
Novel Selective and Low-Toxic Inhibitor of LmCPB2.8ΔCTE (CPB) One Important Cysteine Protease for Leishmania Virulence

 , , , , , , , , , , ,
, , , , , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
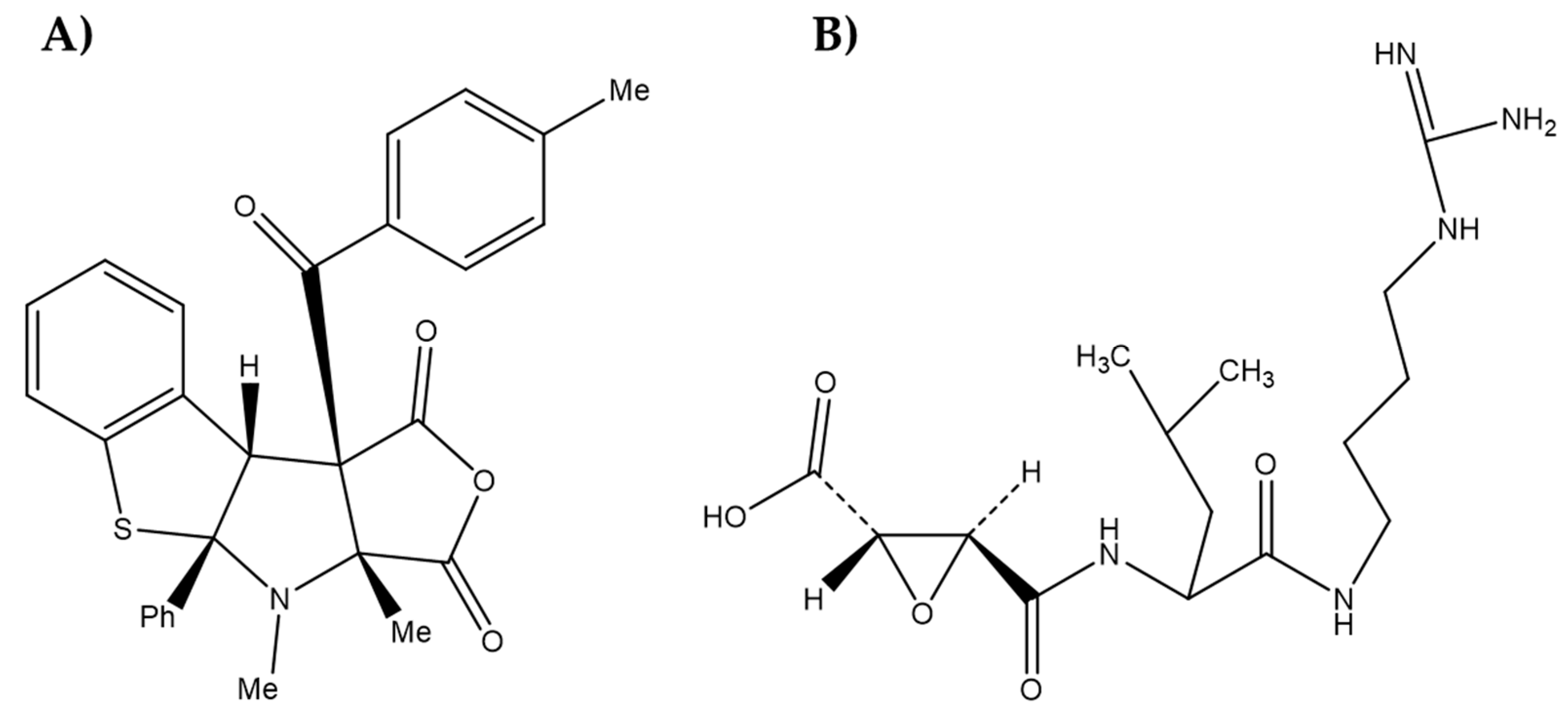
2.1. Synthesis of Guanidines
2.2. Structural Characterization
2.2.1. NMR Measurements
2.2.2. Single Crystal X-ray Diffraction Analysis
2.3. Biological Toxicity Studies
2.3.1. Cell Toxicity
2.3.2. Organ Toxicity on Isolated Tissue Preparations
