
Cross-Talk between p53 and Wnt Signaling in Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
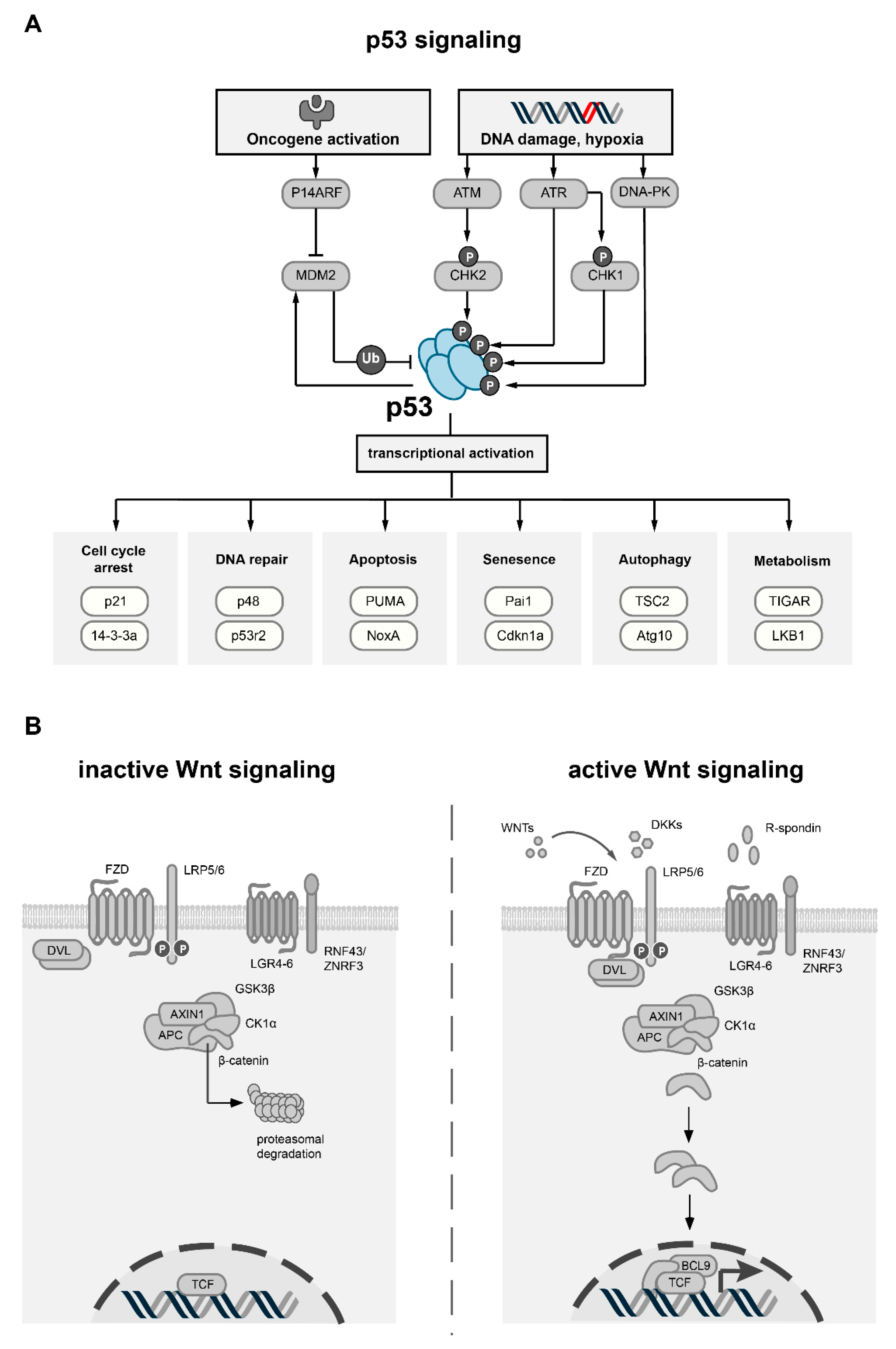
2. p53 and Its Role in Cancer
p53 Associated Proteins and Transcriptional Targets
3. Wnt Signaling
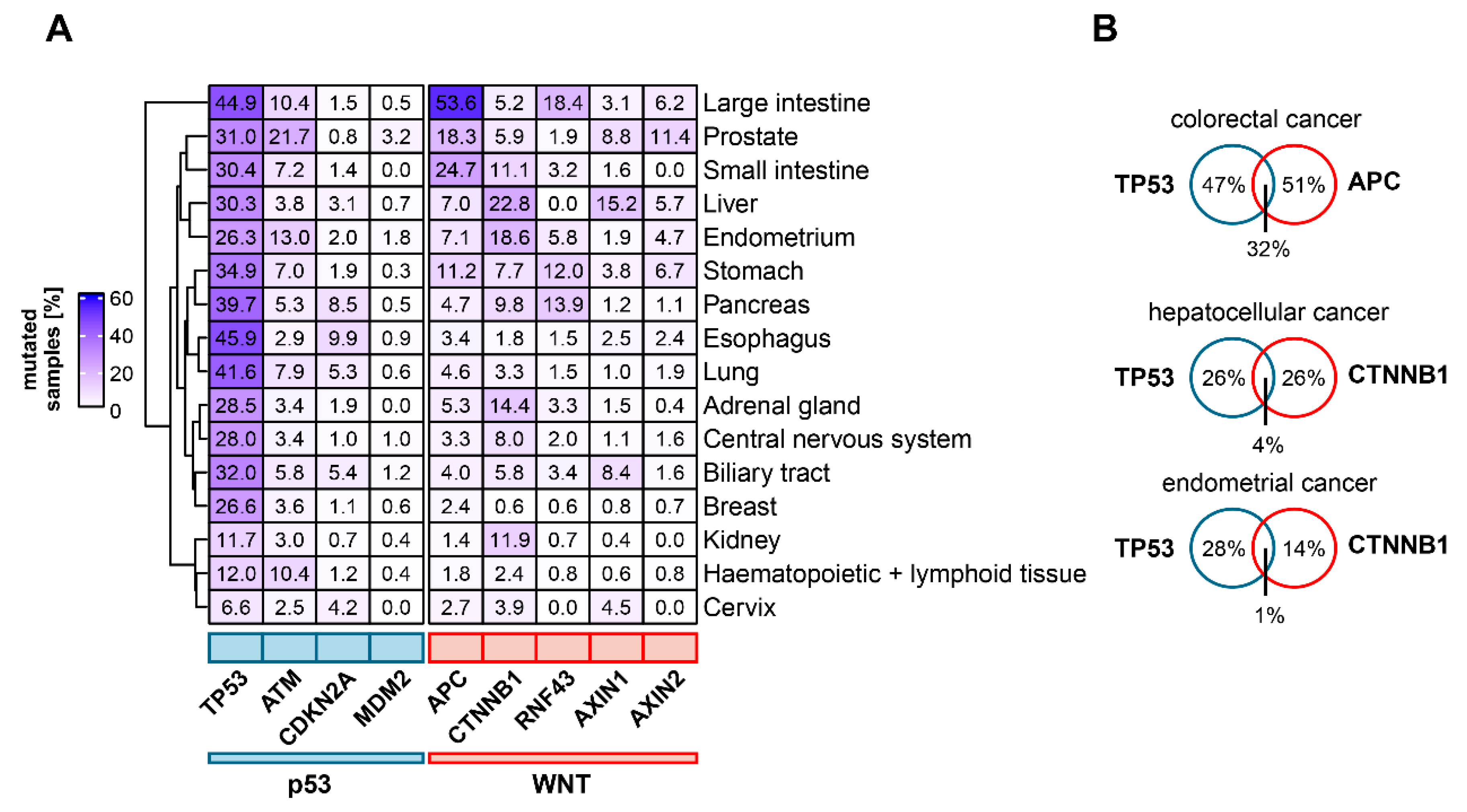
4. The Mutational Landscape of p53 and Wnt Pathway Genes in Cancer
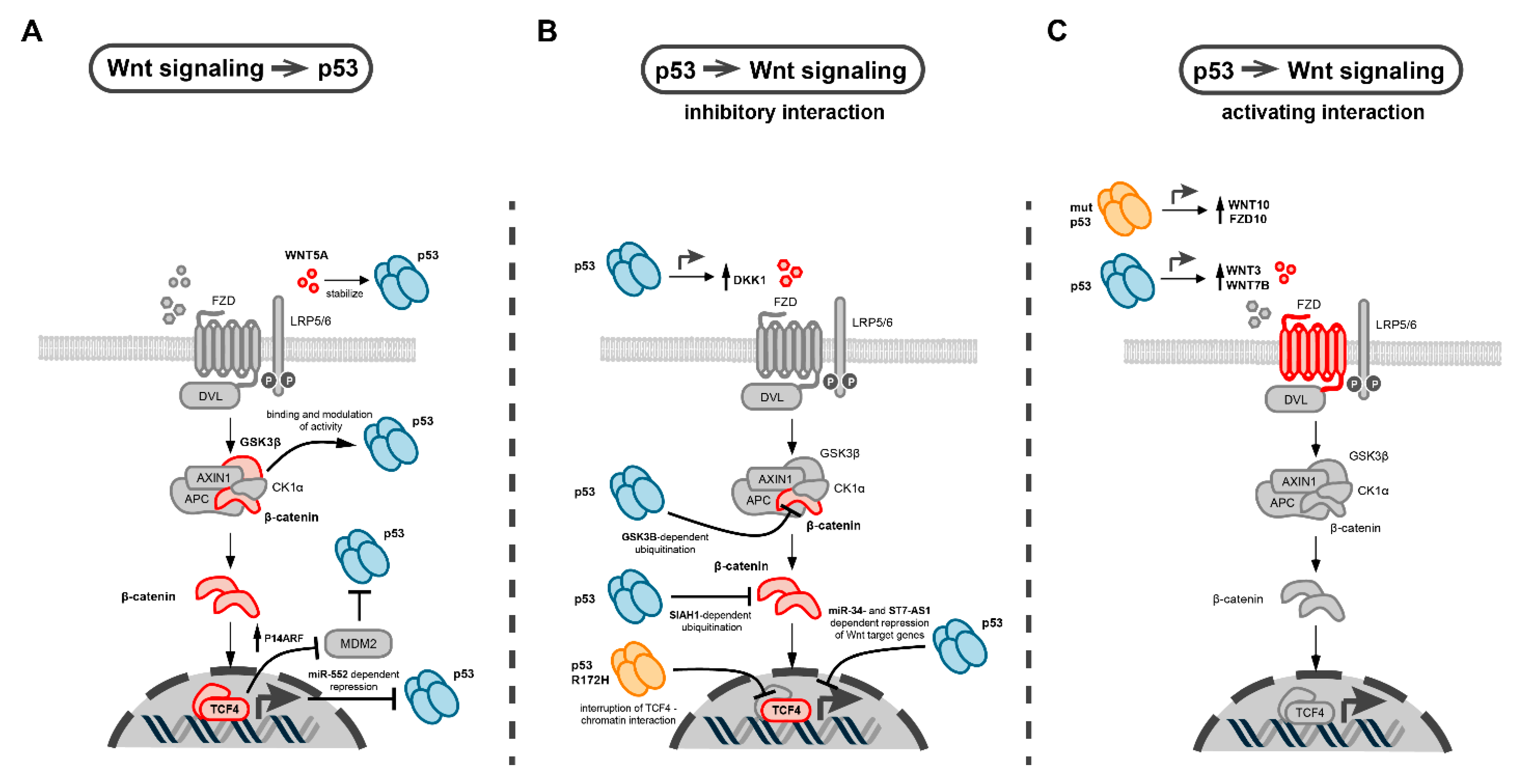
5. Molecular Interactions between p53 and Wnt Pathway in Cancer
5.1. Interactions of p53 with the β-Catenin Destruction Complex
5.2. Interactions of p53 with Secreted WNT Ligands and Antagonists
5.3. Interactions of p53 with Wnt Transcription Factors
5.4. Mutant p53 Specific Interactions with Wnt Signaling
5.5. Interactions of p53 and Wnt Signaling Mediated by Non-Coding RNAs
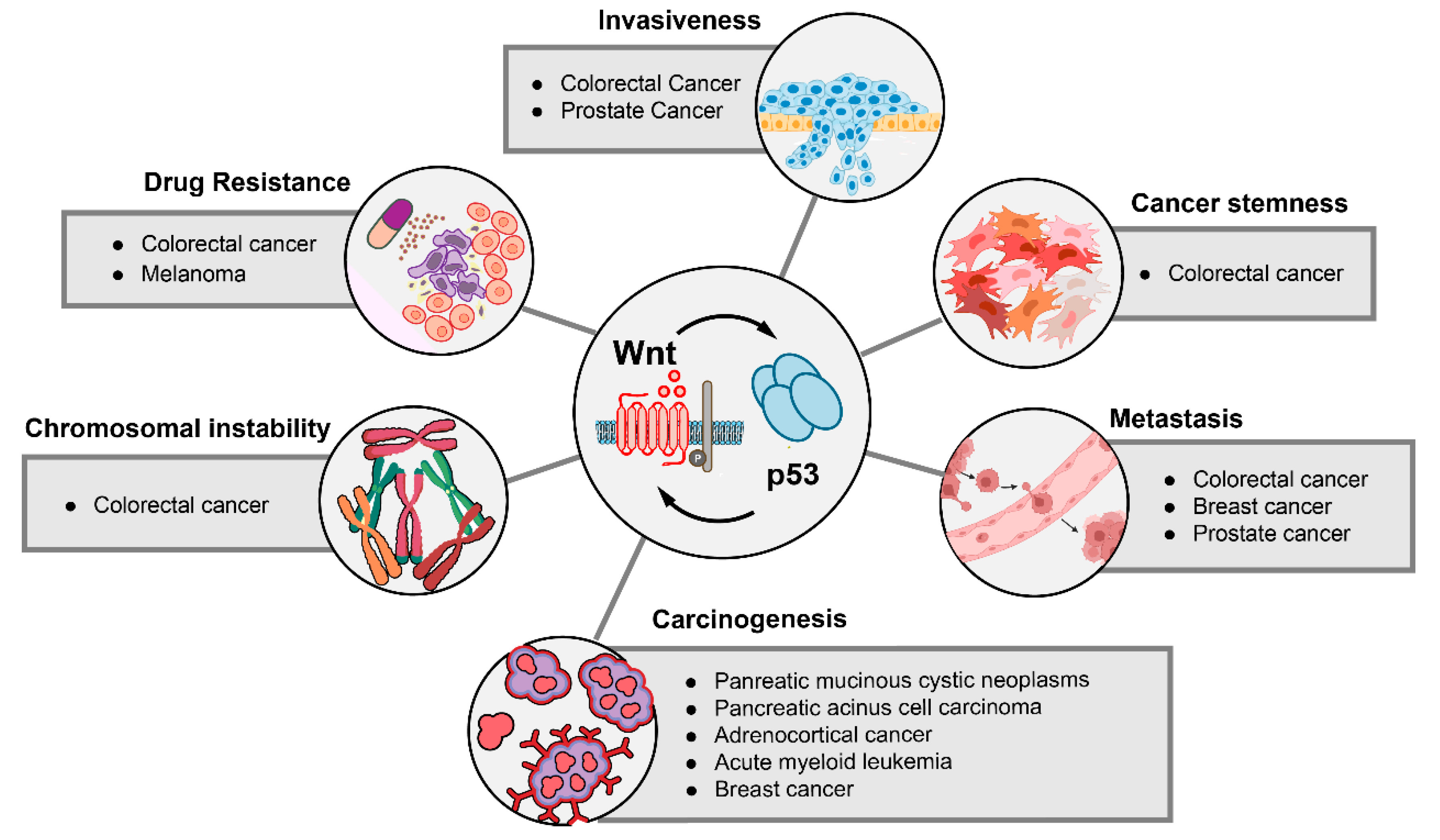
6. The Impact of p53-Wnt Cross-Talk on Cancer Phenotypes
7. Targeting the p53-Wnt Cross-Talk for Cancer Therapy
8. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef]
- Nguyen, L.K.; Kholodenko, B.N. Feedback regulation in cell signalling: Lessons for cancer therapeutics. Semin. Cell Dev. Biol. 2016, 50, 85–94. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. The greatest hits of the human genome: A tour through the most studied genes in biology reveals some surprises. Nature 2017, 551, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A.; Donehower, L.A. Spontaneous and carcinogen−induced tumorigenesis in p53−deficient mice. Nat. Genet. 1993, 5, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.; Flores, E.R.; Miranda, B.; Hsieh, H.M.; Van Oostrom, C.T.M.; Sage, J.; Jacks, T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl. Acad. Sci. USA 2002, 99, 2948–2953. [Google Scholar] [CrossRef]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; Zani, F.; Vousden, K.H. Control of metabolism by p53—Cancer and beyond. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Xu, Y. p53 and stem cells: New developments and new concerns. Trends Cell Biol. 2010, 20, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Valente, L.J.; Gray, D.H.D.; Michalak, E.M.; Pinon-Hofbauer, J.; Egle, A.; Scott, C.L.; Janic, A.; Strasser, A. P53 Efficiently Suppresses Tumor Development in the Complete Absence of Its Cell-Cycle Inhibitory and Proapoptotic Effectors p21, Puma, and Noxa. Cell Rep. 2013, 3, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Fuchs, S.Y.; Adler, V.; Buschmann, T.; Wu, X.; Ronai, Z. Mdm2 association with p53 targets its ubiquitination. Oncogene 1998, 17, 2543–2547. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. P53: An ubiquitous target of anticancer drugs. Int. J. cancer 2002, 98, 161–166. [Google Scholar] [CrossRef]
- D’Orazi, G. Recent Advances in p53. Biomolecules 2021, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Yamaguchi, H.; Higashimoto, Y.; Chao, C.; Xu, Y.; Fornace, A.J.; Appella, E.; Anderson, C.W. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 2003, 278, 37536–37544. [Google Scholar] [CrossRef] [PubMed]
- Nakamizo, A.; Amano, T.; Zhang, W.; Zhang, X.Q.; Ramdas, L.; Liu, T.J.; Bekele, N.; Shono, T.; Sasaki, T.; Benedict, W.F.; et al. Phosphorylation of Thr18 and Ser20 of p53 in Ad-p53—Induced apoptosis. Neuro. Oncol. 2008, 10, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Jabbur, J.R.; Huang, P.; Zhang, W. DNA damage-induced phosphorylation of p53 at serine 20 correlates with p21 and Mdm-2 induction in vivo. Oncogene 2000, 19, 6203–6208. [Google Scholar] [CrossRef]
