

Structural Achievability of an NH–π Interaction between Gln and Phe in a Crystal Structure of a Collagen-like Peptide


Abstract
1. Introduction
2. Materials and Methods
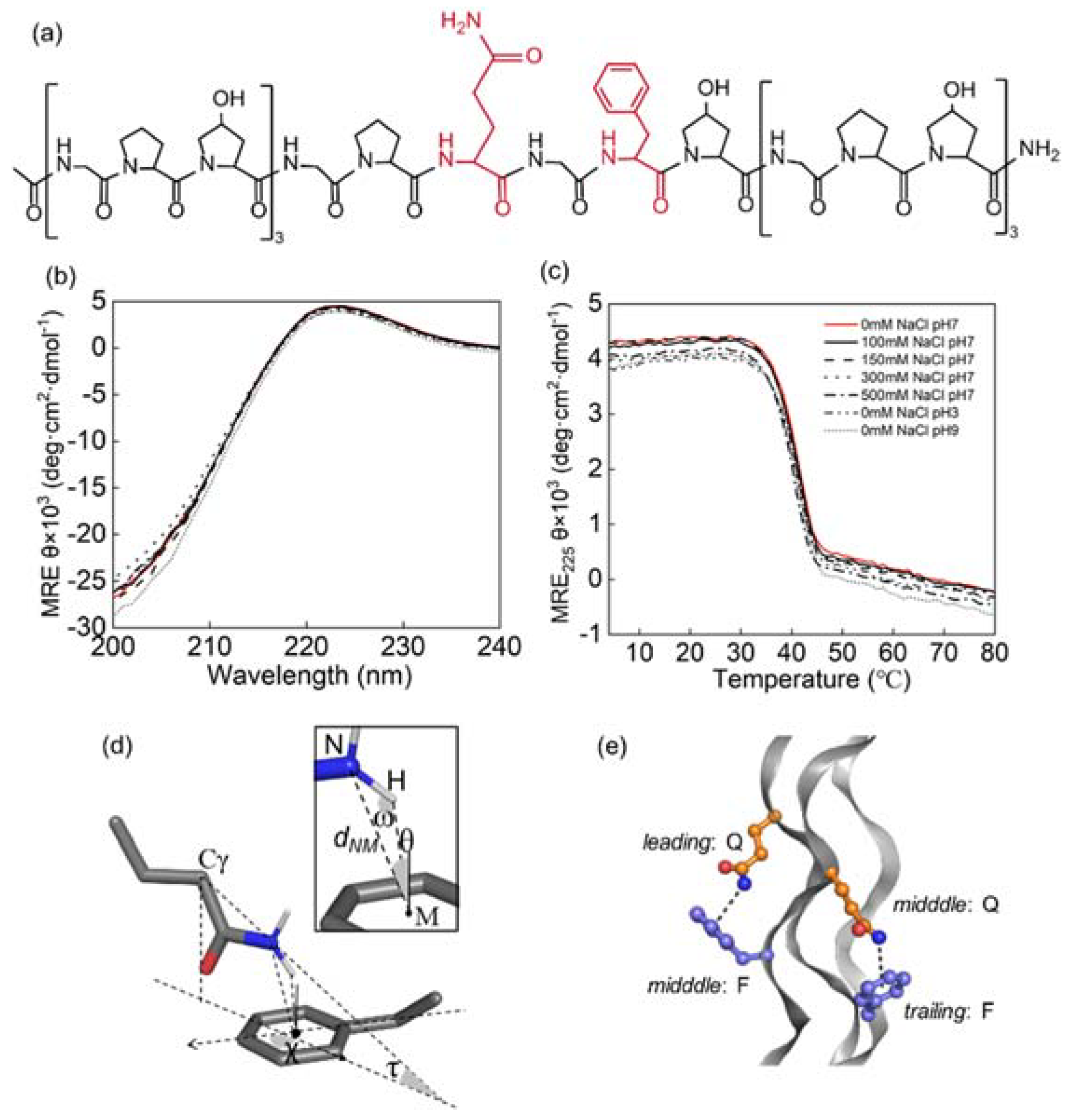
2.1. Peptide Synthesis and Purification
2.2. Crystallization and Data Collection
2.3. Structure Determination and Refinement
2.4. Circular Dichroism (CD) Spectra
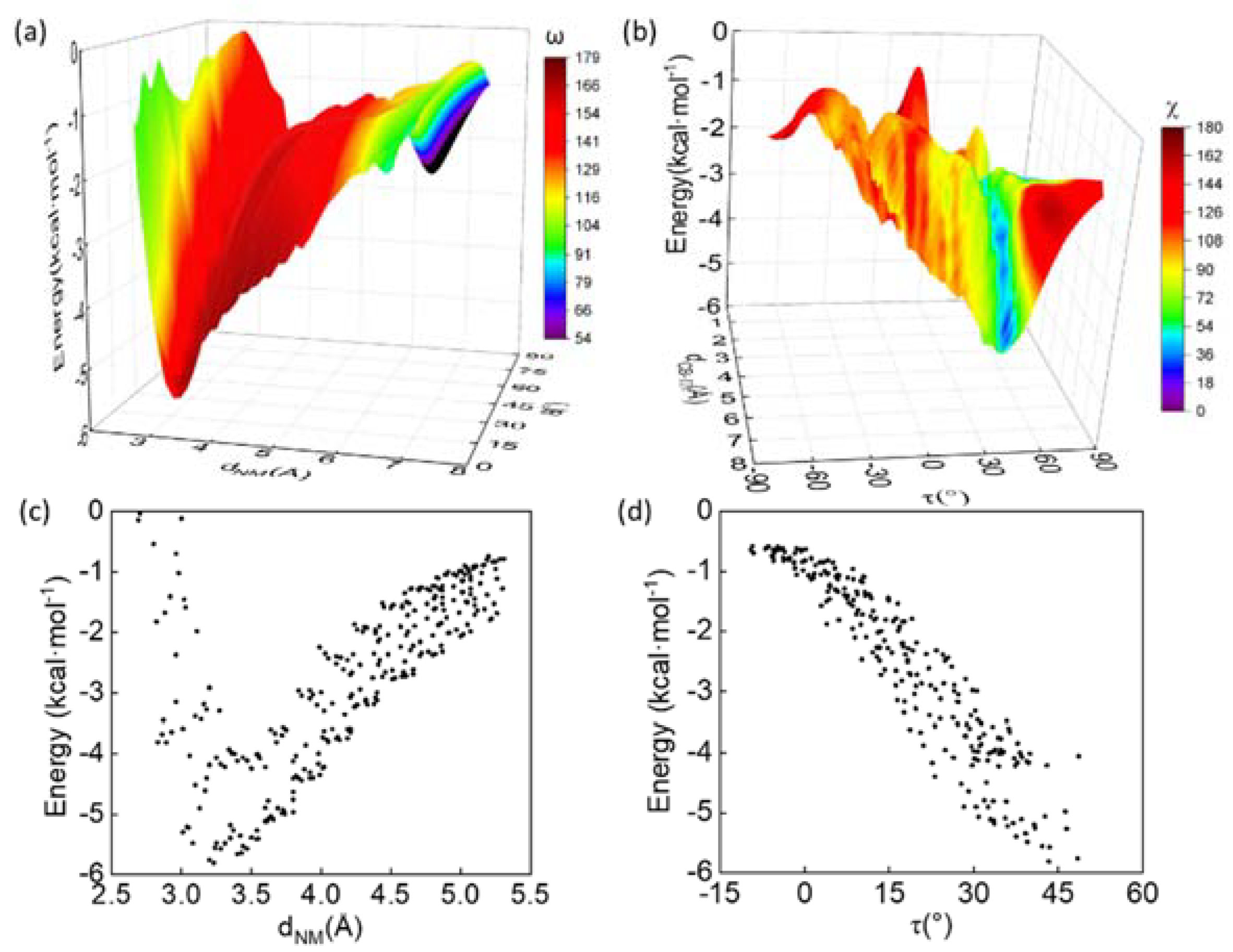
2.5. Quantum Mechanical (QM) Analysis
2.6. Calculation of Geometric Parameters
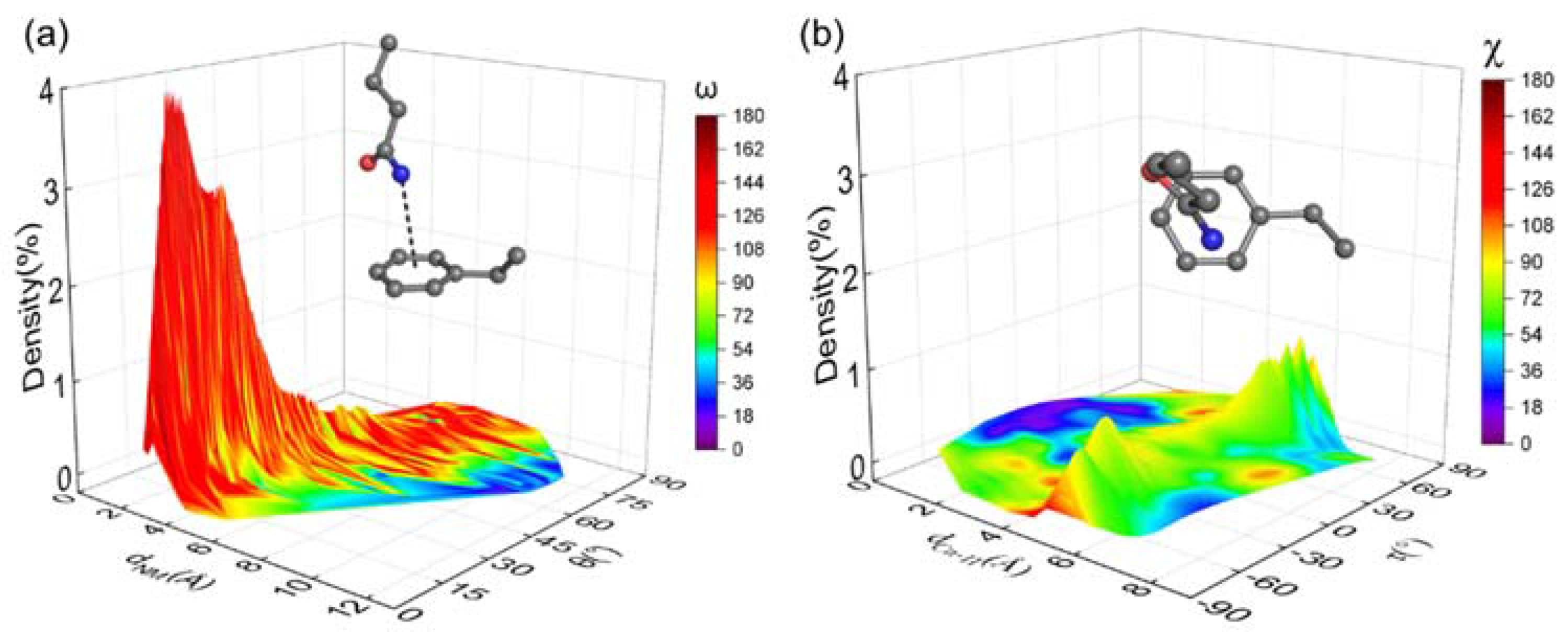
2.7. Molecular Dynamics (MD) Simulation
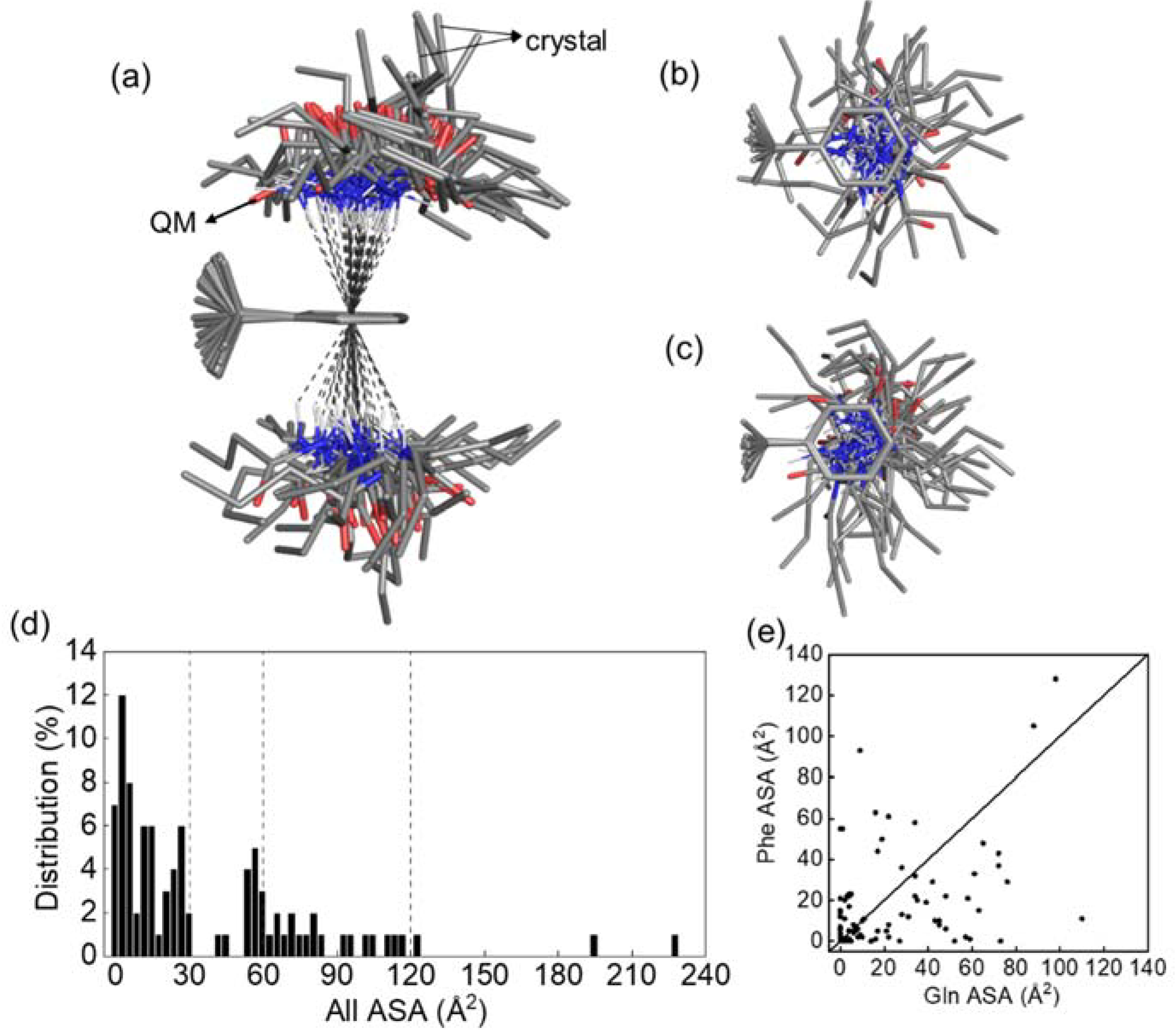
2.8. Database Analysis of Q-F Pair Interaction
3. Results
3.1. Triple Helices Remained Stable under Various Ionic Strengths and pH
3.2. Side Chain Conformations in Crystal Structure
3.3. Energetic Favorability of Amide–π Interactions Characterized with Quantum Methanics
3.4. Balanced Contributions from Various Types of Chemical Forces
3.5. Environmental Dielectric Constants
3.6. Conformational Distribution Sampled by Molecular Dynamics Simulations
3.7. Data Mining of Protein Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steiner, T.; Koellner, G. Hydrogen bonds with pi-acceptors in proteins: Frequencies and role in stabilizing local 3D structures. J. Mol. Biol. 2001, 305, 535–557. [Google Scholar] [CrossRef]
- Andrew, C.D.; Bhattacharjee, S.; Kokkoni, N.; Hirst, J.D.; Jones, G.R.; Doig, A.J. Stabilizing Interactions between Aromatic and Basic Side Chains in α-Helical Peptides and Proteins. Tyrosine Effects on Helix Circular Dichroism. J. Am. Chem. Soc. 2002, 124, 12706–12714. [Google Scholar] [CrossRef]
