar] [CrossRef]
- Moritz, A.; Li, Y.; Guo, A.; Villén, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt–RSK–S6 Kinase Signaling Networks Activated by Oncogenic Receptor Tyrosine Kinases. Sci. Signal. 2010, 3, ra64. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Villen, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; et al. Signaling Networks Assembled by Oncogenic EGFR and C-Met. Proc. Natl. Acad. Sci. USA 2008, 105, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined Preparation and Use of Anti-Diglycine Remnant (K-ε-GG) Antibody Enables Routine Quantification of 10,000s of Ubiquitination Sites in Single Proteomics Experiments. Mol. Cell Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Lumpkin, R.J.; Gu, H.; Zhu, Y.; Leonard, M.; Ahmad, A.S.; Clauser, K.R.; Meyer, J.G.; Bennett, E.J.; Komives, E.A. Site-Specific Identification and Quantitation of Endogenous SUMO Modifications under Native Conditions. Nat. Commun. 2017, 8, 1171. [Google Scholar] [CrossRef] [PubMed]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.-K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite Approach for Comprehensive Mapping of Lysine and N-Terminal Ubiquitination Sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-Wide Analysis of Ubiquitylation Dynamics Reveals a Key Role for PAF15 Ubiquitylation in DNA-Damage Bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Franz-Wachtel, M.; Eisler, S.A.; Krug, K.; Wahl, S.; Carpy, A.; Nordheim, A.; Pfizenmaier, K.; Hausser, A.; Macek, B. Global Detection of Protein Kinase D-Dependent Phosphorylation Events in Nocodazole-Treated Human Cells. Mol. Cell Proteom. 2012, 11, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Schreiber, T.B.; Daub, H. Dual Phosphoproteomics and Chemical Proteomics Analysis of Erlotinib and Gefitinib Interference in Acute Myeloid Leukemia Cells. J. Proteom. 2012, 75, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villén, J. Global Analysis of Phosphorylation and Ubiquitylation Cross-Talk in Protein Degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef]
- Vijay-Kumar, S.; Bugg, C.E.; Cook, W.J. Structure of Ubiquitin Refined at 1.8 Å Resolution. J. Mol. Biol. 1987, 194, 531–544. [Google Scholar] [CrossRef]
- Walser, F.; Mulder, M.P.C.; Bragantini, B.; Burger, S.; Gubser, T.; Gatti, M.; Botuyan, M.V.; Villa, A.; Altmeyer, M.; Neri, D.; et al. Ubiquitin Phosphorylation at Thr12 Modulates the DNA Damage Response. Mol. Cell 2020, 80, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Rodriguez-Mias, R.A.; Villen, J. Phosphorylation of Ubiquitin at Ser65 Affects Its Polymerization, Targets, and Proteome-Wide Turnover. EMBO Rep. 2015, 16, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Aguirre, J.D.; Spratt, D.E.; Bi, Y.; Jeffery, M.; Shaw, G.S.; O’Donoghue, P. Generation of Phospho-Ubiquitin Variants by Orthogonal Translation Reveals Codon Skipping. FEBS Lett. 2016, 590, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Hepowit, N.L.; Pereira, K.N.; Tumolo, J.M.; Chazin, W.J.; MacGurn, J.A. Identification of Ubiquitin Ser57 Kinases Regulating the Oxidative Stress Response in Yeast. eLife 2020, 9, e58155. [Google Scholar] [CrossRef] [PubMed]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like Protein Activation by E1 Enzymes: The Apex for Downstream Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.K.; Lima, C.D. Structure of a Ubiquitin E1-E2 Complex: Insights to E1-E2 Thioester Transfer. Mol. Cell 2013, 49, 884–896. [Google Scholar] [CrossRef]
- Williams, K.M.; Qie, S.; Atkison, J.H.; Salazar-Arango, S.; Alan Diehl, J.; Olsen, S.K. Structural Insights into E1 Recognition and the Ubiquitin-Conjugating Activity of the E2 Enzyme Cdc34. Nat. Commun. 2019, 10, 3296. [Google Scholar] [CrossRef]