- Prives, C. Signaling to p53: Breaking the MDM2-p53 circuit. Cell 1998, 95, 5–8. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Gu, W. P53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. P53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Gi, Y.J.; Chakravarti, D.; Chan, I.L.; Zhang, A.; Xia, X.; Tsai, K.Y.; Flores, E.R. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 2012, 16, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Speidel, D. Transcription-independent p53 apoptosis: An alternative route to death. Trends Cell Biol. 2010, 20, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Nusse, R. Wnt proteins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007864. [Google Scholar] [CrossRef]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef]
- Cadigan, K.M. TCFs and Wnt/β-catenin signaling: More than one way to throw the switch. Curr. Top. Dev. Biol. 2012, 98, 1–34. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Wagle, M.; Tran, K.; Zhan, X.; Dixon, M.A.; Liu, S.; Gros, D.; Korver, W.; Yonkovich, S.; Tomasevic, N.; et al. R-Spondin family members regulate the Wnt pathway by a common mechanism. Mol. Biol. Cell 2008, 19, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.-X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin / Lgr5 / Rnf43 module: Regulator of Wnt signal strength The R-spondin / Lgr5 / Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef]
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264. [Google Scholar] [CrossRef]
- Nusse, R. Wnt signaling and stem cell control. Cell Res. 2008, 18, 523–527. [Google Scholar] [CrossRef]
- Van Camp, J.K.; Beckers, S.; Zegers, D.; Van Hul, W. Wnt Signaling and the Control of Human Stem Cell Fate. Stem Cell Rev. Rep. 2014, 10, 207–229. [Google Scholar] [CrossRef]
- Farin, H.F.; Jordens, I.; Mosa, M.H.; Basak, O.; Korving, J.; Tauriello, D.V.F.; de Punder, K.; Angers, S.; Peters, P.J.; Maurice, M.M.; et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 2016, 530, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Nusse, R.; van Amerongen, R. The Role of Ryk and Ror Receptor Tyrosine Kinases in Wnt Signal Transduction. Cold Spring Harb. Perspect. Biol. 2014, 6, a009175. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Fumoto, K.; Okamoto, T.; Kaibuchi, K.; Kikuchi, A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J. 2010, 29, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.R.M.; Das, A.M.; Helvensteijn, W.; Franken, P.F.; Swagemakers, S.; van der Valk, M.A.; ten Hagen, T.L.M.; Kuipers, E.J.; van Veelen, W.; Smits, R. Wnt5a promotes human colon cancer cell migration and invasion but does not augment intestinal tumorigenesis in Apc1638N mice. Carcinogenesis 2013, 34, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, R.; Berns, A. Knockout mouse models to study Wnt signal transduction. Trends Genet. 2006, 22, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt Signaling and Its Significance Within the Tumor Microenvironment: Novel Therapeutic Insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef]
