




The Role of Membrane Affinity and Binding Modes in Alpha-Synuclein Regulation of Vesicle Release and Trafficking

 , , , and
, , , and
Abstract
:
1. Introduction
2. Methods
2.1. Expression and Purification of Alpha-Synuclein Variants
2.2. Lipid Vesicle Preparation and Characterization
2.3. NMR
2.4. Paramagnetic Relaxation Enhancement Experiments
2.5. Tryptophan Fluorescence
2.6. Exocytosis
2.7. Distribution of Recycling Endosomes
2.8. Expression Levels of Alpha-Synuclein Using Immunocytochemistry
2.9. Effects of Alpha-Synuclein on Enrichment of Lipid Vesicles in Synapsin-1 Condensates
3. Results
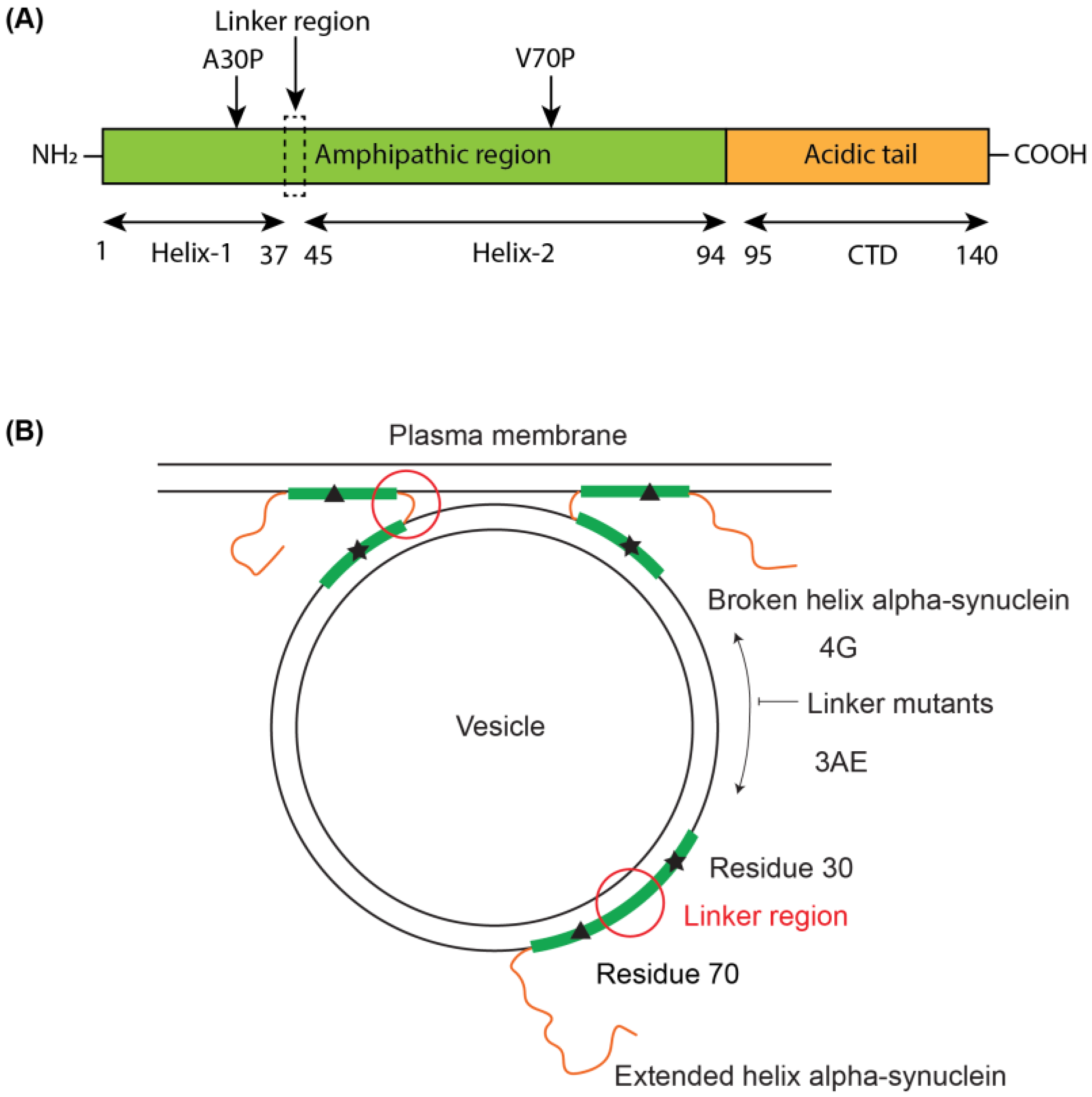
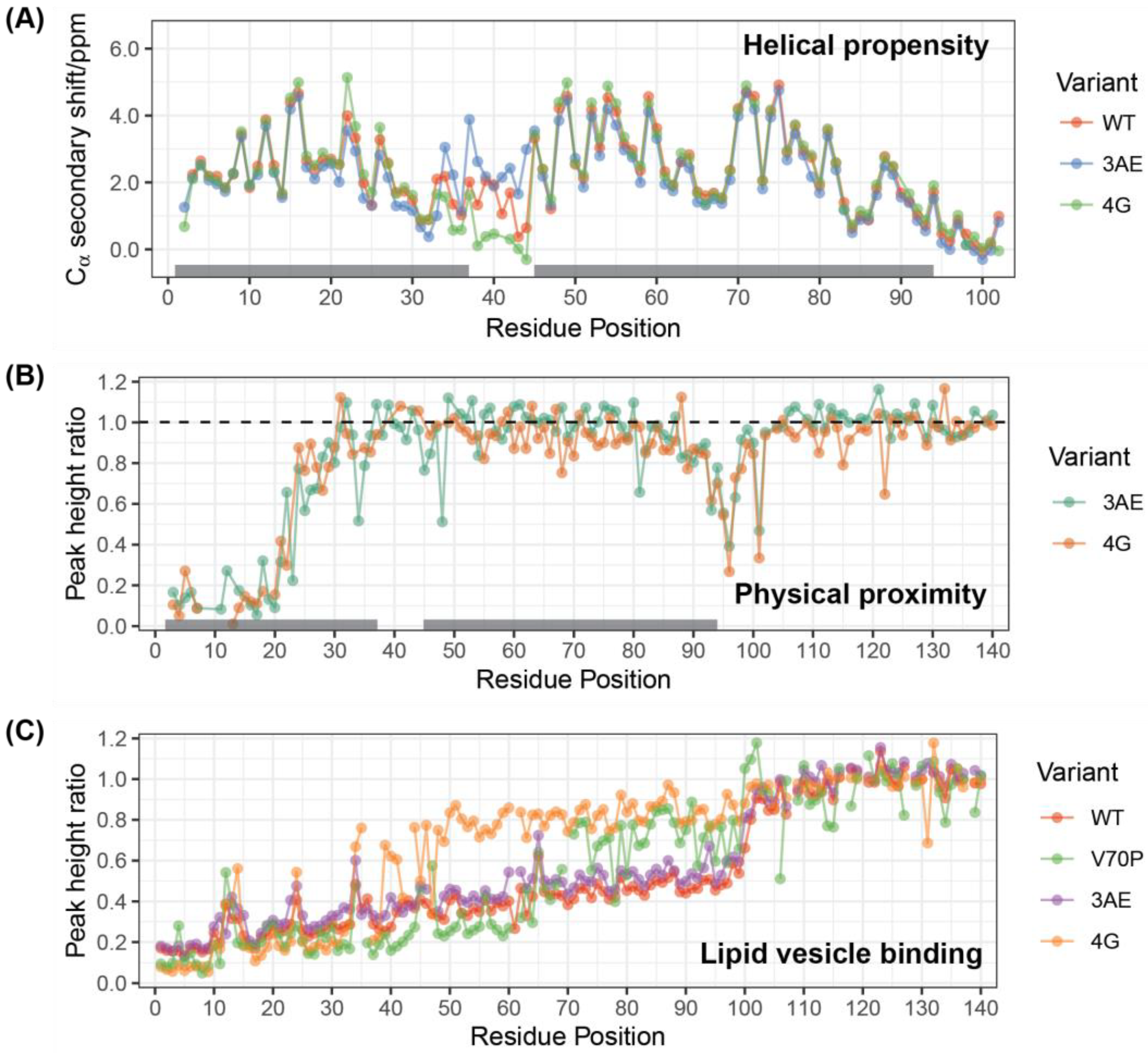
3.1. Linker Mutants Bias Linker Region Helicity in Micelle-Bound State
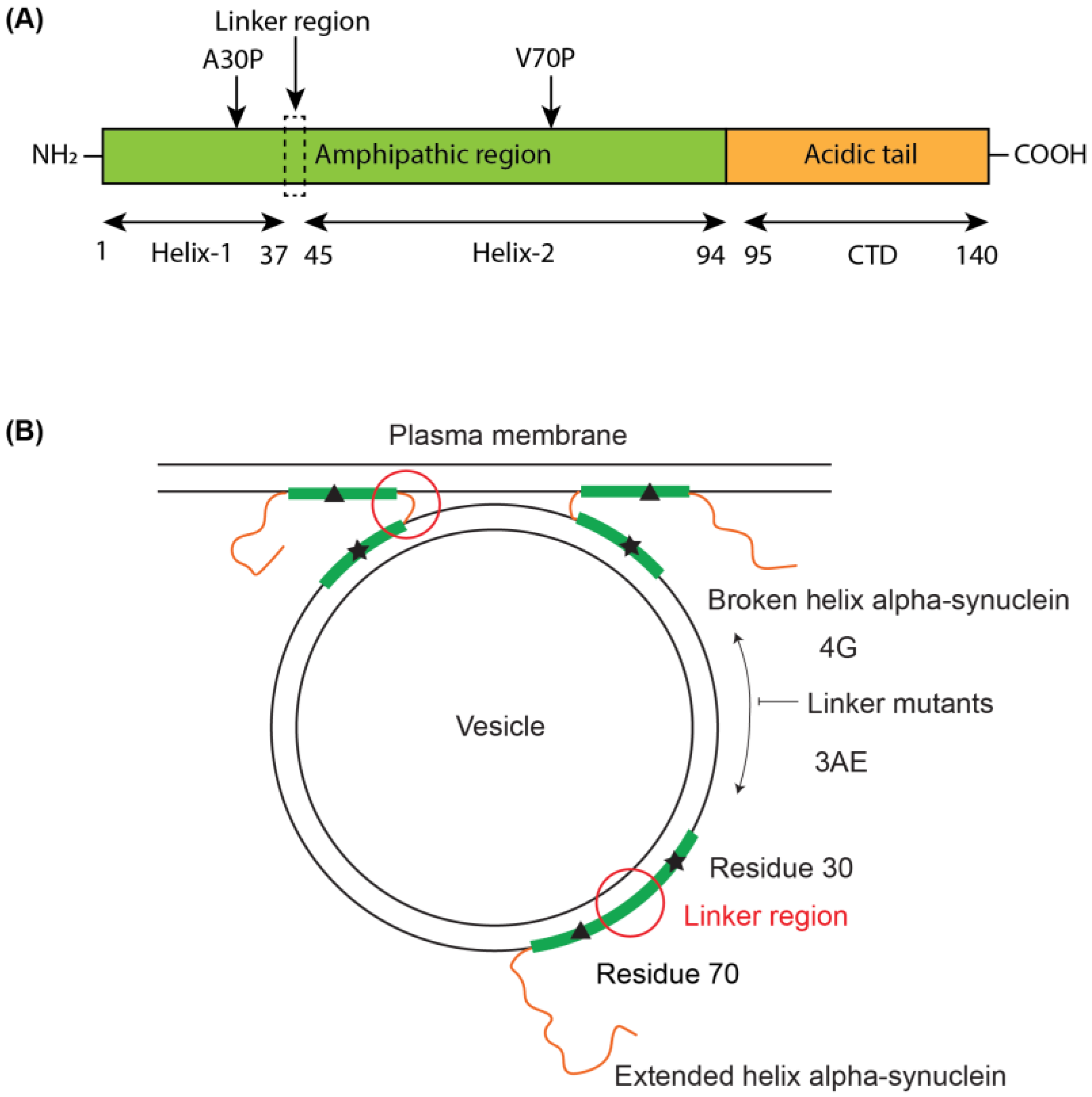
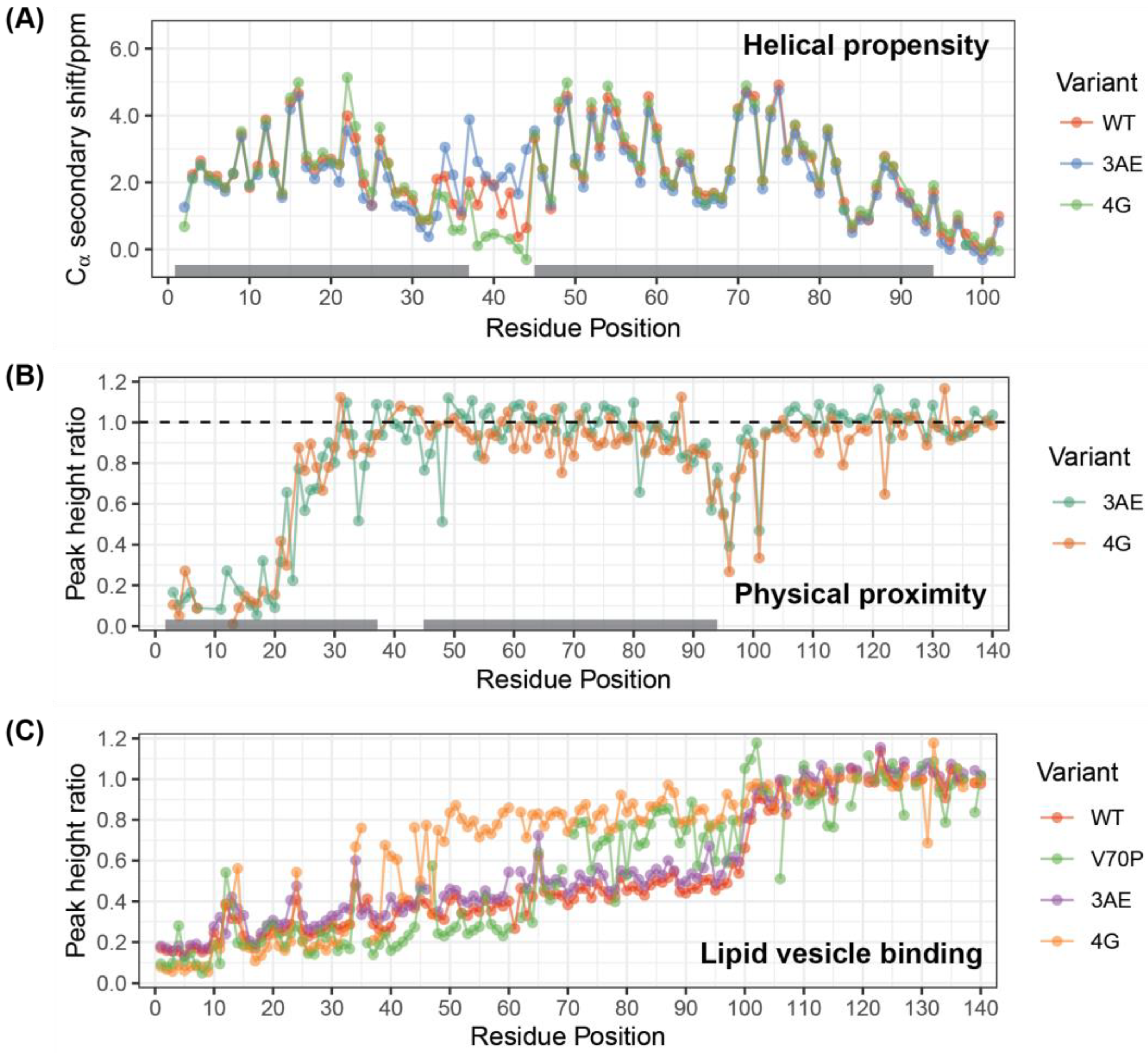
3.2. 4G Mutant Prevents Propagation of the Vesicle-Bound Extended-Helix Conformation
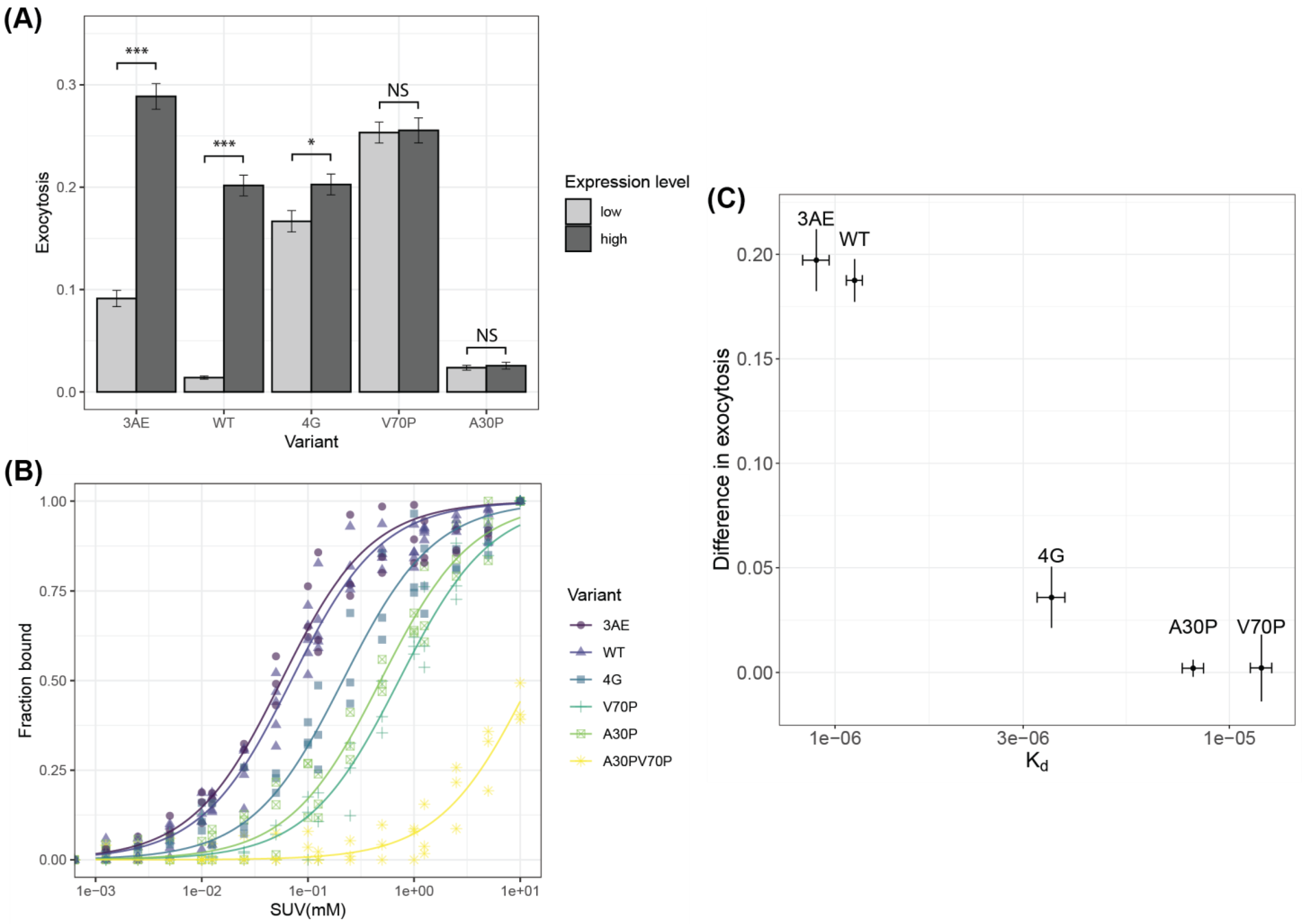
3.3. Linker Mutants Alter Effects of Alpha-Synuclein on Vesicle Release

3.4. Membrane Affinity for Isolated Vesicles Correlates with Potentiation of Exocytosis
3.5. Alpha-Synuclein Can Disperse Vesicles from Condensates In Vitro
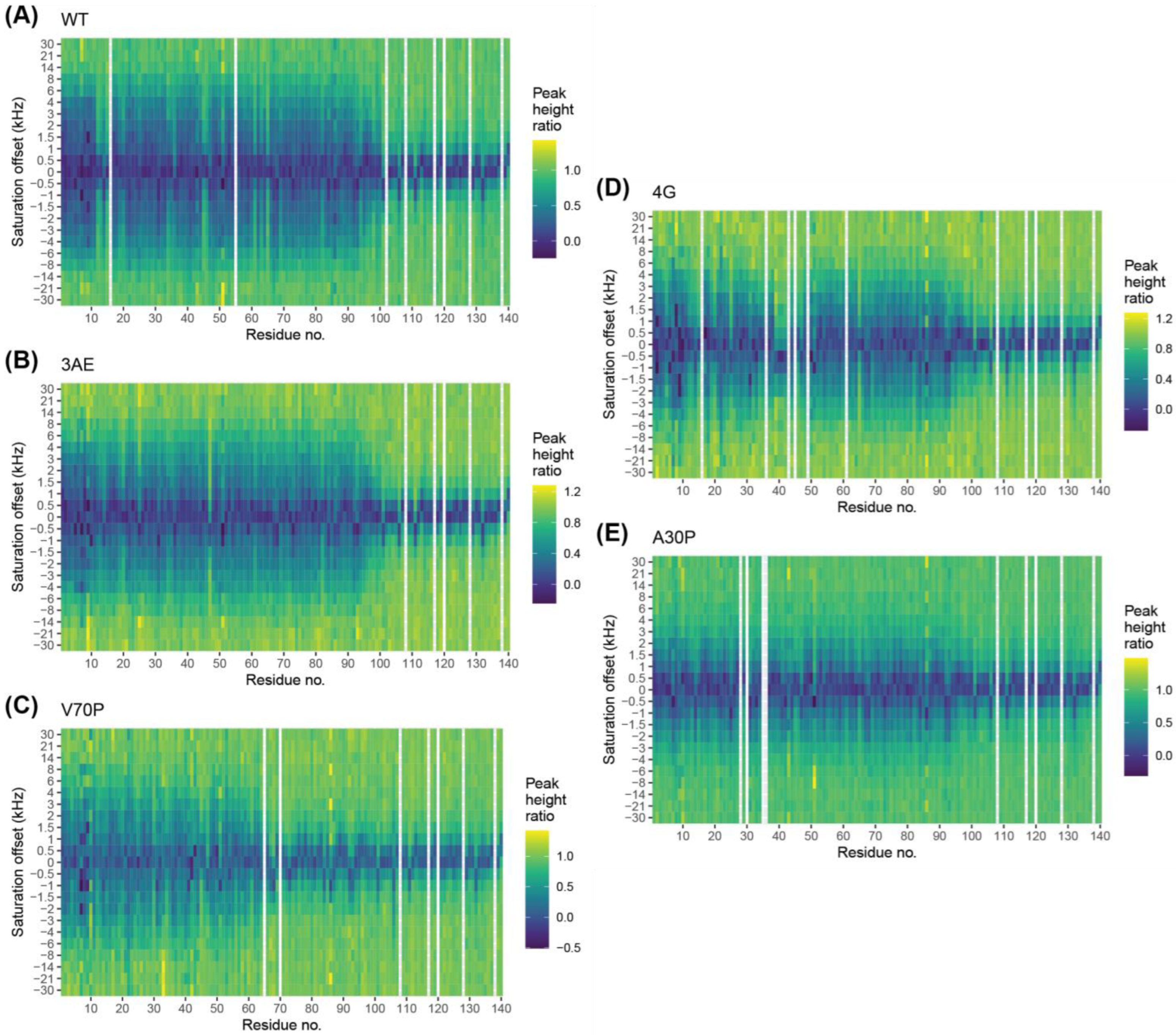
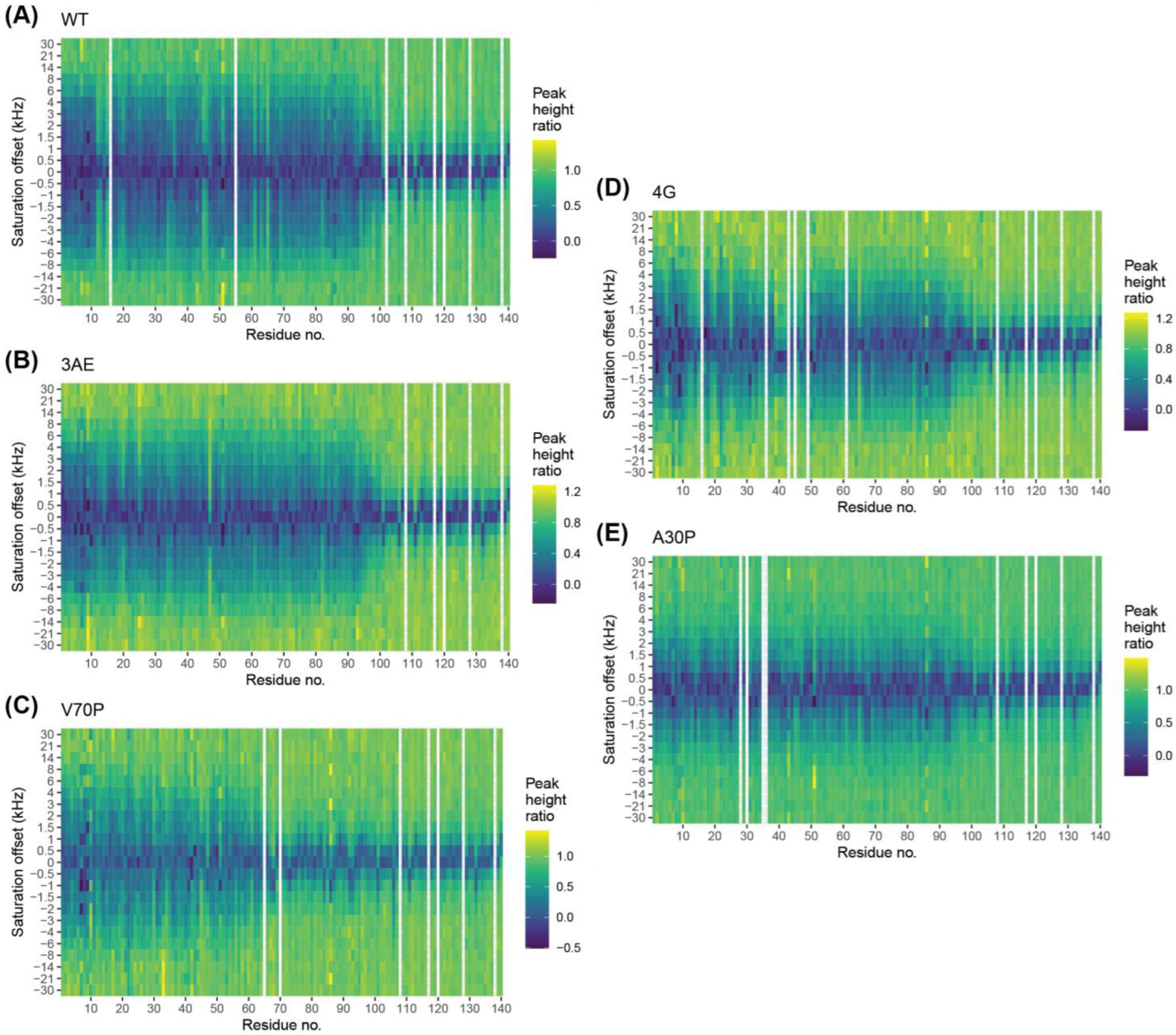
3.6. Bipartite Binding Contributes to Affinity of Alpha-Synuclein to Membranes
3.7. Quantitative Analysis of DEST Data Support Bipartite Binding Model
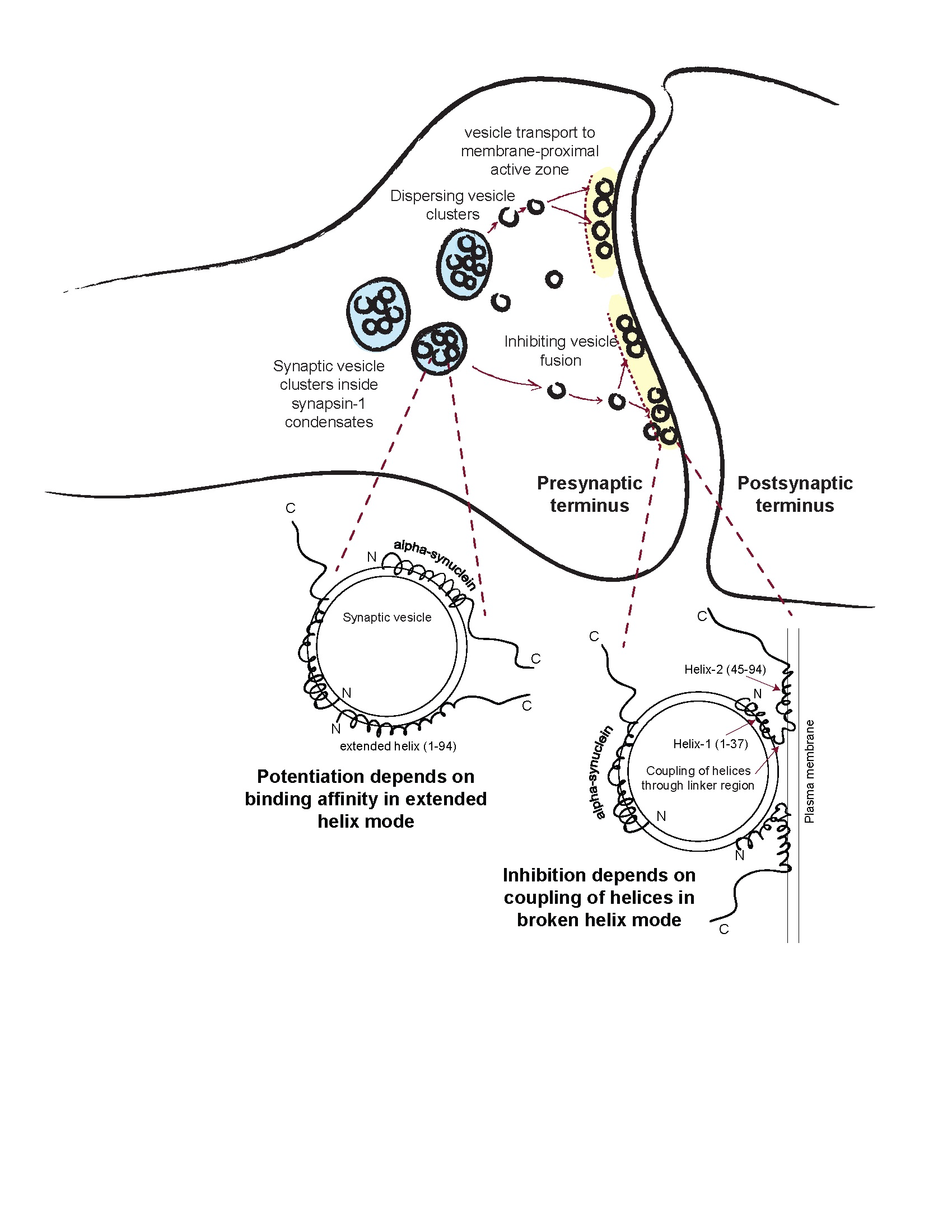
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The Process of Lewy Body Formation, Rather than Simply α-Synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A Neuron-Specific Protein Localized to the Nucleus and Presynaptic Nerve Terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a Novel Protein Regulated during the Critical Period for Song Learning in the Zebra Finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ninan, I.; Antonova, I.; Battaglia, F.; Trinchese, F.; Narasanna, A.; Kolodilov, N.; Dauer, W.; Hawkins, R.D.; Arancio, O. Alpha-Synuclein Produces a Long-Lasting Increase in Neurotransmitter Release. EMBO J. 2004, 23, 4506–4516. [Google Scholar] [CrossRef] [Green Version]
- Gureviciene, I.; Gurevicius, K.; Tanila, H. Role of Alpha-Synuclein in Synaptic Glutamate Release. Neurobiol. Dis. 2007, 28, 83–89. [Google Scholar] [CrossRef]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic Vesicle Depletion Correlates with Attenuated Synaptic Responses to Prolonged Repetitive Stimulation in Mice Lacking Alpha-Synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef] [Green Version]