- Lv, Z.; Rickman, K.A.; Yuan, L.; Williams, K.; Selvam, S.P.; Woosley, A.N.; Howe, P.H.; Ogretmen, B.; Smogorzewska, A.; Olsen, S.K.S. Pombe Uba1-Ubc15 Structure Reveals a Novel Regulatory Mechanism of Ubiquitin E2 Activity. Mol. Cell 2017, 65, 699–714.e6. [Google Scholar] [CrossRef]
- Huang, D.T.; Hunt, H.W.; Zhuang, M.; Ohi, M.D.; Holton, J.M.; Schulman, B.A. Basis for a Ubiquitin-like Protein Thioester Switch Toggling E1–E2 Affinity. Nature 2007, 445, 394–398. [Google Scholar] [CrossRef]
- Lv, Z.; Williams, K.M.; Yuan, L.; Atkison, J.H.; Olsen, S.K. Crystal Structure of a Human Ubiquitin E1–Ubiquitin Complex Reveals Conserved Functional Elements Essential for Activity. J. Biol. Chem. 2018, 293, 18337–18352. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Schindelin, H. Structural Insights into E1-Catalyzed Ubiquitin Activation and Transfer to Conjugating Enzymes. Cell 2008, 134, 268–278. [Google Scholar] [CrossRef]
- Schäfer, A.; Kuhn, M.; Schindelin, H. Structure of the Ubiquitin-activating Enzyme Loaded with Two Ubiquitin Molecules. Acta Cryst. D 2014, 70, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Yuan, L.; Atkison, J.H.; Aldana-Masangkay, G.; Chen, Y.; Olsen, S.K. Domain Alternation and Active Site Remodeling Are Conserved Structural Features of Ubiquitin E1. J. Biol. Chem. 2017, 292, 12089–12099. [Google Scholar] [CrossRef] [PubMed]
- Hann, Z.S.; Ji, C.; Olsen, S.K.; Lu, X.; Lux, M.C.; Tan, D.S.; Lima, C.D. Structural Basis for Adenylation and Thioester Bond Formation in the Ubiquitin E1. Proc. Natl. Acad. Sci. USA 2019, 116, 15475–15484. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Tokgöz, Z.; Bohnsack, R.N.; Haas, A.L. Pleiotropic Effects of ATP·Mg2+ Binding in the Catalytic Cycle of Ubiquitin-Activating Enzyme. J. Biol. Chem. 2006, 281, 14729–14737. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Campbell, S.J.; Edwards, R.A.; Leung, C.C.Y.; Neculai, D.; Hodge, C.D.; Dhe-Paganon, S.; Glover, J.N.M. Molecular Insights into the Function of RING Finger (RNF)-Containing Proteins HRNF8 and HRNF168 in Ubc13/Mms2-Dependent Ubiquitylation. J. Biol. Chem. 2012, 287, 23900–23910. [Google Scholar] [CrossRef]
- Hodge, C.D.; Spyracopoulos, L.; Glover, J.N.M. Ubc13: The Lys63 Ubiquitin Chain Building Machine. Oncotarget 2016, 7, 64471–64504. [Google Scholar] [CrossRef]
- Middleton, A.J.; Zhu, J.; Day, C.L. The RING Domain of RING Finger 12 Efficiently Builds Degradative Ubiquitin Chains. J. Mol. Biol. 2020, 432, 3790–3801. [Google Scholar] [CrossRef]
- Buetow, L.; Gabrielsen, M.; Anthony, N.G.; Dou, H.; Patel, A.; Aitkenhead, H.; Sibbet, G.J.; Smith, B.O.; Huang, D.T. Activation of a Primed RING E3-E2–Ubiquitin Complex by Non-Covalent Ubiquitin. Mol. Cell 2015, 58, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Kamadurai, H.B.; Souphron, J.; Scott, D.C.; Duda, D.M.; Miller, D.J.; Stringer, D.; Piper, R.C.; Schulman, B.A. Insights into Ubiquitin Transfer Cascades from a Structure of a UbcH5B~Ubiquitin-HECTNEDD4L Complex. Mol. Cell 2009, 36, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kinnucan, E.; Wang, G.; Beaudenon, S.; Howley, P.M.; Huibregtse, J.M.; Pavletich, N.P. Structure of an E6AP-UbcH7 Complex: Insights into Ubiquitination by the E2-E3 Enzyme Cascade. Science 1999, 286, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 Enzymes: More than Just Middle Men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, D.M.; Stoll, K.E.; Klevit, R.E. E2s: Structurally Economical and Functionally Replete. Biochem. J. 2011, 433, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhu, J.; Li, H.; Zhu, L. Structural Analysis of Recombinant Human Ubiquitin-Conjugating Enzyme UbcH5c. Acta Pharm. Sin. B 2017, 7, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-D.; Ye, F.; Zhang, D.-Z.; Hu, P.; Ren, H.; Li, S.-L. ITRAQ Quantitative Analysis of Multidrug Resistance Mechanisms in Human Gastric Cancer Cells. J. Biomed. Biotechnol. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Condos, T.E.; Dunkerley, K.M.; Freeman, E.A.; Barber, K.R.; Aguirre, J.D.; Chaugule, V.K.; Xiao, Y.; Konermann, L.; Walden, H.; Shaw, G.S. Synergistic Recruitment of UbcH7~Ub and Phosphorylated Ubl Domain Triggers Parkin Activation. EMBO J. 2018, 37, e100014. [Google Scholar] [CrossRef] [PubMed]