- Azbazdar, Y.; Karabicici, M.; Erdal, E.; Ozhan, G. Regulation of Wnt Signaling Pathways at the Plasma Membrane and Their Misregulation in Cancer. Front. Cell Dev. Biol. 2021, 9, 17. [Google Scholar] [CrossRef]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Sieber, O.M.; Rowan, A.; Setien, F.; Neale, K.; Phillips, R.K.S.; Ward, R.; Esteller, M.; Tomlinson, I.P.M. Promoter hypermethylation leads to decreased APC mRNA expression in familial polyposis and sporadic colorectal tumours, but does not substitute for truncating mutations. Exp. Mol. Pathol. 2008, 85, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Mazzoni, S.M.; Fearon, E.R. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 2014, 355, 1–8. [Google Scholar] [CrossRef]
- Aghabozorgi, A.S.; Bahreyni, A.; Soleimani, A.; Bahrami, A.; Khazaei, M.; Ferns, G.A.; Avan, A.; Hassanian, S.M. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 2019, 157, 64–71. [Google Scholar] [CrossRef]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Wang, X.; Hahn, W.C. Genomic insights into WNT/β-catenin signaling. Trends Pharmacol. Sci. 2014, 35, 103–109. [Google Scholar] [CrossRef]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2020, 21, 5–21. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Hänninen, U.A.; Katainen, R.; Tanskanen, T.; Plaketti, R.M.; Laine, R.; Hamberg, J.; Ristimäki, A.; Pukkala, E.; Taipale, M.; Mecklin, J.P.; et al. Exome-wide somatic mutation characterization of small bowel adenocarcinoma. PLoS Genet. 2018, 14, e1007200. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Matoba, R.; Maekawa, H.; Sakurada, M.; Kushida, T.; Orita, H.; Wada, R.; Sato, K. Detection of gene mutations in gastric cancer tissues using a commercial sequencing panel. Mol. Clin. Oncol. 2019, 11, 455. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Jing, C.; Chang, X.; Ding, D.; Han, T.; Yang, J.; Lu, Z.; Hu, X.; Liu, Z.; Wang, J.; et al. Mutational landscape of gastric cancer and clinical application of genomic profiling based on target next-generation sequencing. J. Transl. Med. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Ando, H.; Nagase, H.; Nishisho, I.; Horii, A.; Miki, Y.; Mori, T.; Utsunomiya, J.; Baba, S.; Petersen, G.; et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc. Natl. Acad. Sci. USA 1992, 89, 4452–4456. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-M.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Murillo-Garzón, V.; Kypta, R. WNT signalling in prostate cancer. Nat. Rev. Urol. 2017, 14, 683–696. [Google Scholar] [CrossRef]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 2418. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Sakai, E.; Echizen, K.; Yamada, Y.; Oshima, H.; Han, T.S.; Ohki, R.; Fujii, S.; Ochiai, A.; Robine, S.; et al. Intestinal cancer progression by mutant p53 through the acquisition of invasiveness associated with complex glandular formation. Oncogene 2017, 36, 5885–5896. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Luo, S.; Deng, C.; Luo, T.; Yin, W.; Zhang, H.; Zhang, Y.; Zhang, X.; Lan, Y.; Ping, Y.; et al. Identifying mutual exclusivity across cancer genomes: Computational approaches to discover genetic interaction and reveal tumor vulnerability. Brief. Bioinform. 