- Martín, E.D.; González-García, C.; Milán, M.; Fariñas, I.; Ceña, V. Stressor-Related Impairment of Synaptic Transmission in Hippocampal Slices from Alpha-Synuclein Knockout Mice. Eur. J. Neurosci. 2004, 20, 3085–3091. [Google Scholar] [CrossRef]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice Lacking Alpha-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Senior, S.L.; Ninkina, N.; Deacon, R.; Bannerman, D.; Buchman, V.L.; Cragg, S.J.; Wade-Martins, R. Increased Striatal Dopamine Release and Hyperdopaminergic-like Behaviour in Mice Lacking Both Alpha-Synuclein and Gamma-Synuclein. Eur. J. Neurosci. 2008, 27, 947–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavich, L.; Tanila, H.; Vepsäläinen, S.; Jäkälä, P. Role of Alpha-Synuclein in Presynaptic Dopamine Recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, J.B.; Hatami, A.; David, H.; Masliah, E.; Roberts, K.; Evans, C.E.; Levine, M.S. Alterations in Corticostriatal Synaptic Plasticity in Mice Overexpressing Human Alpha-Synuclein. Neuroscience 2009, 159, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, S.; Fornai, F.; Kwon, H.-B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-Knockout Mice for Alpha- and Beta-Synucleins: Effect on Synaptic Functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [Green Version]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-Robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional Alterations to the Nigrostriatal System in Mice Lacking All Three Members of the Synuclein Family. J. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef] [Green Version]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Joshi, P.R.; Cepeda, C.; Masliah, E.; Levine, M.S. Alpha-Synuclein Overexpression in Mice Alters Synaptic Communication in the Corticostriatal Pathway. J. Neurosci. Res. 2010, 88, 1764–1776. [Google Scholar] [CrossRef] [Green Version]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. Alpha-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.A.; Tabarean, I.; Tang, Y.; Cartier, A.; Masliah, E.; Roy, S. A Pathologic Cascade Leading to Synaptic Dysfunction in Alpha-Synuclein-Induced Neurodegeneration. J. Neurosci. 2010, 30, 8083–8095. [Google Scholar] [CrossRef] [Green Version]
- Janezic, S.; Threlfell, S.; Dodson, P.D.; Dowie, M.J.; Taylor, T.N.; Potgieter, D.; Parkkinen, L.; Senior, S.L.; Anwar, S.; Ryan, B.; et al. Deficits in Dopaminergic Transmission Precede Neuron Loss and Dysfunction in a New Parkinson Model. Proc. Natl. Acad. Sci. USA 2013, 110, E4016-25. [Google Scholar] [CrossRef]