- Dove, K.K.; Olszewski, J.L.; Martino, L.; Duda, D.M.; Wu, X.S.; Miller, D.J.; Reiter, K.H.; Rittinger, K.; Schulman, B.A.; Klevit, R.E. Structural Studies of HHARI/UbcH7~Ub Reveal Unique E2~Ub Conformational Restriction by RBR RING1. Structure 2017, 25, 890–900.e5. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, E.; Dursun, B.; Lesage, S.; Hanagasi, H.A.; Sevinc, G.; Honore, A.; Bilgic, B.; Gürvit, H.; Dogu, O.; Kaleagası, H.; et al. Genetic Bases and Phenotypes of Autosomal Recessive Parkinson Disease in a Turkish Population: Autosomal Recessive Parkinson Disease in Turkey. Eur. J. Neurol. 2012, 19, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Sakata, E.; Satoh, T.; Yamamoto, S.; Yamaguchi, Y.; Yagi-Utsumi, M.; Kurimoto, E.; Tanaka, K.; Wakatsuki, S.; Kato, K. Crystal Structure of UbcH5b~Ubiquitin Intermediate: Insight into the Formation of the Self-Assembled E2~Ub Conjugates. Structure 2010, 18, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Bosanac, I.; Phu, L.; Pan, B.; Zilberleyb, I.; Maurer, B.; Dixit, V.M.; Hymowitz, S.G.; Kirkpatrick, D.S. Modulation of K11-Linkage Formation by Variable Loop Residues within UbcH5A. J. Mol. Biol. 2011, 408, 420–431. [Google Scholar] [CrossRef]
- Page, R.C.; Pruneda, J.N.; Amick, J.; Klevit, R.E.; Misra, S. Structural Insights into the Conformation and Oligomerization of E2~Ubiquitin Conjugates. Biochemistry 2012, 51, 4175–4187. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Lissounov, A.; Christensen, D.E.; Hoyt, D.W.; Klevit, R.E. A UbcH5/Ubiquitin Noncovalent Complex Is Required for Processive BRCA1-Directed Ubiquitination. Mol. Cell 2006, 21, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Bocik, W.E.; Sircar, A.; Gray, J.J.; Tolman, J.R. Mechanism of Polyubiquitin Chain Recognition by the Human Ubiquitin Conjugating Enzyme Ube2g2. J. Biol. Chem. 2011, 286, 3981–3991. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.S.; Ellison, M.J.; Barber, K.R.; Williams, R.S.; Huzil, J.T.; McKenna, S.; Ptak, C.; Glover, M.; Shaw, G.S. Structure of a Conjugating Enzyme-Ubiquitin Thiolester Intermediate Reveals a Novel Role for the Ubiquitin Tail. Structure 2001, 9, 897–904. [Google Scholar] [CrossRef]
- Huang, H.; Ceccarelli, D.F.; Orlicky, S.; St-Cyr, D.J.; Ziemba, A.; Garg, P.; Plamondon, S.; Auer, M.; Sidhu, S.; Marinier, A.; et al. E2 Enzyme Inhibition by Stabilization of a Low-Affinity Interface with Ubiquitin. Nat. Chem. Biol. 2014, 10, 156–163. [Google Scholar] [CrossRef]
- Dove, K.K.; Stieglitz, B.; Duncan, E.D.; Rittinger, K.; Klevit, R.E. Molecular Insights into RBR E3 Ligase Ubiquitin Transfer Mechanisms. EMBO Rep. 2016, 17, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Plechanovová, A.; Jaffray, E.G.; Tatham, M.H.; Naismith, J.H.; Hay, R.T. Structure of a RING E3 Ligase and Ubiquitin-Loaded E2 Primed for Catalysis. Nature 2012, 489, 115–120. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Rajput, A.; Sanishvili, R.; Dobaczewska, M.K.; Ware, C.F.; Mace, P.D.; Riedl, S.J. Structure of a HOIP/E2~ubiquitin Complex Reveals RBR E3 Ligase Mechanism and Regulation. Nature 2016, 529, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, S.; Bhattacharyya, M.; Feiler, C.; Rape, M.; Kuriyan, J. Crystal Structure of a Ube2S-Ubiquitin Conjugate. PLoS ONE 2016, 11, e0147550. [Google Scholar] [CrossRef] [PubMed]
- Pruneda, J.N.; Smith, F.D.; Daurie, A.; Swaney, D.L.; Villén, J.; Scott, J.D.; Stadnyk, A.W.; Le Trong, I.; Stenkamp, R.E.; Klevit, R.E.; et al. E2~Ub Conjugates Regulate the Kinase Activity of Shigella Effector OspG during Pathogenesis. EMBO J. 2014, 33, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Middleton, A.J.; Day, C.L. The Molecular Basis of Lysine 48 Ubiquitin Chain Synthesis by Ube2K. Sci. Rep. 2015, 5, 16793. [Google Scholar] [CrossRef] [PubMed]