2019, 20, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Guo, H.; Zhu, Y.; Xia, Y.; Cui, J.; Shi, K.; Fan, Y.; Shi, B.; Chen, S. TP53 alterations of hormone-naïve prostate cancer in the Chinese population. Prostate Cancer Prostatic Dis. 2020, 24, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Wang, X.W.; Budhu, A.; Kim, Y.H.; Kwon, S.M.; Tang, Z.; Sun, Z.; Harris, C.C.; Thorgeirsson, S.S. Association of TP53 Mutations With Stem Cell-Like Gene Expression and Survival of Patients With Hepatocellular Carcinoma. Gastroenterology 2011, 140, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Oyama, T.; Kusano, T.; Osaki, T.; Nakanishi, R.; Shirakusa, T. Mutations of the p53 Gene as a Predictor of Poor Prognosis in Patients With Non-Small-Cell Lung Cancer. JNCI J. Natl. Cancer Inst. 1993, 85, 2018–2023. [Google Scholar] [CrossRef]
- Hamelin, R.; Laurent-Puig, P.; Olschwang, S.; Jego, N.; Asselain, B.; Remvikos, Y.; Girodet, J.; Salmon, R.J.; Thomas, G. Association of p53 mutations with short survival in colorectal cancer. Gastroenterology 1994, 106, 42–48. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Curtin, K.; Ma, K.N.; Edwards, S.; Schaffer, D.; Leppert, M.F.; Slattery, M.L. Prognostic significance of p53 mutations in colon cancer at the population level. Int. J. cancer 2002, 99, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Wada, C.; Ishida, H.; Wang, C.; Egawa, S.; Yokoyama, E.; Kameya, T.; Koshiba, K. p53 Mutations and Prognosis in Bladder Tumors. J. Urol. 1995, 153, 1097–1104. [Google Scholar] [CrossRef]
- Huszno, J.; Grzybowska, E. TP53 mutations and SNPs as prognostic and predictive factors in patients with breast cancer (Review). Oncol. Lett. 2018, 16, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Zhang, X.M.; Zhang, Z.; Zheng, L.H.; Jindal, S.; Liu, Y.J. Association of p53 expression with poor prognosis in patients with triple-negative breast invasive ductal carcinoma. Medicine 2019, 98, e15449. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Christie, M.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Love, C.; Xu, Z.Z.; Molloy, P.L.; Jones, I.T.; McLaughlin, S.; et al. Wild-type APC predicts poor prognosis in microsatellite-stable proximal colon cancer. Br. J. Cancer 2015, 113, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.A.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed]
- Conlin, A.; Smith, G.; Carey, F.A.; Wolf, C.R.; Steele, R.J.C. The prognostic significance of K-ras, p53, and APC mutations in colorectal carcinoma. Gut 2005, 54, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Surun, A.; Varlet, P.; Brugières, L.; Lacour, B.; Faure-Conter, C.; Leblond, P.; Bertozzi-Salomon, A.I.; Berger, C.; André, N.; Sariban, E.; et al. Medulloblastomas associated with an APC germline pathogenic variant share the good prognosis of CTNNB1-mutated medulloblastomas. Neuro. Oncol. 2020, 22, 128–138. [Google Scholar] [CrossRef]
- Kastritis, E.; Murray, S.; Kyriakou, F.; Horti, M.; Tamvakis, N.; Kavantzas, N.; Patsouris, E.S.; Noni, A.; Legaki, S.; Dimopoulos, M.A.; et al. Somatic mutations of adenomatous polyposis coli gene and nuclear b-catenin accumulation have prognostic significance in invasive urothelial carcinomas: Evidence for Wnt pathway implication. Int. J. Cancer 2009, 124, 103–108. [Google Scholar] [CrossRef]