- Sudhof, T.C.; Rizo, J. Synaptic Vesicle Exocytosis. Cold Spring Harb Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, S.O.; Betz, W.J. Synaptic Vesicle Pools. Nat. Rev. Neurosci. 2005, 6, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Alabi, A.A.; Tsien, R.W. Synaptic Vesicle Pools and Dynamics. Cold Spring Harb Perspect. Biol. 2012, 4, a013680. [Google Scholar] [CrossRef] [Green Version]
- Vargas, K.J.; Schrod, N.; Davis, T.; Fernandez-Busnadiego, R.; Taguchi, Y.V.; Laugks, U.; Lucic, V.; Chandra, S.S. Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep. 2017, 18, 161–173. [Google Scholar] [CrossRef]
- Dikiy, I.; Fauvet, B.; Jovičić, A.; Mahul-Mellier, A.-L.; Desobry, C.; El-Turk, F.; Gitler, A.D.; Lashuel, H.A.; Eliezer, D. Semisynthetic and in Vitro Phosphorylation of Alpha-Synuclein at Y39 Promotes Functional Partly Helical Membrane-Bound States Resembling Those Induced by PD Mutations. ACS Chem. Biol. 2016, 11, 2428–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S. Novel Regulation of Parkin Function through C-Abl-Mediated Tyrosine Phosphorylation: Implications for Parkinson’s Disease. J. Neurosci. 2011, 31, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The C-Abl Inhibitor, Nilotinib, Protects Dopaminergic Neurons in a Preclinical Animal Model of Parkinson’s Disease. Sci. Rep. 2014, 4, 4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.S.; Lee, Y.; Shin, J.H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the C-Abl Protein Tyrosine Kinase Inhibits Parkin’s Ubiquitination and Protective Function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell, R.; Browne, G. Conformational Properties of α-Synuclein in Its Free and Lipid-Associated States. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef]
- Johnson, M.; Geeves, M.A.; Mulvihill, D.P. Production of Amino-Terminally Acetylated Recombinant Proteins in E. Coli. Methods Mol. Biol. 2013, 981, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Coulton, A.T.; Geeves, M.A.; Mulvihill, D.P. Targeted Amino-Terminal Acetylation of Recombinant Proteins in E. Coli. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both Familial Parkinson’s Disease Mutations Accelerate α-Synuclein Aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho-Cerqueira, E.; Carmo-Gonçalves, P.; Sá Pinheiro, A.; Cortines, J.; Follmer, C. α-Synuclein as an Intrinsically Disordered Monomer - Fact or Artefact? FEBS J. 2013, 280, 4915–4927. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, J.W.; Kelly, R.B. Lipids of Synaptic Vesicles: Relevance to the Mechanism of Membrane Fusion. Biochemistry 1981, 20, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular Anatomy of a Trafficking Organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breckenridge, W.C.C.; Morgan, I.G.G.; Zanetta, J.P.P.; Vincendon, G. Adult Rat Brain Synaptic Vesicles Ii. Lipid Composition. Biochim. Biophys. Acta 1973, 320, 681–686. [Google Scholar] [CrossRef]
- Rouser, G.; Siakotos, A.N.; Fleischer, S. Quantitative Analysis of Phospholipids by Thin-Layer Chromatography and Phosphorus Analysis of Spots. Lipids 1966, 1, 85–86. [Google Scholar] [CrossRef]
- Rouser, G.; Fleischer, S.; Yamamoto, A. Two Dimensional Thin Layer Chromatographic Separation of Polar Lipids and Determination of Phospholipids by Phosphorus Analysis of Spots. Lipids 1970, 5, 494–496. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN Data Model for NMR Spectroscopy: Development of a Software Pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, N.L.; Ying, J.; Torchia, D.A.; Clore, G.M. Probing Exchange Kinetics and Atomic Resolution Dynamics in High-Molecular-Weight Complexes Using Dark-State Exchange Saturation Transfer NMR Spectroscopy. Nat. Protoc. 2012, 7, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- McConnell, H.M. Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 1958, 28, 430–431. [Google Scholar] [CrossRef] [Green Version]
- Helgstrand, M.; Härd, T.; Allard, P. Simulations of NMR Pulse Sequences during Equilibrium and Non-Equilibrium Chemical Exchange. J. Biomol. NMR 2000, 18, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Burstein, E.A.; Emelyanenko, V.I. Log-Normal Description of Fluorescence Spectra of Organic Fluorophores. Photochem. Photobiol. 1996, 64, 316–320. [Google Scholar] [CrossRef]
- Burstein, E.A.; Abornev, S.M.; Reshetnyak, Y.K. Decomposition of Protein Tryptophan Fluorescence Spectra into Log-Normal Components. I. Decomposition Algorithms. Biophys. J. 2001, 81, 1699–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siano, D.B.; Metzler, D.E. Band Shapes of the Electronic Spectra of Complex Molecules. J. Chem. Phys. 1969, 51, 1856–1861. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Jayasinghe, S.; White, S.H. How to Measure and Analyze Tryptophan Fluorescence in Membranes Properly, and Why Bother? Anal. Biochem. 2000, 285, 235–245. [Google Scholar] [CrossRef]