- Budhidarmo, R.; Nakatani, Y.; Day, C.L. RINGs Hold the Key to Ubiquitin Transfer. Trends Biochem. Sci. 2012, 37, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, M.; Walden, H. Structural Basis of Generic versus Specific E2–RING E3 Interactions in Protein Ubiquitination. Protein Sci. 2019, 28, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Polo, S.; Maspero, E. HECT E3 Ligases: A Tale With Multiple Facets. Front. Physiol. 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.; Distel, B. Regulating the Human HECT E3 Ligases. Cell Mol. Life Sci. 2018, 75, 3121–3141. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 Reactivity Profile Reveals Parkin and HHARI to Be RING/HECT Hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 Ubiquitin Ligases: New Structures, New Insights, New Questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, L.; Li, Z.; Lane, W.S.; Chen, J. ATM Activates P53 by Regulating MDM2 Oligomerization and E3 Processivity. EMBO J. 2009, 28, 3857–3867. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.; Kutchukian, P.S.; Verdine, G.L.; Huber, R.; Holak, T.A.; Lee, K.W.; Popowicz, G.M. Structure of the Stapled P53 Peptide Bound to Mdm2. J. Am. Chem. Soc. 2012, 134, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.A.; Worrall, E.G.; Lin, Y.; Landre, V.; Pettersson, S.; Blackburn, E.; Walkinshaw, M.; Muller, P.; Vojtesek, B.; Ball, K.; et al. Phosphomimetic Mutation of the N-Terminal Lid of MDM2 Enhances the Polyubiquitination of P53 through Stimulation of E2-Ubiquitin Thioester Hydrolysis. J. Mol. Biol. 2015, 427, 1728–1747. [Google Scholar] [CrossRef] [PubMed]
- Worrall, E.G.; Wawrzynow, B.; Worrall, L.; Walkinshaw, M.; Ball, K.L.; Hupp, T.R. Regulation of the E3 Ubiquitin Ligase Activity of MDM2 by an N-Terminal Pseudo-Substrate Motif. J. Chem. Biol. 2009, 2, 113–129. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A Phosphatidylinositol 3-Kinase/Akt Pathway Promotes Translocation of Mdm2 from the Cytoplasm to the Nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Zhou, B.P.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M.-C. HER-2/Neu Induces P53 Ubiquitination via Akt-Mediated MDM2 Phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Leal, J.F.M.; Seger, R.; Taya, Y.; Oren, M. Cross-Talk between Akt, P53 and Mdm2: Possible Implications for the Regulation of Apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xiong, J.; Yi, S.; Zhang, H.; Zhou, S.; Gu, L.; Zhou, M. FKBP12 Enhances Sensitivity to Chemotherapy-Induced Cancer Cell Apoptosis by Inhibiting MDM2. Oncogene 2017, 36, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Nihira, N.T.; Ogura, K.; Shimizu, K.; North, B.J.; Zhang, J.; Gao, D.; Inuzuka, H.; Wei, W. Acetylation-Dependent Regulation of MDM2 E3 Ligase Activity Dictates Its Oncogenic Function. Sci. Signal. 2017, 10, eaai8026. [Google Scholar] [CrossRef]
- Wang, X.; Taplick, J.; Geva, N.; Oren, M. Inhibition of P53 Degradation by Mdm2 Acetylation. FEBS Lett. 2004, 561, 195–201. [Google Scholar] [CrossRef]
- Kales, S.C.; Ryan, P.E.; Nau, M.M.; Lipkowitz, S. Cbl and Human Myeloid Neoplasms: The Cbl Oncogene Comes of Age. Cancer Res. 2010, 70, 4789–4794. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Buetow, L.; Hock, A.; Sibbet, G.J.; Vousden, K.H.; Huang, D.T. Structural Basis for Autoinhibition and Phosphorylation-Dependent Activation of c-Cbl. Nat. Struct. Mol. Biol. 2012, 19, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Loh, M.L.; Sakai, D.S.; Flotho, C.; Kang, M.; Fliegauf, M.; Archambeault, S.; Mullighan, C.G.; Chen, L.; Bergstraesser, E.; Bueso-Ramos, C.E.; et al. Mutations in CBL Occur Frequently in Juvenile Myelomonocytic Leukemia. Blood 2009, 114, 1859–1863. [Google Scholar] [CrossRef]