2.4. Inhibitory Activity on the Enzyme LmCPB2.8ΔCTE (CPB)
2.4.1. Enzyme Kinetic Assay
2.4.2. Proteases Assays
2.5. Leishmanicidal Activity
2.5.1. Parasites
2.5.2. Antileishmanial In Vivo Assay
2.5.3. Toxicity Assay for BALB/c Mice
2.5.4. Statistical Analysis
2.5.5. Ethics Statement
2.6. Docking Investigation
3. Results and Discussion
3.1. Structural Characterization
3.2. Biological Assays
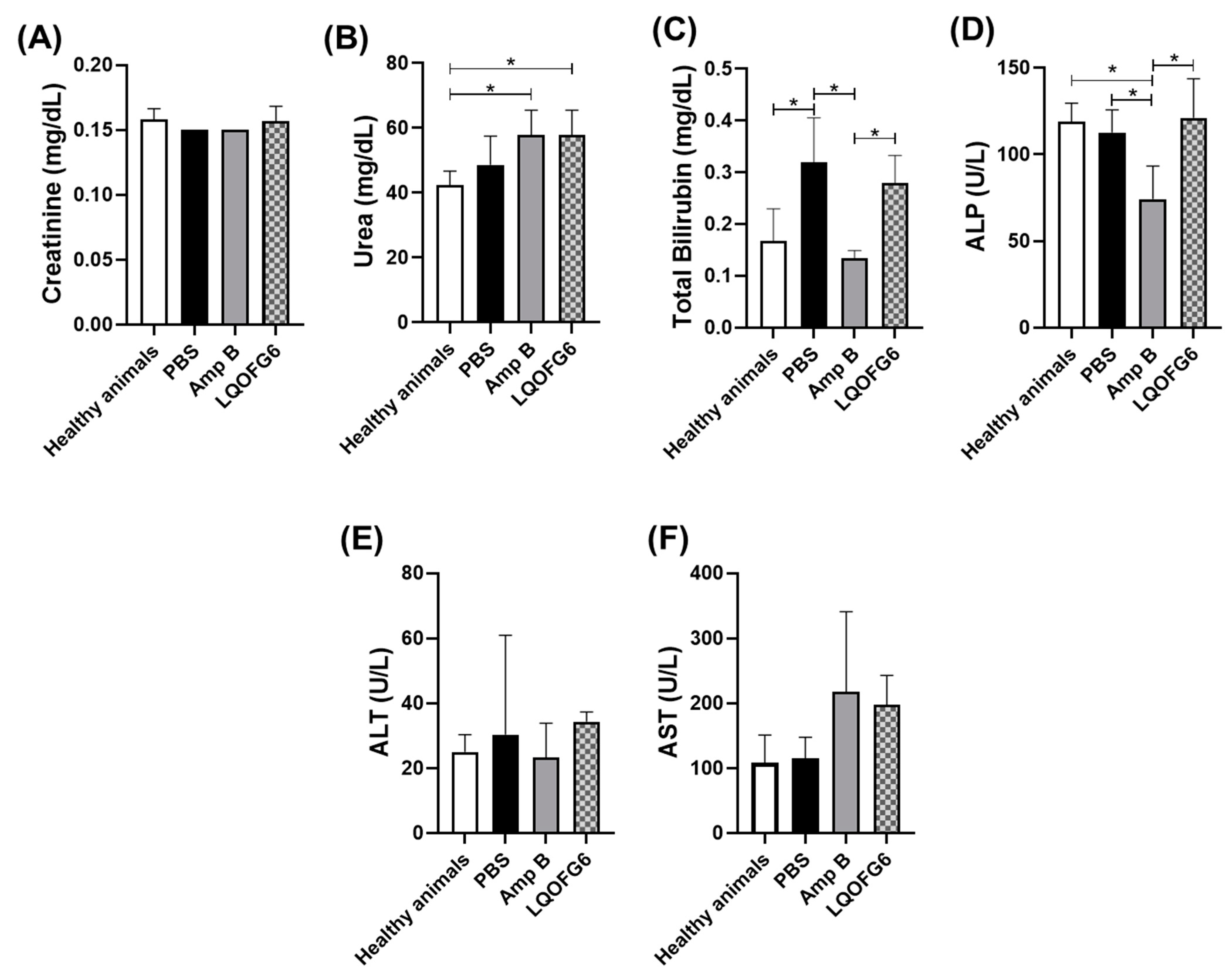