- Burstein, E.A.; Vedenkina, N.S.; Ivkova, M.N. Fluorescence and the Location of Tryptophan Residues in Protein Molecules. Photochem. Photobiol. 1973, 18, 263–279. [Google Scholar] [CrossRef]
- Dikiy, I.; Eliezer, D. N-Terminal Acetylation Stabilizes N-Terminal Helicity in Lipid- and Micelle-Bound α-Synuclein and Increases Its Affinity for Physiological Membranes. J. Biol. Chem. 2014, 289, 3652–3665. [Google Scholar] [CrossRef]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-Terminal Acetylation of α-Synuclein Induces Increased Transient Helical Propensity and Decreased Aggregation Rates in the Intrinsically Disordered Monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of N-Terminal Acetylation of α-Synuclein on Its Random Coil and Lipid Binding Properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, C.M.; Lee, J.C. Tryptophan Probes at the Alpha-Synuclein and Membrane Interface. J. Phys. Chem. B 2010, 114, 4615–4622. [Google Scholar] [CrossRef] [Green Version]
- Gosse, J.A.; Wagenknecht-Wiesner, A.; Holowka, D.; Baird, B. Transmembrane Sequences Are Determinants of Immunoreceptor Signaling. J. Immunol. 2005, 175, 2123–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkes, M.M.; Wilson, J.D.; Baird, B.; Holowka, D. Activation of Cdc42 Is Necessary for Sustained Oscillations of Ca2+and PIP2 Stimulated by Antigen in RBL Mast Cells. Biol. Open 2014, 3, 700–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.D.; Shelby, S.A.; Holowka, D.; Baird, B. Rab11 Regulates the Mast Cell Exocytic Response. Traffic 2016, 17, 1027–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramezani, M.; Wilkes, M.M.; Das, T.; Holowka, D.; Eliezer, D.; Baird, B. Regulation of Exocytosis and Mitochondrial Relocalization by Alpha-Synuclein in a Mammalian Cell Model. NPJ Parkinsons Dis. 2019, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Milovanovic, D.; Wu, Y.; Bian, X.; De Camilli, P. A Liquid Phase of Synapsin and Lipid Vesicles. Science 2018, 361, 604–607. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Hohng, S.; Ha, T. A Practical Guide to Single-Molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Li, C.H.; Tam, P.K.S. An Iterative Algorithm for Minimum Cross Entropy Thresholding. Pattern Recognit. Lett. 1998, 19, 771–776. [Google Scholar] [CrossRef]
- Snead, D. Mechanisms of Inhibition of Synaptic Vesicle Exocytosis by Complexin; Weill Medical College of Cornell University: New York, NY, USA, 2015. [Google Scholar]