- Grand, F.H.; Hidalgo-Curtis, C.E.; Ernst, T.; Zoi, K.; Zoi, C.; McGuire, C.; Kreil, S.; Jones, A.; Score, J.; Metzgeroth, G.; et al. Frequent CBL Mutations Associated with 11q Acquired Uniparental Disomy in Myeloproliferative Neoplasms. Blood 2009, 113, 6182–6192. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.S.; Reddy, M.M.; Croteau, N.J.; Walz, C.; Weisbach, H.; Podar, K.; Band, H.; Carroll, M.; Reiter, A.; Larson, R.A.; et al. Novel Oncogenic Mutations of CBL in Human Acute Myeloid Leukemia That Activate Growth and Survival Pathways Depend on Increased Metabolism. J. Biol. Chem. 2010, 285, 32596–32605. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Buetow, L.; Gabrielsen, M.; Lilla, S.; Sibbet, G.J.; Sumpton, D.; Zanivan, S.; Hedley, A.; Clark, W.; Huang, D.T. E3 Ligase-Inactivation Rewires CBL Interactome to Elicit Oncogenesis by Hijacking RTK–CBL–CIN85 Axis. Oncogene 2021, 40, 2149–2164. [Google Scholar] [CrossRef] [PubMed]
- Meisner, H.; Conway, B.R.; Hartley, D.; Czech, M.P. Interactions of Cbl with Grb2 and Phosphatidylinositol 3’-Kinase in Activated Jurkat Cells. Mol. Cell Biol. 1995, 15, 3571–3578. [Google Scholar] [CrossRef] [PubMed]
- Miura-Shimura, Y.; Duan, L.; Rao, N.L.; Reddi, A.L.; Shimura, H.; Rottapel, R.; Druker, B.J.; Tsygankov, A.; Band, V.; Band, H. Cbl-Mediated Ubiquitinylation and Negative Regulation of Vav. J. Biol. Chem. 2003, 278, 38495–38504. [Google Scholar] [CrossRef] [PubMed]
- Buitrago, L.; Langdon, W.Y.; Sanjay, A.; Kunapuli, S.P. Tyrosine Phosphorylated C-Cbl Regulates Platelet Functional Responses Mediated by Outside-in Signaling. Blood 2011, 118, 5631–5640. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.-C.; Klevit, R.E. Structure of a BRCA1– BARD1 Heterodimeric RING–RING Complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Morris, J.R.; Pangon, L.; Boutell, C.; Katagiri, T.; Keep, N.H.; Solomon, E. Genetic Analysis of BRCA1 Ubiquitin Ligase Activity and Its Relationship to Breast Cancer Susceptibility. Hum. Mol. Genet. 2006, 15, 599–606. [Google Scholar] [CrossRef]
- Madaan, K.; Kaushik, D.; Verma, T. Hydroxyurea: A Key Player in Cancer Chemotherapy. Expert Rev. Anticancer Ther. 2012, 12, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Hinton, C.V.; Fitzgerald, L.D.; Thompson, M.E. Phosphatidylinositol 3-Kinase/Akt Signaling Enhances Nuclear Localization and Transcriptional Activity of BRCA1. Exp. Cell Res. 2007, 313, 1735–1744. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ismail, N.A.S.; Baines, D.L.; Wilson, S.M. The Phosphorylation of Endogenous Nedd4-2 In Na+—Absorbing Human Airway Epithelial Cells. Eur. J. Pharmacol. 2014, 732, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-H.; Dinudom, A.; Sanchez-Perez, A.; Kumar, S.; Cook, D.I. Akt Mediates the Effect of Insulin on Epithelial Sodium Channels by Inhibiting Nedd4-2*. J. Biol. Chem. 2007, 282, 29866–29873. [Google Scholar] [CrossRef]
- Arroyo, J.P.; Lagnaz, D.; Ronzaud, C.; Vázquez, N.; Ko, B.S.; Moddes, L.; Ruffieux-Daidié, D.; Hausel, P.; Koesters, R.; Yang, B.; et al. Nedd4-2 Modulates Renal Na +-Cl− Cotransporter via the Aldosterone-SGK1-Nedd4-2 Pathway. J. Am. Soc. Nephrol. 2011, 22, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, J.; You, G. The Mechanistic Links between Insulin and Human Organic Anion Transporter 4. Int. J. Pharm. 2019, 555, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; You, G. SGK1/Nedd4-2 Signaling Pathway Regulates the Activity of Human Organic Anion Transporters 3. Biopharm. Drug Dispos. 2017, 38, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Snyder, P.M.; Olson, D.R.; Kabra, R.; Zhou, R.; Steines, J.C. CAMP and Serum and Glucocorticoid-Inducible Kinase (SGK) Regulate the Epithelial Na+ Channel through Convergent Phosphorylation of Nedd4-2. J. Biol. Chem. 2004, 279, 45753–45758. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Alarcón, C.; Sapkota, G.; Rahman, S.; Chen, P.-Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massagué, J. Ubiquitin Ligase Nedd4L Targets Activated Smad2/3 to Limit TGF-β Signaling. Mol. Cell 2009, 36, 457–468. [Google Scholar] [CrossRef]
