

Elucidating the Structural Impacts of Protein InDels



Abstract
1. Introduction
Previous and Current Work
2. Methods
2.1. Identifying InDels in the PDB
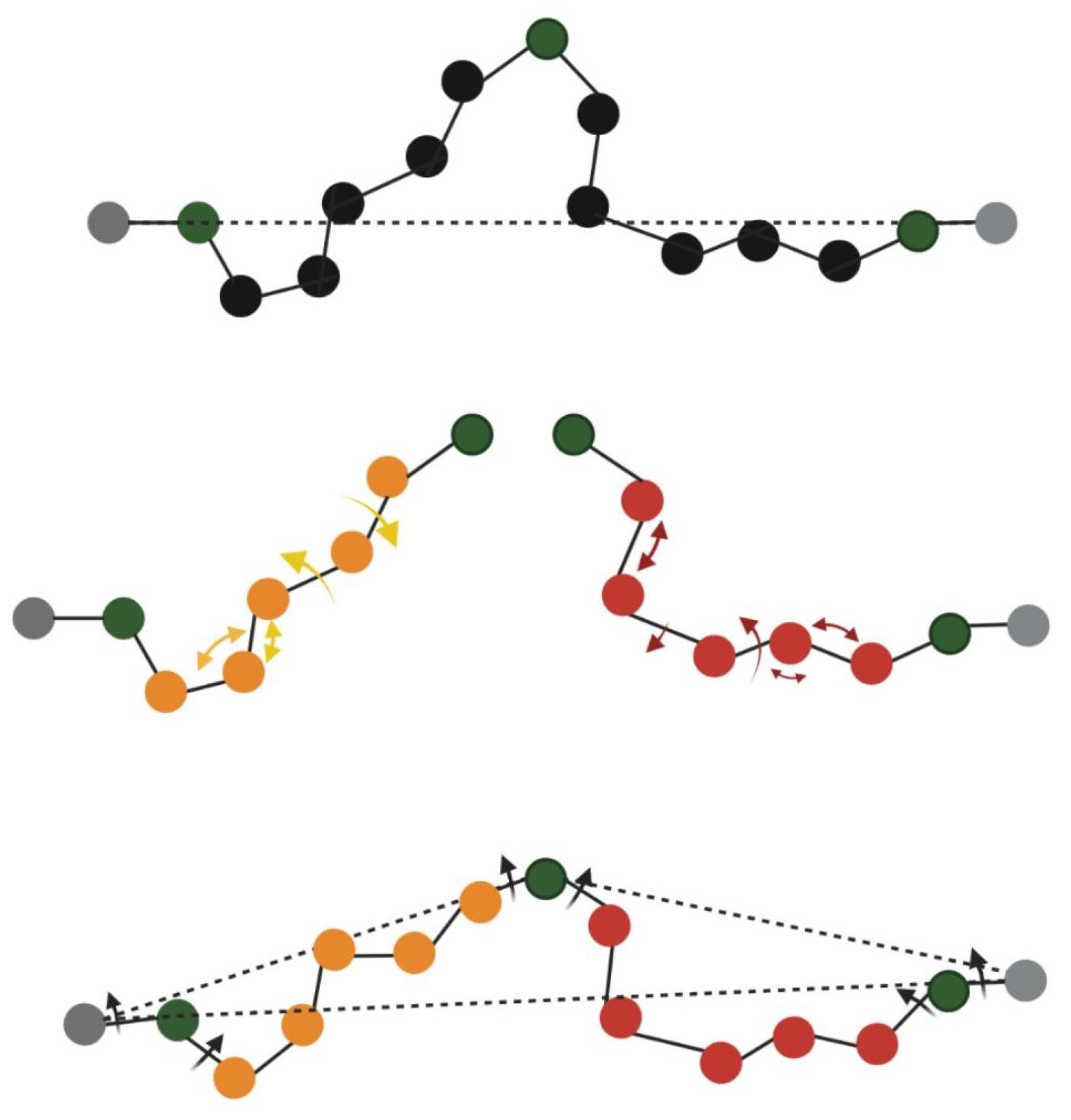
2.2. Creating InDels and Refining the Resulting Structures
- Close the loop using geometric modeling without applying a refinement step;
- Employ kinematic closure to refine the loop region.
- Close the gap without refinement of resulting structure using geometric modeling.
- Perform all-atom refinement on the resulting structure for the purpose of energy minimization.