3.2.1. Cell and Organ Toxicity
3.2.2. Inhibition of Leishmania Cysteine Protease
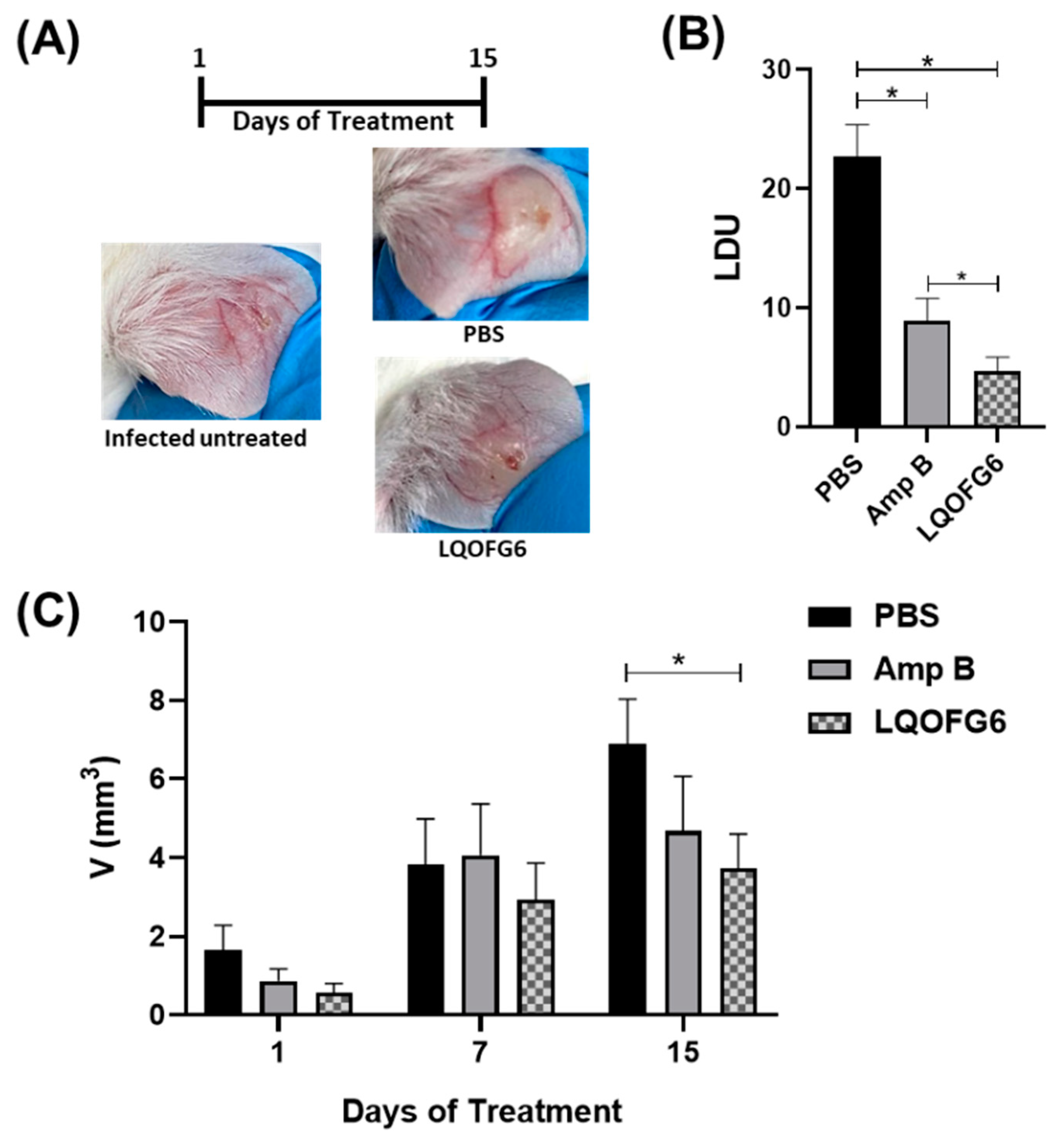
3.2.3. Leishmanicidal Activity
Reduction of Parasite Load by LQOF-G6 in BALB/c Mice Infected with L. amazonensis
Investigation of Hepatic or Renal Disturbance
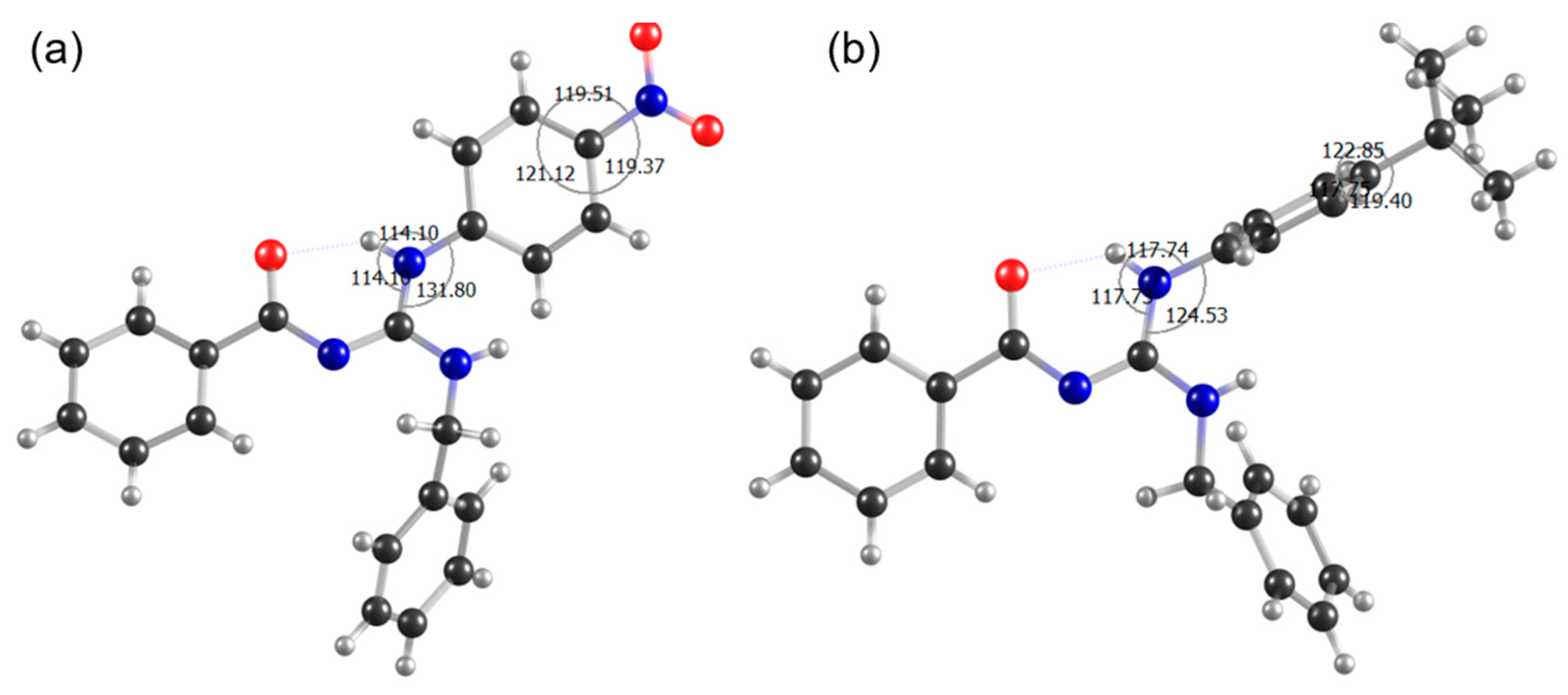
3.3. Conformational Analysis
3.3.1. Investigation of Spatial Coupling by NOESY
3.3.2. Solid-state NMR
3.4. Docking Investigation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; Boer, M.D.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Scala, A.; Micale, N.; Piperno, A.; Rescifina, A.; Schirmeister, T.; Kesselring, J.; Grassi, G. Targeting of the Leishmania mexicana cysteine protease CPB2.8DCTE by decorated fused benzo[b] thiophene scaffold. RSC Adv. 2016, 6, 30628–30635. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization Home Page. Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 4 April 2020).