- Blaber, M.; Zhang, X.J.; Matthews, B.W. Structural Basis of Amino Acid Alpha Helix Propensity. Science 1993, 260, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Scholtz, J.M. A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Marius Clore, G.; Iwahara, J. Theory, Practice, and Applications of Paramagnetic Relaxation Enhancement for the Characterization of Transient Low-Population States of Biological Macromolecules and Their Complexes. Chem. Rev. 2009, 109, 4108–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clore, G.M. Practical Aspects of Paramagnetic Relaxation Enhancement in Biological Macromolecules, 1st ed.; Qin, P.Z., Warncke, K., Eds.; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Rao, J.N.; Kim, Y.E.; Park, L.S.; Ulmer, T.S. Effect of Pseudorepeat Rearrangement on Alpha-Synuclein Misfolding, Vesicle Binding, and Micelle Binding. J. Mol. Biol. 2009, 390, 516–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct Observation of the Three Regions in α-Synuclein That Determine Its Membrane-Bound Behaviour. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholak, E.; Bugge, K.; Khondker, A.; Gauger, K.; Pedraz-Cuesta, E.; Pedersen, M.E.; Bucciarelli, S.; Vestergaard, B.; Pedersen, S.F.; Rheinstädter, M.C.; et al. Avidity within the N-Terminal Anchor Drives α-Synuclein Membrane Interaction and Insertion. FASEB J. 2020, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin Inhibits the Sarcoplasmic or Endoplasmic Reticulum Ca-ATPase Family of Calcium Pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [CrossRef]
- Chen, Y.; Barkley, M.D. Toward Understanding Tryptophan Fluorescence in Proteins. Biochemistry 1998, 37, 9976–9982. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Jensen, P.H.; Marsh, D. Alpha-Synuclein Association with Phosphatidylglycerol Probed by Lipid Spin Labels. Biochemistry 2003, 42, 12919–12926. [Google Scholar] [CrossRef]
- Hoffmann, C.; Sansevrino, R.; Morabito, G.; Logan, C.; Vabulas, R.M.; Ulusoy, A.; Ganzella, M.; Milovanovic, D. Synapsin Condensates Recruit Alpha-Synuclein. J. Mol. Biol. 2021, 433, 166961. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Paya, C.; Sanz-Hernandez, M.; De Simone, A. Plasticity of Membrane Binding by the Central Region of α-Synuclein. Front. Mol. Biosci. 