- Mecozzi, S.; West, A.P., Jr.; Dougherty, D.A. Cation-pi interactions in aromatics of biological and medicinal interest: Electrostatic potential surfaces as a useful qualitative guide. Proc. Natl. Acad. Sci. USA 1996, 93, 10566–10571. [Google Scholar] [CrossRef]
- Hughes, R.M.; Waters, M.L. Arginine methylation in a beta-hairpin peptide: Implications for Arg-pi interactions, DeltaCp(o), and the cold denatured state. J. Am. Chem. Soc. 2006, 128, 12735–12742. [Google Scholar] [CrossRef]
- Chen, C.C.; Hsu, W.; Hwang, K.C.; Hwu, J.R.; Lin, C.C.; Horng, J.C. Contributions of cation-pi interactions to the collagen triple helix stability. Arch. Biochem. Biophys. 2011, 508, 46–53. [Google Scholar] [CrossRef]
- Burley, S.K.; Petsko, G.A. Amino-aromatic interactions in proteins. FEBS Lett. 1986, 203, 139–143. [Google Scholar] [CrossRef]
- Cheng, J.; Kang, C.; Zhu, W.; Luo, X.; Puah, C.M.; Chen, K.; Shen, J.; Jiang, H. N-Methylformamide−Benzene Complex as a Prototypical Peptide N−H···π Hydrogen-Bonded System: Density Functional Theory and MP2 Studies. J. Org. Chem. 2003, 68, 7490–7495. [Google Scholar] [CrossRef]
- Tóth, G.; Murphy, R.F.; Lovas, S. Investigation of Aromatic-Backbone Amide Interactions in the Model Peptide Acetyl-Phe-Gly-Gly-N-Methyl Amide Using Molecular Dynamics Simulations and Protein Database Search. J. Am. Chem. Soc. 2001, 123, 11782–11790. [Google Scholar] [CrossRef]
- Rich, A.; Crick, F.H.C. The molecular structure of collagen. J. Mol. Biol. 1961, 3, 483–484. [Google Scholar] [CrossRef]
- Bella, J.; Eaton, M.; Brodsky, B.; Berman, H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 A resolution. Science 1994, 266, 75–81. [Google Scholar] [CrossRef]