- Travaglino, A.; Raffone, A.; Raimondo, D.; Reppuccia, S.; Ruggiero, A.; Arena, A.; Casadio, P.; Zullo, F.; Insabato, L.; Seracchioli, R.; et al. Prognostic significance of CTNNB1 mutation in early stage endometrial carcinoma: A systematic review and meta-analysis. Arch. Gynecol. Obstet. 2022, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.C.; Shao, Y.Y.; Lee, Y.H.; Hsieh, M.S.; Hsiao, C.H.; Lin, H.H.; Kao, H.F.; Ma, Y.Y.; Yen, F.C.; Cheng, A.L.; et al. β-Catenin (CTNNB1) Mutations Are Not Associated with Prognosis in Advanced Hepatocellular Carcinoma. Oncology 2014, 87, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, W.; Shao, J.; Zhao, J.; Chen, C. Analysis of the Clinicopathologic Characteristics of Lung Adenocarcinoma With CTNNB1 Mutation. Front. Genet. 2020, 10, 1367. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Damalas, A.; Ben-Ze’ev, A.; Simcha, I.; Shtutman, M.; Martinez Leal, J.F.; Zhurinsky, J.; Geiger, B.; Oren, M. Excess β-catenin promotes accumulation of transcriptionally active p53. EMBO J. 1999, 18, 3054–3063. [Google Scholar] [CrossRef]
- Damalas, A.; Kahan, S.; Shtutman, M.; Ben-Ze’ev, A.; Oren, M. Deregulated β-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. EMBO J. 2001, 20, 4912–4922. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Sadot, E.; Geiger, B.; Oren, M.; Ben-Ze’ev, A. Down-Regulation of β-Catenin by Activated p53. Mol. Cell. Biol. 2001, 21, 6768–6781. [Google Scholar] [CrossRef]
- Ban, K.C.; Singh, H.; Krishnan, R.; Seow, H.F. GSK-3β phosphorylation and alteration of β-catenin in hepatocellular carcinoma. Cancer Lett. 2003, 199, 201–208. [Google Scholar] [CrossRef]
- Watcharasit, P.; Bijur, G.N.; Zmijewski, J.W.; Song, L.; Zmijewska, A.; Chen, X.; Johnson, G.V.W.; Jope, R.S. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc. Natl. Acad. Sci. USA 2002, 99, 7951–7955. [Google Scholar] [CrossRef]
- Eom, T.Y.; Jope, R.S. GSK3β N-terminus binding to p53 promotes its acetylation. Mol. Cancer 2009, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Zhuang, L.; Leong, H.S.; Iyer, N.G.; Liu, E.T.; Yu, Q. Pharmacologic modulation of glycogen synthase kinase-3β promotes p53-dependent apoptosis through a direct bax-mediated mitochondrial pathway in colorectal cancer cells. Cancer Res. 2005, 65, 9012–9020. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Altieri, D.C. Activation of p53-dependent apoptosis by acute ablation of glycogen synthase kinase-3β in colorectal cancer cells. Clin. Cancer Res. 2005, 11, 4580–4588. [Google Scholar] [CrossRef][Green Version]
- Elyada, E.; Pribluda, A.; Goldstein, R.E.; Morgenstern, Y.; Brachya, G.; Cojocaru, G.; Snir-Alkalay, I.; Burstain, I.; Haffner-Krausz, R.; Jung, S.; et al. CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 2011, 470, 409–413. [Google Scholar] [CrossRef]
- Matsuzawa, S.I.; Reed, J.C. Siah-1, SIP, and Ebi collaborate in a novel pathway for β-catenin degradation linked to p53 responses. Mol. Cell 2001, 7, 915–926. [Google Scholar] [CrossRef]
- Dimitrova, Y.N.; Li, J.; Lee, Y.T.; Rios-Esteves, J.; Friedman, D.B.; Choi, H.J.; Weis, W.I.; Wang, C.Y.; Chazin, W.J. Direct ubiquitination of β-catenin by Siah-1 and regulation by the exchange factor TBL1. J. Biol. Chem. 2010, 285, 13507–13516. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Stevens, J.; Rote, C.A.; Yost, H.J.; Hu, Y.; Neufeld, K.L.; White, R.L.; Matsunami, N. Siah-1 mediates a novel β-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol. Cell 2001, 7, 927–936. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y.; Kong, T.; Fan, F.; Jiang, Y. Hypoxia-induced β-catenin downregulation involves p53-dependent activation of Siah-1. Cancer Sci. 2011, 102, 1322–1328. [Google Scholar] [CrossRef]