2022, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A. Comparison of Structure and Dynamics of Micelle-Bound Human Alpha-Synuclein and Parkinson Disease Variants. J. Biol. Chem. 2005, 280, 43179–43187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussell, R.; Eliezer, D. Effects of Parkinson’s Disease-Linked Mutations on the Structure of Lipid-Associated Alpha-Synuclein. Biochemistry 2004, 43, 4810–4818. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, N.L.; Ying, J.; Ghirlando, R.; Torchia, D.A.; Clore, G.M. Atomic-Resolution Dynamics on the Surface of Amyloid-β Protofibrils Probed by Solution NMR. Nature 2011, 480, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Busch, D.J.; Oliphint, P.A.; Walsh, R.B.; Banks, S.M.L.; Woods, W.S.; George, J.M.; Morgan, J.R. Acute Increase of α-Synuclein Inhibits Synaptic Vesicle Recycling Evoked during Intense Stimulation. Mol. Biol. Cell 2014, 25, 3926–3941. [Google Scholar] [CrossRef]
- Ben Gedalya, T.; Loeb, V.; Israeli, E.; Altschuler, Y.; Selkoe, D.J.; Sharon, R. Alpha-Synuclein and Polyunsaturated Fatty Acids Promote Clathrin-Mediated Endocytosis and Synaptic Vesicle Recycling. Traffic 2009, 10, 218–234. [Google Scholar] [CrossRef] [Green Version]
- Schechter, M.; Atias, M.; Abd Elhadi, S.; Davidi, D.; Gitler, D.; Sharon, R. α-Synuclein Facilitates Endocytosis by Elevating the Steady-State Levels of Phosphatidylinositol 4,5-Bisphosphate. J. Biol. Chem. 2020, 295, 18076–18090. [Google Scholar] [CrossRef]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins Regulate the Kinetics of Synaptic Vesicle Endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef]
- Scott, D.; Roy, S. α-Synuclein Inhibits Intersynaptic Vesicle Mobility and Maintains Recycling-Pool Homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein Promotes Dilation of the Exocytotic Fusion Pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Ruipérez, V.; López, I.; Villanueva, J.; Gutierrez, L.M.; Davletov, B. Alpha-Synuclein Sequesters Arachidonic Acid to Modulate SNARE-Mediated Exocytosis. EMBO Rep. 2010, 11, 528–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Kim, S.; Varkey, J.; Lou, X.; Song, J.-K.; Diao, J.; Langen, R.; Shin, Y.-K. Nonaggregated α-Synuclein Influences SNARE-Dependent Vesicle Docking via Membrane Binding. Biochemistry 2014, 53, 3889–3896. [Google Scholar] [CrossRef]
- Eliezer, D. Protein Folding and Aggregation in in Vitro Models of Parkinson’s Disease: Structure and Function of α–Synuclein. In Parkinson’s Disease: Molecular and Therapeutic Insights from Model Systems; Nass, R., Przedborski, S., Eds.; Academic Press: New York, NY, USA, 2008; pp. 575–595. [Google Scholar]