- Fraser, R.D.B.; MacRae, T.P.; Suzuki, E. Chain conformation in the collagen molecule. J. Mol. Biol. 1979, 129, 463–481. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.M.; Kirkpatrick, A.; Brodsky, B. Amino Acid Propensities for the Collagen Triple-Helix. Biochem. 2000, 39, 14960–14967. [Google Scholar] [CrossRef]
- Shah, N.K.; Ramshaw, J.A.; Kirkpatrick, A.; Shah, C.; Brodsky, B. A host-guest set of triple-helical peptides: Stability of Gly-X-Y triplets containing common nonpolar residues. Biochemistry 1996, 35, 10262–10268. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.; Brodsky, B. Prediction of collagen stability from amino acid sequence. J. Biol. Chem. 2005, 280, 19343–19349. [Google Scholar] [CrossRef]
- Ramshaw, J.A.; Shah, N.K.; Brodsky, B. Gly-X-Y tripeptide frequencies in collagen: A context for host-guest triple-helical peptides. J. Struct. Biol. 1998, 122, 86–91. [Google Scholar] [CrossRef]
- Beck, K.; Chan, V.C.; Shenoy, N.; Kirkpatrick, A.; Ramshaw, J.A.; Brodsky, B. Destabilization of osteogenesis imperfecta collagen-like model peptides correlates with the identity of the residue replacing glycine. Proc. Natl. Acad. Sci. USA 2000, 97, 4273–4278. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.; Kirkpatrick, A.; Brodsky, B. Peptide investigations of pairwise interactions in the collagen triple-helix. J. Mol. Biol. 2002, 316, 385–394. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.M.; Kirkpatrick, A.; Brodsky, B. Electrostatic Interactions Involving Lysine Make Major Contributions to Collagen Triple-Helix Stability. Biochemistry 2005, 44, 1414–1422. [Google Scholar] [CrossRef]