- Pohl, P.; Joshi, R.; Petrvalska, O.; Obsil, T.; Obsilova, V. 14-3-3-Protein Regulates Nedd4-2 by Modulating Interactions between HECT and WW Domains. Commun. Biol. 2021, 4, 899. [Google Scholar] [CrossRef] [PubMed]
- Hallows, K.R.; Bhalla, V.; Oyster, N.M.; Wijngaarden, M.A.; Lee, J.K.; Li, H.; Chandran, S.; Xia, X.; Huang, Z.; Chalkley, R.J.; et al. Phosphopeptide Screen Uncovers Novel Phosphorylation Sites of Nedd4-2 That Potentiate Its Inhibition of the Epithelial Na+ Channel. J. Biol. Chem. 2010, 285, 21671–21678. [Google Scholar] [CrossRef] [PubMed]
- Gwoździńska, P.; Buchbinder, B.A.; Mayer, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadász, I. Hypercapnia Impairs ENaC Cell Surface Stability by Promoting Phosphorylation, Polyubiquitination and Endocytosis of β-ENaC in a Human Alveolar Epithelial Cell Line. Front. Immunol. 2017, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.M.; Chirieleison, S.M.; Abbott, D.W. An IκB Kinase-Regulated Feedforward Circuit Prolongs Inflammation. Cell Rep. 2015, 12, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.M.; Chen, Y.; Xiao, T.S.; Abbott, D.W. Phosphorylation of the E3 Ubiquitin Protein Ligase ITCH Diminishes Binding to Its Cognate E2 Ubiquitin Ligase. J. Biol. Chem. 2018, 293, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.-Y.; Thomas, A.; Zhou, M.; Zhang, Y.E. Phosphorylation of SMURF2 by ATM Exerts a Negative Feedback Control of DNA Damage Response. J. Biol. Chem. 2020, 295, 18485–18493. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Gupta, A.; Naiya, T.; Das, G.; Neogi, R.; Datta, S.; Mukherjee, S.; Das, S.K.; Ray, K.; Ray, J. Molecular Pathogenesis of Parkinson’s Disease: Identification of Mutations in the Parkin Gene in Indian Patients. Parkinsonism Relat. Disord. 2006, 12, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.-C.; Lin, S.-Z.; Chiang, M.-F.; Pang, C.-Y.; Chen, S.-Y.; Hsin, Y.-L.; Thajeb, P.; Lee, Y.-J.; Li, H. Early-Onset Parkinson’s Disease in a Chinese Population: 99mTc-TRODAT-1 SPECT, Parkin Gene Analysis and Clinical Study. Parkinsonism Relat. Disord. 2005, 11, 173–180. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Birsa, N.; Norkett, R.; Wauer, T.; Mevissen, T.E.T.; Wu, H.-C.; Foltynie, T.; Bhatia, K.; Hirst, W.D.; Komander, D.; Plun-Favreau, H.; et al. Lysine 27 Ubiquitination of the Mitochondrial Transport Protein Miro Is Dependent on Serine 65 of the Parkin Ubiquitin Ligase. J. Biol. Chem. 2014, 289, 14569–14582. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 Is Activated by Mitochondrial Membrane Potential Depolarization and Stimulates Parkin E3 Ligase Activity by Phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Heo, J.-M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining Roles of PARKIN and Ubiquitin Phosphorylation by PINK1 in Mitochondrial Quality Control Using a Ubiquitin Replacement Strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.-M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227.e8. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Ma, C.; Liu, Z.; Shi, J.; Zheng, G.; Zhang, C.; Zhang, Z. Antimony Exposure Promotes Bladder Tumor Cell Growth by Inhibiting PINK1-Parkin-Mediated Mitophagy. Ecotoxicol. Environ. Saf. 2021, 221, 112420. [Google Scholar] [CrossRef] [PubMed]