- Georgieva, E.R.; Ramlall, T.F.; Borbat, P.P.; Freed, J.H.; Eliezer, D. The Lipid-Binding Domain of Wild Type and Mutant Alpha-Synuclein: Compactness and Interconversion between the Broken and Extended Helix Forms. J. Biol. Chem. 2010, 285, 28261–28274. [Google Scholar] [CrossRef] [Green Version]
- Dikiy, I.; Eliezer, D. Folding and Misfolding of Alpha-Synuclein on Membranes. Biochim. Biophys. Acta 2012, 1818, 1013–1018. [Google Scholar] [CrossRef] [Green Version]
- Snead, D.; Eliezer, D. A-Synuclein Function and Dysfunction on Cellular Membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef] [Green Version]
- Man, W.K.; Tahirbegi, B.; Vrettas, M.D.; Preet, S.; Ying, L.; Vendruscolo, M.; De Simone, A.; Fusco, G. The Docking of Synaptic Vesicles on the Presynaptic Membrane Induced by α-Synuclein Is Modulated by Lipid Composition. Nat. Commun. 2021, 12, 927. [Google Scholar] [CrossRef]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Kaminski Schierle, G.S.; Vendruscolo, M.; Veglia, G.; Dobson, C.M.; et al. Structural Basis of Synaptic Vesicle Assembly Promoted by α-Synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Kd | SEM | p-Value |
|---|---|---|---|
| WT | |||
| A30P | |||
| V70P | |||
| A30P/V70P | |||
| 3AE | |||
| 4G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, T.; Ramezani, M.; Snead, D.; Follmer, C.; Chung, P.; Lee, K.Y.; Holowka, D.A.; Baird, B.A.; Eliezer, D. The Role of Membrane Affinity and Binding Modes in Alpha-Synuclein Regulation of Vesicle Release and Trafficking. Biomolecules 2022, 12, 1816. https://doi.org/10.3390/biom12121816
Das T, Ramezani M, Snead D, Follmer C, Chung P, Lee KY, Holowka DA, Baird BA, Eliezer D. The Role of Membrane Affinity and Binding Modes in Alpha-Synuclein Regulation of Vesicle Release and Trafficking. Biomolecules. 2022; 12(12):1816. https://doi.org/10.3390/biom12121816
Chicago/Turabian StyleDas, Tapojyoti, Meraj Ramezani, David Snead, Cristian Follmer, Peter Chung, Ka Yee Lee, David A. Holowka, Barbara A. Baird, and David Eliezer. 2022. "The Role of Membrane Affinity and Binding Modes in Alpha-Synuclein Regulation of Vesicle Release and Trafficking" Biomolecules 12, no. 12: 1816. https://doi.org/10.3390/biom12121816
APA StyleDas, T., Ramezani, M., Snead, D., Follmer, C., Chung, P., Lee, K. Y., Holowka, D. A., Baird, B. A., & Eliezer, D. (2022). The Role of Membrane Affinity and Binding Modes in Alpha-Synuclein Regulation of Vesicle Release and Trafficking. Biomolecules, 12(12), 1816. https://doi.org/10.3390/biom12121816