- Chiang, C.H.; Horng, J.C. Cation-pi Interaction Induced Folding of AAB-Type Collagen Heterotrimers. J. Phys. Chem. B 2016, 120, 1205–1211. [Google Scholar] [CrossRef]
- Chen, C.C.; Hsu, W.; Kao, T.C.; Horng, J.C. Self-assembly of short collagen-related peptides into fibrils via cation-pi interactions. Biochemistry 2011, 50, 2381–2383. [Google Scholar] [CrossRef]
- Chiang, C.H.; Fu, Y.H.; Horng, J.C. Formation of AAB-Type Collagen Heterotrimers from Designed Cationic and Aromatic Collagen-Mimetic Peptides: Evaluation of the C-Terminal Cation-pi Interactions. Biomacromolecules 2017, 18, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.R.; Hulgan, S.A.H.; Peterson, C.M.; Li, I.C.; Gonzalez, K.J.; Hartgerink, J.D. Predicting the stability of homotrimeric and heterotrimeric collagen helices. Nat. Chem. 2021, 13, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.R.; Alizadehmojarad, A.A.; Kolomeisky, A.B.; Hartgerink, J.D. Charge-Free, Stabilizing Amide-pi Interactions Can Be Used to Control Collagen Triple-Helix Self-Assembly. Biomacromolecules 2021, 22, 2137–2147. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Cryst. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Zheng, H.; Lu, C.; Lan, J.; Fan, S.; Nanda, V.; Xu, F. How electrostatic networks modulate specificity and stability of collagen. Proc. Natl. Acad. Sci. USA 2018, 115, 6207–6212. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. Psi4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef]