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Res 2017, 6, 750. [Google Scholar] [CrossRef]
- Handler, M.Z.; Patel, P.; Kapila, R.; AlQubati, Y.; Schwartz, R.A. Cutaneous and mucocutaneous leishmaniasis: Differential diagnosis, diagnosis, histopathology, and management. J. Am. Acad. Dermatol. 2015, 73, 911–926. [Google Scholar] [CrossRef]
- Sánchez, C.I.S.; Pastor, E.R.; Veja, R.C. Lingual mucocutaneous leishmaniasis. Acta Otorrinolaringol. 2021, 72, 403–404. [Google Scholar] [CrossRef]
- Suqati, A.A.; Pudszuhn, A.; Hofmann, V.M. Mucocutaneous leishmaniasis: Case report and literature review of a rare endonasal infection. Pan Afr. Med. J. 2020, 36, 292. [Google Scholar] [CrossRef] [PubMed]
- Aronson, N.E.; Joya, C.A. Cutaneous leishmaniasis: Updates in diagnosis and management. Infect. Dis. Clin. 2019, 33, 101–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seblova, V.; Myskova, J.; Hlavacova, J.; Votypka, J.; Antoniou, M.; Volf, P. Natural hybrid of Leishmania infantum/L. donovani: Development in Phlebotomus tobbi, P. perniciosus and Lutzomyia longipalpis and comparison with non-hybrid strains differing in tissue tropism. Parasites Vectors 2015, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Bi, K.; Chen, Y.; Zhao, S.; Kuang, Y.; Wu, C.H.J. Current visceral leishmaniasis research: A research review to inspire future study. BioMed Res. Int. 2018, 2018, 9872095. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, L.S.; Braga, M.G.; Costa, D.A.S.; Simões, T.C.; Lula, M.D.; Silveira, M.R. Lethality among individuals infected with visceral leishmaniasis in Brazil: A retrospective study (2007–2018). Parasitol. Res. 2022, 121, 725–736. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Home Page. Fifteenth Meeting of the Strategic and Technical Advisory Group for Neglected TROPICAL Diseases. 2022. Available online: https://www.who.int/publications/i/item/9789240048065 (accessed on 3 July 2022).