- Roverato, N.D.; Sailer, C.; Catone, N.; Aichem, A.; Stengel, F.; Groettrup, M. Parkin Is an E3 Ligase for the Ubiquitin-like Modifier FAT10, Which Inhibits Parkin Activation and Mitophagy. Cell Rep. 2021, 34, 108857. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, J.J.; Han, S.-A.; Fan, Y.; Guo, L.-S.; Aziz, K.; Nowsheen, S.; Kim, S.S.; Park, S.-Y.; Luo, Q.; et al. The AMPK–Parkin Axis Negatively Regulates Necroptosis and Tumorigenesis by Inhibiting the Necrosome. Nat. Cell Biol. 2019, 21, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.S.; Lee, Y.; Shin, J.-H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the C-Abl Protein Tyrosine Kinase Inhibits Parkin’s Ubiquitination and Protective Function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Bowman, J.; Rodgers, M.A.; Shi, M.; Amatya, R.; Hostager, B.; Iwai, K.; Gao, S.-J.; Jung, J.U. Posttranslational Modification of HOIP Blocks Toll-Like Receptor 4-Mediated Linear-Ubiquitin-Chain Formation. mBio 2015, 6, e01777-15. [Google Scholar] [CrossRef]
- Sanada, T.; Kim, M.; Mimuro, H.; Suzuki, M.; Ogawa, M.; Oyama, A.; Ashida, H.; Kobayashi, T.; Koyama, T.; Nagai, S.; et al. The Shigella Flexneri Effector OspI Deamidates UBC13 to Dampen the Inflammatory Response. Nature 2012, 483, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Zhang, X.; Jin, M.; Xu, L.; Wang, C.; Xia, Z.; Zhu, Y. Complex Structure of OspI and Ubc13: The Molecular Basis of Ubc13 Deamidation and Convergence of Bacterial and Host E2 Recognition. PLoS Pathog. 2013, 9, e1003322. [Google Scholar] [CrossRef] [PubMed]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of Ubiquitin Promotes Serine Ubiquitination and Impairs Conventional Ubiquitination. Cell 2016, 167, 1636–1649.e13. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | UniProt | Acetylation + | Phosphorylation + | Ubiquitination + | Disease |
|---|---|---|---|---|---|---|
| Ubiquitin | ||||||
| UBC | Ubiquitin | P0CG48 | K6, K11, K27, K33, K48, K63 | T7, T12, T14, S20, T22, T55, S57, Y59, S65, T66 | M1, K6, K11, K27, K29, K33, K48, K63 | Multiple myeloma, leukemia, colorectal cancer |
| E1 Activating Enzyme | ||||||
| UBA1 | UBE1 | P22314 | K68, K89, K185, K385, K465, K470, K526, K528, K593, K604, K627, K657, K671, K746, K838, K843, K884, K980, K984, K1024 | S3, S4, S7, S13, S21, S24, S28, S31, S46, Y55, S56, Y60, S74, S140, T191, Y273, T274, S284, Y286, S293, S305, S309, T318, Y388, S460, Y560, Y590, T600, T603, T615, S628, T682, S781, T789, S793, T800, S803, S810, S816, S820, S824, S835, S855, S866, Y873, Y978, S1044 | K68, K89, K97, K185, K296, K299, K304, K322, K385, K411, K416, K443, K465, K468, K470, K526, K528, K593, K604, K627, K635, K657, K671, K746, K802, K806, K830, K838, K843, K889, K923, K980 | Melanoma, lymphoma, leukemia, breast, colorectal, and gastric cancers |
| E2 Conjugating Enzymes | ||||||
| UBE2A | UBE2A, RAD6A | P49459 | K66 | S120, S142, S148 | K66, K75 | Breast, lung cancer |
| UBE2B | UBE2B, RAD6B | P63146 | K66 | T69, S120 | K66 | Leukemia |
| UBE2C | UBE2C, UbcH10 | O00762 | K18, K119, K121, K164 | S3, T11, S51, S87, T160 | K80, K119, K121, K164, K172 | Leukemia |
| UBE2D1 | UbcH5a | P51668 | K144 | S83, Y145 | K144 | Lung cancer |
| UBE2D2 | UbcH5b | P62837 | K8, K144 | S83, S94, T98 | K8, K101, K128, K144 | Lung cancer |
| UBE2D3 | UbcH5c | P61077 | K8, K133, K144 | S11, S83, S94, T98 | K8, K101, K128, K133, K144 | Breast, lung cancers, leukemia |
| UBE2D4 | UbcH5d | Q9Y2X8 | K8, K144 | K144 | Leukemia, multiple myeloma | |
| UBE2E1 | UbcH6 | P51965 | K43, K50, K136 | S2, S6, S9, T28, S46, Y77 | K24, K43, K50, K54, K72, K136 | Breast, colorectal cancers, leukemia |
| UBE2E2 | UbcH8 (ISG15) | Q96LR5 | K48, K52 | T3, S11, S13, T14, S15, S18, S19, T49, S54, Y85 | K62, K144 | Breast, colorectal, lung, gastric cancers, leukemia |
| UBE2E3 | UbcH9 | Q969T4 | K50, K58 | S3, S8, S12, S19, Y91 | K39, K58, K68, K150 | Breast, lung, gastric cancers, leukemia |
| UBE2F | UBE2F, NCE2 (NEDD8) | Q969M7 | K7, K9 | S31, T85, S124, Y179 | K7 | Breast, brain, lung, gastric cancers |
| UBE2G1 | Ubc7 | P62253 | T2, S6, Y65, T76, Y102, Y104 | K19, K63, K73, K101, K106, K163 | Multiple myeloma | |
| UBE2G2 | Ubc7 | P60604 | K7, K142, K153, K156, K161 | Leukemia | ||