- Collaborative Computational Project, N. The CCP4 suite: Programs for protein crystallography. Acta Cryst. D Biol. Cryst. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Wennberg, C.L.; Murtola, T.; Pall, S.; Abraham, M.J.; Hess, B.; Lindahl, E. Direct-Space Corrections Enable Fast and Accurate Lorentz-Berthelot Combination Rule Lennard-Jones Lattice Summation. J. Chem. Theory Comput. 2015, 11, 5737–5746. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, J. Validating the CHARMM36m protein force field with LJ-PME reveals altered hydrogen bonding dynamics under elevated pressures. Commun. Chem. 2021, 4, 99. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone phi, psi and Side-Chain chi(1) and chi(2) Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: Recent improvements to a PDB sequence culling server. Nucleic Acids Res. 2005, 33, W94–W98. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef]
- Huang, J.; Meuwly, M. Explicit Hydrogen-Bond Potentials and Their Application to NMR Scalar Couplings in Proteins. J. Chem. Theory Comput. 2010, 6, 467–476. [Google Scholar] [CrossRef]
- Sheu, S.-Y.; Yang, D.-Y.; Selzle, H.L.; Schlag, E.W. Energetics of hydrogen bonds in peptides. Proc. Natl. Acad. Sci. USA 2003, 100, 12683–12687. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Woo, S.M.; Siu, T.; Cortopassi, W.A.; Duarte, F.; Paton, R.S. Cation–π interactions in protein-ligand binding: Theory and data-mining reveal different roles for lysine and arginine. Chem. Sci. 2018, 9, 2655–2665. [Google Scholar] [CrossRef] [PubMed]
- Rezac, J.; Riley, K.E.; Hobza, P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids. Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Pairs | dNM (Å) | θ (°) | ω (°) | dCγ-Π (Å) | τ (°) | χ(°) |