- Webster, M.R.; Fane, M.E.; Alicea, G.M.; Basu, S.; Kossenkov, A.V.; Marino, G.E.; Douglass, S.M.; Kaur, A.; Ecker, B.L.; Gnanapradeepan, K.; et al. Paradoxical Role for Wild-Type p53 in Driving Therapy Resistance in Melanoma. Mol. Cell 2020, 77, 633–644.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shou, J.; Chen, X. Dickkopf-1, an inhibitor of the Wnt signaling pathway, is induced by p53. Oncogene 2000, 19, 1843–1848. [Google Scholar] [CrossRef]
- Tang, N.; Cai, X.; Peng, L.; Liu, H.; Chen, Y. TCP1 regulates Wnt7b/β-catenin pathway through P53 to influence the proliferation and migration of hepatocellular carcinoma cells. Signal Transduct. Target. Ther. 2020, 5, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.H.; Ro, E.J.; Yoon, J.S.; Mizutani, T.; Kang, D.W.; Park, J.C.; Il Kim, T.; Clevers, H.; Choi, K.Y. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat. Commun. 2020, 11, 5321. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Li, M.; Michalowski, A.M.; Zhang, X.; Liao, H.; Chen, L.; Xu, Y.; Wu, X.; Huang, J. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zou, Y.; Nowotschin, S.; Kim, S.Y.; Li, Q.V.; Soh, C.L.; Su, J.; Zhang, C.; Shu, W.; Xi, Q.; et al. The p53 Family Coordinates Wnt and Nodal Inputs in Mesendodermal Differentiation of Embryonic Stem Cells. Cell Stem Cell 2017, 20, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.; Johne, C.; Spiesbach, K.; Haugwitz, U.; Tschöp, K.; Wasner, M.; Klein-Hitpass, L.; Möröy, T.; Mössner, J.; Engeland, K. Identification of Tcf-4 as a transcriptional target of p53 signalling. Oncogene 2004, 23, 3376–3384. [Google Scholar] [CrossRef] [PubMed]
- Cagatay, T.; Ozturk, M. P53 Mutation As a Source of Aberrant Β-Catenin Accumulation in Cancer Cells. Oncogene 2002, 21, 7971–7980. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Rizvi, S.M.A.; Madigan, M.C.; Cozzi, P.J.; Power, C.A.; Qu, C.F.; Morgenstern, A.; Apostolidis, C.; Russell, P.J.; Allen, B.J.; et al. Modulation of beta-catenin-mediated TCF-signalling in prostate cancer cell lines by wild-type and mutant p53. Prostate 2007, 67, 1384–1396. [Google Scholar] [CrossRef]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef]
- Imedio, L.; Cristóbal, I.; Rubio, J.; Santos, A.; Rojo, F.; García-Foncillas, J. MicroRNAs in rectal cancer: Functional significance and promising therapeutic value. Cancers 2020, 12, 2040. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Calin, G.A.; Lopez-Berestein, G.; Sood, A.K. MiRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 2016, 6, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Kaushik, A.C.; Zhang, J. The emerging role of major regulatory RNAs in cancer control. Front. Oncol. 2019, 9, 920. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; De Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.H.; Kim, N.H.; Park, C.; Lee, I.; Kim, H.S.; Yook, J.I. miRNA-34 intrinsically links p53 tumor suppressor and Wnt signaling p53 Tumor Suppressor and Canonical Wnt Signaling in Cancer. Cell Cycle 2012, 117, 1273–1281. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Kim, N.G.; Lee, I.; Choi, H.S.; Li, X.Y.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Na, J.M.; et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 2011, 4, ra71. [Google Scholar] [CrossRef]