- Moraes, L.; Santos, L.A.; Arruda, L.B.; Silva, M.P.P.; Silva, M.O.; Silva, J.A.G.; Ramos, A.; dos Santos, M.B.; Torres, F.G.; Orge, C.; et al. High seroprevalence of leishmania infantum is linked to immune activation in people with HIV: A two-stage cross-sectional study in Bahia, Brazil. medRxiv 2021, 1, 1–75. [Google Scholar] [CrossRef]
- Yesilova, Y.; Surucu, H.A.; Ardic, N.; Aksoy, M.; Yesilova, A.; Oghumu, S.; Satoskar, A.R. Meglumine antimoniate is more effective than sodium stibogluconate in the treatment of cutaneous leishmaniasis. J. Dermatol. Treatment 2015, 27, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, M.P.; Croft, S.L. Management of trypanosomiasis and leishmaniasis. Br. Med. Bull. 2012, 104, 175–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Kumar, M.; Singh, R.K. Leishmaniasis: Current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 2012, 5, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Luca, L.; Ferro, S.; Buemi, M.R.; Monforte, A.; Gitto, R.; Schirmeister, T.; Maes, L.; Rescifina, A.; Micale, N. Discovery of benzimidazole-based Leishmania mexicana cysteine protease CPB2.8ΔCTE inhibitors as potential therapeutics for leishmaniasis. Chem. Biol. Drug Des. 2018, 92, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, S.J.; Pollock, K.G.; Hilley, J.D.; Meldal, M.; St Hilaire, P.; Juliano, M.A.; Juliano, L.; Mottram, J.C.; Coombs, G.H. Expression and characterization of a recombinant cysteine proteinase of Leishmania mexicana. Biochem. J. 2000, 347, 383–388. [Google Scholar] [CrossRef]
- Klemba, M.; Goldberg, D.E. Biological roles of proteases in parasitic protozoa. Annu. Rev. Biochem. 2002, 71, 275. [Google Scholar] [CrossRef]
- Clementino, L.C.; Fernandes, G.F.S.; Prokopczyk, I.M.; Laurindo, W.C.; Toyama, D.; Motta, B.P.; Baviera, A.M.; Henrique-Silva, F.; Santos, J.L.D.; Graminha, M.A. Design, synthesis and biological evaluation of N-oxide derivatives with potent in vivo antileishmanial activity. PLoS ONE 2021, 16, e0259008. [Google Scholar] [CrossRef]
- Chen, G.; Seukep, A.J.; Guo, M. Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs. Mar. Drugs 2020, 18, 545. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Ventakash, T.; Suresh, P.S. Study on the interaction of the bromodomain inhibitor JQ1 with human serum albumin by spectroscopic and molecular docking studies. J. Mol. Struct. 2022, 1273, 134374. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef] [PubMed]
- do Espírito Santo, R.; Velásquez, Á.M.A.; Passianoto, L.V.G.; Sepulveda, A.A.L.; da Costa Clementino, L.; Assis, R.P.; Baviera, A.M.; Kalaba, P.; Dos Santos, F.N.; Éberlin, M.N.; et al. N,N′N″-Trisubstituted Guanidines: Synthesis, characterization, and evaluation of their leishmanicidal activity. Eur. J. Med. Chem. 2019, 171, 116–128. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Huebschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXS v 2016/4; University of Göttingen: Göttingen, Germany, 2015. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Nair, M.G. An efficient and economical MTT assay for determining the antioxidant activity of plant natural product extracts and pure compounds. J. Nat. Prod. 2010, 73, 1193–1195. [Google Scholar] [CrossRef]