| UBE2H | UbcH2, E2-20K | P62256 | K8, K60, K64, K147 | S2, S3, S5, T13, S65, S166, S169 | K17, K60, K64, K147 | Breast cancer |
| UBE2I | Ubc9 (SUMO) | P63279 | K30, K48, K59, K65, K74, K146 | T35, S70, S71 | K18, K49, K59, K65, K74 | Bone cancer, leukemia |
| UBE2J1 | NCUBE1 | Q9Y385 | K8, K17 | Y5, S9, S51, S184, S251, S266, T267, S268, T282, T295, Y307, Y312 | K8, K143, K164, K177, K186, K194 | Breast, lung cancers, leukemia, multiple myeloma |
| UBE2J2 | NCUBE2 | Q8N2K1 | K18 | Y31, Y46 | K18, K64, K139, K152, K154, K168 | Esophageal cancer |
| UBE2K | E2-25K, HIP-2 | P61086 | K14, K18, K72, K142, K164, K165 | T49, S158 *, S159, Y162 *, T163 *, S185 | K14, K18, K24, K28, K61, K72, K97, K142, K164, K165 | Leukemia, lung cancer |
| UBE2L3 | UbcH7 | P68036 | K9, K20, K64, K73, K82, K96, K131, K138, K145 | Y75, Y129, Y147 | K9, K20, K48, K64, K67, K71, K73, K82, K96, K100, K131, K138, K145, K150 | Lung, colorectal cancers, lymphoma, leukemia |
| UBE2L6 | UbcH8 (ISG15) | O14933 | K138 | S26, S153 | K9, K16, K17, K96, K138 | Breast cancer, leukemia, neuroblastoma |
| UBE2M | Ubc12 (NEDD8) | P61081 | K3, K8, K36, K45, K72 | S6, T20, S23, S28, T46, S50, S52 *, Y86, Y172, Y177 | K3, K8, K26, K36, K45, K61, K72, K75, K81, K92, K94 | Breast, lung cancers, leukemia, lymphoma |
| UBE2N | Ubc13 | P61088 | K10, K24, K53, K82, K92, K94 | Y34, S45, T139, T144, Y147 | K10, K24, K68, K74, K82, K92, K94 | Breast, lung, gastric, colorectal cancers, leukemia, lymphoma |
| UBE2O | E2-230K, KIAA1734 | Q9C0C9 | None in UBC | None in UBC | K953, K990, K1038 | Leukemia, lymphoma |
| UBE2Q1 | UBE2Q, NICE5 | Q7Z7E8 | K403 | Y264, S391, Y393, S394, S401, S404, Y415 | K307, K390, K403 | Breast cancer |
| UBE2Q2 | UBE2Q2 | Q8WVN8 | S357, Y368, T369 | Breast, gastric cancers | ||
| UBE2R1 | Cdc34, UBCH3B | P49427 | K167, K173 | Y68, S71, T89, T162, Y190, S203, S222, S231, T233, S236 | K11, K18, K63, K157, K167, K182, K193 | Bone, colorectal, cervical cancers |
| UBE2R2 | Cdc34B, UBC3B | Q712K3 | Y190, S202, Y207, Y228, S233, S238 | K11, K18, K63, K157, K159, K167, K173, K182, K193, K195 | Leukemia | |
| UBE2S | UBE2S, E2-24K | Q16763 | K68, K82 | S73, Y78, T81, T152, S173, S175, T180 | K18, K32, K63, K68, K76, K100, K117, K197, K198 | Prostate, gastric cancers, leukemia |
| UBE2T | UBE2T | Q9NPD8 | K28, K91, K191 | T72, S172, S177, T178, S184 | K28, K48, K91, K136, K141, K156, K182, K191, K192 | Breast, lung cancers, leukemia, multiple myeloma |
| UBE2U | UBE2U | Q5VVX9 | None in UBC | |||
| UBE2V1 | UEV1 | Q13404 | K10, K24, K30, K74 | S7, T86, S106, Y145, S146 | K10, K68, K74, K131 | Breast, lung cancers, leukemia |
| UBE2V2 | UEV2, MMS2 | Q15819 | K8, K66, K72, K108 | S4, T5, S79, S102 | K8, K66, K72, K108, K129, K133 | Breast, colorectal cancers, leukemia |
| UBE2W | UBC16 | Q96B02 | S29, S33 | K10 | ||
| UBE2Z | UBE2Z, USE1(FAT10) | Q9H832 | K166, K238 | None in UBC | K113, K166, K238 | Leukemia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacoursiere, R.E.; Hadi, D.; Shaw, G.S. Acetylation, Phosphorylation, Ubiquitination (Oh My!): Following Post-Translational Modifications on the Ubiquitin Road. Biomolecules 2022, 12, 467. https://doi.org/10.3390/biom12030467
Lacoursiere RE, Hadi D, Shaw GS. Acetylation, Phosphorylation, Ubiquitination (Oh My!): Following Post-Translational Modifications on the Ubiquitin Road. Biomolecules. 2022; 12(3):467. https://doi.org/10.3390/biom12030467
Chicago/Turabian StyleLacoursiere, Rachel E., Dania Hadi, and Gary S. Shaw. 2022. "Acetylation, Phosphorylation, Ubiquitination (Oh My!): Following Post-Translational Modifications on the Ubiquitin Road" Biomolecules 12, no. 3: 467. https://doi.org/10.3390/biom12030467
APA StyleLacoursiere, R. E., Hadi, D., & Shaw, G. S. (2022). Acetylation, Phosphorylation, Ubiquitination (Oh My!): Following Post-Translational Modifications on the Ubiquitin Road. Biomolecules, 12(3), 467. https://doi.org/10.3390/biom12030467