- Kwak, B.; Kim, D.U.; Kim, T.O.; Kim, H.S.; Kim, S.W. MicroRNA-552 links Wnt signaling to p53 tumor suppressor in colorectal cancer. Int. J. Oncol. 2018, 53, 1800–1808. [Google Scholar] [CrossRef]
- Sheng, J.; He, X.; Yu, W.; Chen, Y.; Long, Y.; Wang, K.; Zhu, S.; Liu, Q. p53-targeted lncRNA ST7-AS1 acts as a tumour suppressor by interacting with PTBP1 to suppress the Wnt/β-catenin signalling pathway in glioma. Cancer Lett. 2021, 503, 54–68. [Google Scholar] [CrossRef]
- Borges, K.S.; Pignatti, E.; Leng, S.; Kariyawasam, D.; Ruiz-Babot, G.; Ramalho, F.S.; Taketo, M.M.; Carlone, D.L.; Breault, D.T. Wnt/β-catenin activation cooperates with loss of p53 to cause adrenocortical carcinoma in mice. Oncogene 2020, 39, 5282–5291. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Cummings, M.C.; Harrison, D.J. Interaction between murine germline mutations in p53 and APC predisposes to pancreatic neoplasia but not to increased intestinal malignancy. Oncogene 1995, 11, 1913–1920. [Google Scholar] [PubMed]
- Kuo, T.L.; Weng, C.C.; Kuo, K.K.; Chen, C.Y.; Wu, D.C.; Hung, W.C.; Cheng, K.H. APC haploinsufficiency coupled with p53 loss sufficiently induces mucinous cystic neoplasms and invasive pancreatic carcinoma in mice. Oncogene 2016, 35, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, A.; Fernald, A.A.; Wang, J.; Davis, E.M.; Karrison, T.; Anastasi, J.; Le Beau, M.M. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood 2014, 123, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Méniel, V.; Hay, T.; Douglas-Jones, A.; Sansom, O.J.; Clarke, A.R. Mutations in Apc and p53 synergize to promote mammary neoplasia. Cancer Res. 2005, 65, 410–416. [Google Scholar]
- Gunther, E.J.; Moody, S.E.; Belka, G.K.; Hahn, K.T.; Innocent, N.; Dugan, K.D.; Cardiff, R.D.; Chodosh, L.A. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev. 2003, 17, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.E.; van Boxtel, R.; de Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Van Noesel, C.J.M.; Peppelenbosch, M.P.; Korkmaz, K.S.; Molendijk, I.; Dekker, E.; Morreau, H.; Van Pelt, G.W.; et al. The BMP pathway either enhances or inhibits the Wnt pathway depending on the SMAD4 and p53 status in CRC. Br. J. Cancer 2014, 112, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; van Miltenburg, M.H.; Slagter, M.; de Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ye, L.; Xin, Z.; Wang, M.; Lin, C.; Huang, S.; Guo, W.; Lai, Y.; Du, H.; Li, J.; et al. FZD8, a target of p53, promotes bone metastasis in prostate cancer by activating canonical Wnt/β-catenin signaling. Cancer Lett. 2017, 402, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Garzón, V.; Gorroño-Etxebarria, I.; Åkerfelt, M.; Puustinen, M.C.; Sistonen, L.; Nees, M.; Carton, J.; Waxman, J.; Kypta, R.M. Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat. Commun. 2018, 9, 1747. [Google Scholar] [CrossRef]
- Leibold, J.; Ruscetti, M.; Cao, Z.; Ho, Y.J.; Baslan, T.; Zou, M.; Abida, W.; Feucht, J.; Han, T.; Barriga, F.M.; et al. Somatic tissue engineering in mouse models reveals an actionable role for WNT pathway alterations in prostate cancer metastasis. Cancer Discov. 2020, 10, 1038–1057. [Google Scholar] [CrossRef]
- Cheng, J.; Dwyer, M.; Okolotowicz, K.J.; Mercola, M.; Cashman, J.R. A novel inhibitor targets both WNT signaling and ATM/P53 in colorectal cancer. Cancer Res. 2018, 78, 5072–5083. [Google Scholar] [CrossRef]