- Labuda, R.; Bacher, M.; Gratzl, H.; Doppler, M.; Parich, A.; Aufy, M.; Lemmens-Gruber, R.; Schuhmacher, R.; Rychli, K.; Wagner, M.; et al. Luteapyrone, a novel ƴ-pyrone isolated from the filamentous fungus metapochonia lutea. Molecules 2021, 26, 6589. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.H.P.; Nussenzweig, V. A strain of Trypanosoma cruzi highly virulent for mice. Folia Clinica et Biologica 1953, 20, 191–208. [Google Scholar]
- Velásquez, A.M.A.; Ribeiro, W.C.; Venn, V.; Castelli, S.; Camargo, M.S.; Assis, R.P.; De Souza, R.A.; Ribeiro, A.R.; Passalacqua, T.G.; Da Rosa, J.A.; et al. Efficacy of a binuclear cyclopalladated compound therapy for cutaneous leishmaniasis in the murine model of infection with Leishmania amazonensis and its inhibitory effect on topoisomerase 1B. Antimicrob. Agents Chemother. 2017, 61, e00688-17. [Google Scholar] [CrossRef] [Green Version]
- Weigend, R. Ahlrichs Balanced basis set of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wires Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.; Passalacqua, T.G.; Dutra, L.A.; Fonseca, J.N.V.; Nascimento, R.F.Q.; Imamura, K.B.; Andrade, C.R.; Santos, J.L.; Graminha, M.A.S. In vivo antileishmanial activity and histopathological evaluation in Leishmania infantum infected hamsters after treatment with a furoxan derivative. Biomed. Pharmacother. 2017, 95, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, G.A.; Ayres, M.; Chelly, F.; Kilbourn, R.G. Effects of NG-methyl-L-arginine, NG-nitro-L-arginine, and aminoguanidine on constitutive and inducible nitric oxide synthase in rat aorta. Biochem. Biophys. Res. Commun. 1994, 199, 147–154. [Google Scholar] [CrossRef]
- Wideman, R.F.; Bowen, O.T.; Erf, G.F.; Chapman, M.E. Influence of aminoguanidine, an inhibitor of inducible nitric oxide synthase, on the pulmonary hypertensive response to microparticle injections in broilers. Poult. Sci. 2006, 85, 511–527. [Google Scholar] [CrossRef]
- Sadek, B.; Alisch, R.; Buschauer, A.; Elz, S. Synthesis, and dual histamine H1 and H2 receptor antagonist activity of cyanoguanidine derivatives. Molecules 2013, 18, 14186–14202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottram, J.C.; Brooks, D.R.; Coombs, G.H. Roles of cysteine proteinases of trypanosomes and leishmania in host-parasite interactions. Curr. Opin. Microbiol. 1998, 1, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Brinen, L.S.; Kerr, I.D.; Hansell, E.; Doyle, P.S.; McKerrow, J.H.; Roush, W.R. In vitro and in vivo studies of the trypanocidal properties of WRR-483 against trypanosoma cruzi. PLoS Negl. Trop. Dis. 2010, 4, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ran, J.; Jiang, T. Urea. In Urea Transporters; Subcellular Biochemistry; Yang, B., Sands, J., Eds.; Springer: Dordrecht, The Netherland, 2014; pp. 7–29. [Google Scholar] [CrossRef]
- Ramos, H.; Valdivieso, E.; Gamargo, M.; Dagger, F.; Cohen, B.E. Amphotericin B kills unicellular leishmanias by forming aqueous pores permeable to small cations and anions. J. Membr. Biol. 1996, 152, 65–75. [Google Scholar] [CrossRef]
- Ellis, M.E.; al-Hokail, A.A.; Clink, H.M.; Padmos, M.A.; Ernst, P.; Spence, D.G.; Tharpe, W.N.; Hillier, V.F. Double-blind randomized study of the effect of infusion rates on toxicity of Amphotericin B. Antimicrob. Agents Chemother. 1992, 36, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaña-Fouce, R.; Reguera, R.M.; Cubría, J.C.; Ordóñez, D. The pharmacology of leishmaniasis. Gen. Pharmacol. Vasc. Syst. 1998, 30, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Stauber, L.A. Host resistance to the khartoum strain of leishmania donovani. Rice Inst. Pam.-Rice Univ. Stud. 1958, 45, 80–96. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Ajay; Murcko, M.A. Computational Methods to Predict Binding Free Energy in Ligand-Receptor Complexes. J. Med. Chem. 1995, 38, 4953–4967. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Treatment |