|---|---|---|---|---|---|---|---|
| QM | Acetamide-Methylbenzene | 3.2 | 8.2 | 149.1 | 4.9 | 46.7 | 65.9 |
| Crystal | La:Q-Ma:F | 3.3 | 8.4 | 159.2 | 5.3 | 61.1 | 93.0 |
| Ma:Q-Ta:F | 3.5 | 20.9 | 141.9 | 5.5 | 62.3 | 107.8 | |
| MD b | La:Q-Ma:F | 3.3 | 15.9 | 145.4 | 5.6 | 72.3 | 43.8 |
| 5.2 | −50.5 | 115.1 | |||||
| Ma:Q-Ta:F | 3.2 | 10.5 | 139.9 | 5.5 | 73.0 | 113.4 | |
| 5.5 | −78.4 | 122.4 | |||||
| Data mining c | Q-F | 3.5 ± 0.23 | 14.1 ± 6.1 | 146.3 ± 14.1 | 3.9 ± 0.8 | 72.1 ± 19.8 | 103.9 ± 37.1 |
| 3.9 ± 0.6 | −16.0 ± 14.9 | 122.2 ± 43.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Xu, Y.; Lan, J.; Fan, S.; Huang, J.; Xu, F. Structural Achievability of an NH–π Interaction between Gln and Phe in a Crystal Structure of a Collagen-like Peptide. Biomolecules 2022, 12, 1433. https://doi.org/10.3390/biom12101433
Zhang R, Xu Y, Lan J, Fan S, Huang J, Xu F. Structural Achievability of an NH–π Interaction between Gln and Phe in a Crystal Structure of a Collagen-like Peptide. Biomolecules. 2022; 12(10):1433. https://doi.org/10.3390/biom12101433
Chicago/Turabian StyleZhang, Ruixue, You Xu, Jun Lan, Shilong Fan, Jing Huang, and Fei Xu. 2022. "Structural Achievability of an NH–π Interaction between Gln and Phe in a Crystal Structure of a Collagen-like Peptide" Biomolecules 12, no. 10: 1433. https://doi.org/10.3390/biom12101433
APA StyleZhang, R., Xu, Y., Lan, J., Fan, S., Huang, J., & Xu, F. (2022). Structural Achievability of an NH–π Interaction between Gln and Phe in a Crystal Structure of a Collagen-like Peptide. Biomolecules, 12(10), 1433. https://doi.org/10.3390/biom12101433