- da Silva Siqueira, E.; Concato, V.M.; Tomiotto-Pellissier, F.; Silva, T.F.; da Silva Bortoleti, B.T.; Gonçalves, M.D.; Costa, I.N.; Junior, W.A.V.; Pavanelli, W.R.; Panis, C.; et al. Trans-chalcone induces death by autophagy mediated by p53 up-regulation and β-catenin down-regulation on human hepatocellular carcinoma HuH7.5 cell line. Phytomedicine 2021, 80, 153373. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Ko, H.J.; Chiou, S.J.; Lai, Y.L.; Hou, C.C.; Javaria, T.; Huang, Z.Y.; Cheng, T.S.; Hsu, T.I.; Chuang, J.Y.; et al. Nbm-bmx, an hdac8 inhibitor, overcomes temozolomide resistance in glioblastoma multiforme by downregulating the β-catenin/c-myc/sox2 pathway and upregulating p53-mediated mgmt inhibition. Int. J. Mol. Sci. 2021, 22, 5907. [Google Scholar] [CrossRef]
- Ju, J.; Schmitz, J.C.; Song, B.; Kudo, K.; Chu, E. Regulation of p53 expression in response to 5-fluorouracil in human cancer RKO cells. Clin. Cancer Res. 2007, 13, 4245–4251. [Google Scholar] [CrossRef]
- Sun, X.X.; Dai, M.S.; Lu, H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J. Biol. Chem. 2007, 282, 8052–8059. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Futur. Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef]
- Hui, W.; Liu, S.; Zheng, J.; Fang, Z.; Ding, Q.; Feng, C. Nutlin-3a as a novel anticancer agent for adrenocortical carcinoma with CTNNB1 mutation. Cancer Med. 2018, 7, 1440–1449. [Google Scholar] [CrossRef]
- Wang, M.; Wang, X.; Li, Y.; Xiao, Q.; Cui, X.H.; Xiao, G.D.; Wang, J.C.; Xu, C.W.; Ren, H.; Liu, D. Nutlin-3-Induced Sensitization of Non-Small Cell Lung Cancer Stem Cells to Axitinib-Induced Apoptosis Through Repression of Akt1/Wnt Signaling. Oncol. Res. 2019, 27, 987. [Google Scholar] [CrossRef]
- Qin, J.J.; Wang, W.; Li, X.; Deokar, H.; Buolamwini, J.K.; Zhang, R. Inhibiting β-Catenin by β-Carboline-type MDM2 inhibitor for pancreatic cancer therapy. Front. Pharmacol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Huang, J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol. Ther. 2021, 220. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S.; Bayram, T.; Van De Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Bolhaqueiro, A.C.F.; Ponsioen, B.; Bakker, B.; Klaasen, S.J.; Kucukkose, E.; van Jaarsveld, R.H.; Vivié, J.; Verlaan-Klink, I.; Hami, N.; Spierings, D.C.J.; et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 2019, 51, 824–834. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Q.; Werner, J.; Venkatachalam, N.; Boonekamp, K.E.; Ebert, M.P.; Zhan, T. Cross-Talk between p53 and Wnt Signaling in Cancer. Biomolecules 2022, 12, 453. https://doi.org/10.3390/biom12030453
Xiao Q, Werner J, Venkatachalam N, Boonekamp KE, Ebert MP, Zhan T. Cross-Talk between p53 and Wnt Signaling in Cancer. Biomolecules. 2022; 12(3):453. https://doi.org/10.3390/biom12030453
Chicago/Turabian StyleXiao, Qiyun, Johannes Werner, Nachiyappan Venkatachalam, Kim E. Boonekamp, Matthias P. Ebert, and Tianzuo Zhan. 2022. "Cross-Talk between p53 and Wnt Signaling in Cancer" Biomolecules 12, no. 3: 453. https://doi.org/10.3390/biom12030453
APA StyleXiao, Q., Werner, J., Venkatachalam, N., Boonekamp, K. E., Ebert, M. P., & Zhan, T. (2022). Cross-Talk between p53 and Wnt Signaling in Cancer. Biomolecules, 12(3), 453. https://doi.org/10.3390/biom12030453