|---|---|
| 1 | Infected and treated with PBS + 1% DMSO |
| 2 | Infected and treated with Amp B 2 mg/kg/day |
| 3 | Infected and treated with LQOF-G6 2 mg/kg/day |
| 4 | Uninfected and untreated |
| Sample | Instrument | Source | Temp. | Detector Distance | Time/Frame | #Frames | Frame Width | CCDC |
|---|---|---|---|---|---|---|---|---|
| [K] | [mm] | [s] | [°] | |||||
| LQOF-G6 | Bruker D8 Venture | Mo | 120 | 40 | 60 | 3506 | 0.3 | 2,144,089 |
| LQOF-G1 | Bruker D8 Venture | Mo | 100 | 34 | 15 | 4215 | 0.5 | 1,992,228 |
| Molecule | Entry | Atoms | Angle/° (A) | Atoms | Length (A) |
|---|---|---|---|---|---|
| LQOF-G6 | 1 | C2A-N1A-C1A-N3A | 0.9 (3) | H(C3A)-H(N3A) | 2.90 |
| 2 | C1A-N1A-C2A-C3A | 81.2(3) | H(C3A)-H(N1A) | 3.07 | |
| 3 | C1A-N1A-C2A-C7A | −98.6(2) | H(C7A)-H(N3A) | 3.43 | |
| 4 | N2A-C12A-C13A-C14A | 8.3(3) | H(C7A)-H(N1A) | 2.93 | |
| LQOF-G1 | 1 | C2A-N1A-C1A-N3A | 2.9 (3) | H(C3A)-H(N3A) | 4.46 |
| 2 | C1A-N1A-C2A-C3A | −36.4 (3) | H(C3A)-H(N1A) | 2.30 | |
| 3 | C1A-N1A-C2A-C7A | 14.0 (2) | H(C7A)-H(N3A) | 1.88 | |
| 4 | N2A-C12A-C13A-C14A | 3.7 (3) | H(C7A)-H(N1A) | 3.43 |
| Ligand | Affinity/kcal.mol−1 | Inhibition Index/% |
|---|---|---|
| LQOF-G1 | −7.9 | - |
| LQOF-G2 | −8.3 | - |
| LQOF-G6 | −8.8 | 73 |
| LQOF-G32 | −8.2 | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, V.P.; da Silva Mela, M.F.; Anjos, L.R.d.; Saraiva, L.F.; Arenas Velásquez, A.M.; Kalaba, P.; Fabisiková, A.; Clementino, L.d.C.; Aufy, M.; Studenik, C.; et al. Novel Selective and Low-Toxic Inhibitor of LmCPB2.8ΔCTE (CPB) One Important Cysteine Protease for Leishmania Virulence. Biomolecules 2022, 12, 1903. https://doi.org/10.3390/biom12121903
Moreira VP, da Silva Mela MF, Anjos LRd, Saraiva LF, Arenas Velásquez AM, Kalaba P, Fabisiková A, Clementino LdC, Aufy M, Studenik C, et al. Novel Selective and Low-Toxic Inhibitor of LmCPB2.8ΔCTE (CPB) One Important Cysteine Protease for Leishmania Virulence. Biomolecules. 2022; 12(12):1903. https://doi.org/10.3390/biom12121903
Chicago/Turabian StyleMoreira, Vitor Partite, Michele Ferreira da Silva Mela, Luana Ribeiro dos Anjos, Leonardo Figueiredo Saraiva, Angela M. Arenas Velásquez, Predrag Kalaba, Anna Fabisiková, Leandro da Costa Clementino, Mohammed Aufy, Christian Studenik, and et al. 2022. "Novel Selective and Low-Toxic Inhibitor of LmCPB2.8ΔCTE (CPB) One Important Cysteine Protease for Leishmania Virulence" Biomolecules 12, no. 12: 1903. https://doi.org/10.3390/biom12121903
APA StyleMoreira, V. P., da Silva Mela, M. F., Anjos, L. R. d., Saraiva, L. F., Arenas Velásquez, A. M., Kalaba, P., Fabisiková, A., Clementino, L. d. C., Aufy, M., Studenik, C., Gajic, N., Prado-Roller, A., Magalhães, A., Zehl, M., Figueiredo, I. D., Baviera, A. M., Cilli, E. M., Graminha, M. A. S., Lubec, G., & Gonzalez, E. R. P. (2022). Novel Selective and Low-Toxic Inhibitor of LmCPB2.8ΔCTE (CPB) One Important Cysteine Protease for Leishmania Virulence. Biomolecules, 12(12), 1903. https://doi.org/10.3390/biom12121903