2.3. Rigidity Analysis
2.4. Rigidity Analysis to Measure InDel Effect
3. Results and Discussion
3.1. Baseline Similarity Score Validation
3.2. Comparing PDB and Computed InDel Mutants
3.3. Using Rigidity Analysis to Measure the Effect of an InDel
3.4. Evaluating the Magnitude of InDel Effect
3.5. Statistical Validation
3.6. Effects of InDels Local to Mutation Site
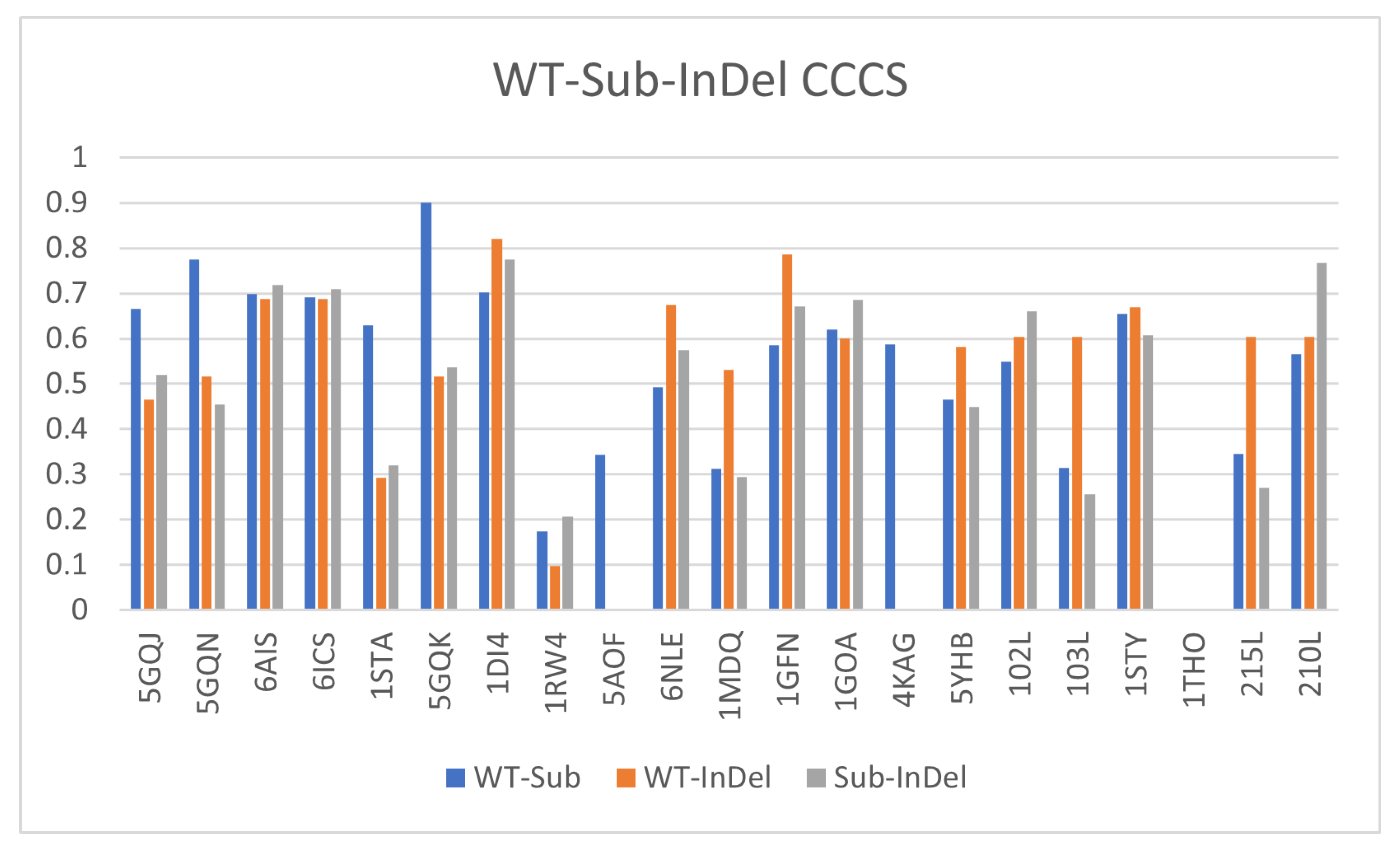
3.7. Comparing InDels and Substitutions
3.8. Correlating TLCCS and RDSM Scores with Experimental Data
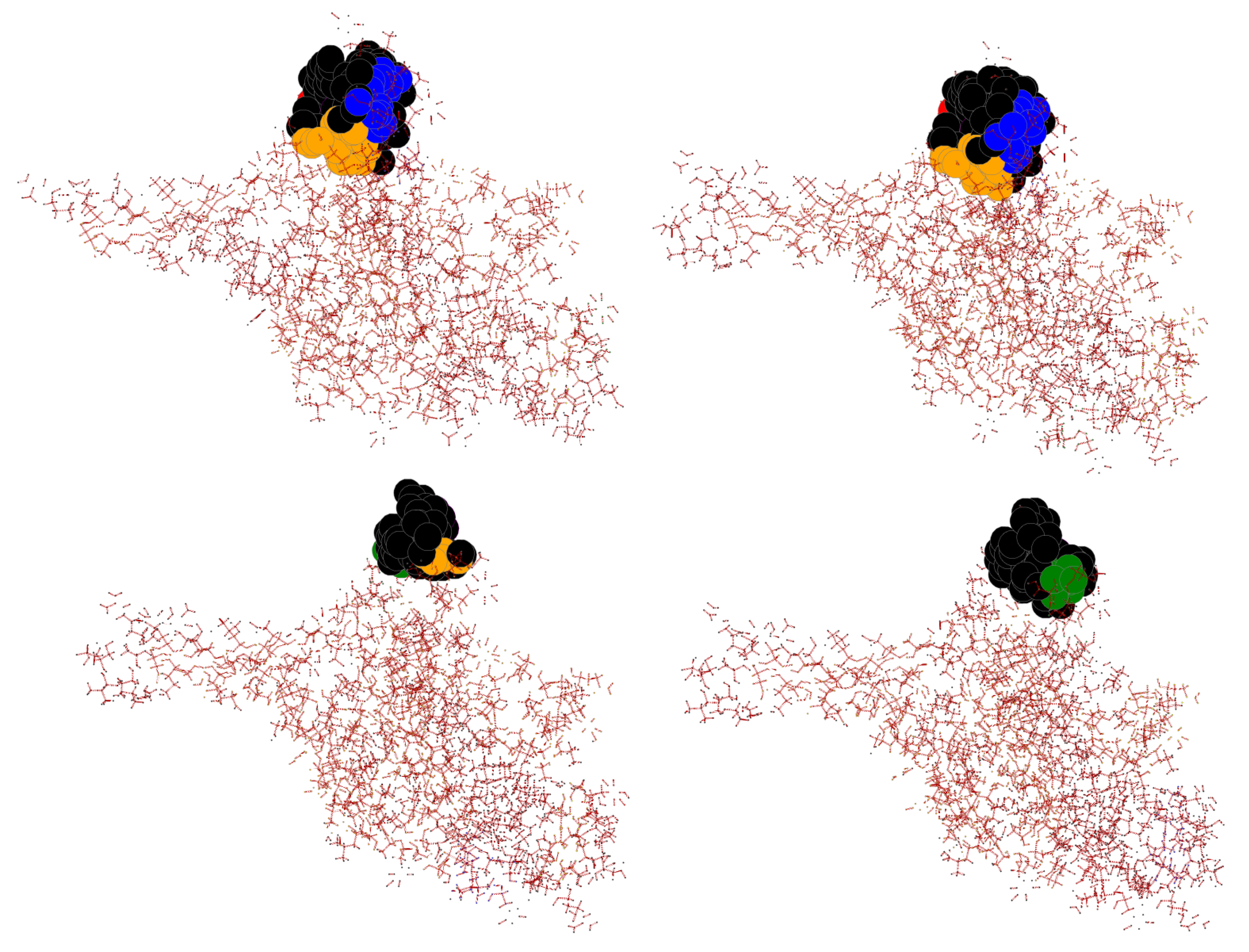
3.9. Case Study of InDel vs. Substitution Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gonzalez, C.E.; Roberts, P.; Ostermeier, M. Fitness effects of single amino acid insertions and deletions in TEM-1 β-lactamase. J. Mol. Biol. 2019, 431, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Berrondo, M.; Gray, J.J. Computed structures of point deletion mutants and their enzymatic activities. Proteins Struct. Funct. Bioinform. 2011, 79, 2844–2860. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, S.; Argos, P. Analysis of insertions/deletions in protein structures. J. Mol. Biol. 1992, 224, 461–471. [Google Scholar] [CrossRef]
- Hormozdiari, F.; Salari, R.; Hsing, M.; Schönhuth, A.; Chan, S.K.; Sahinalp, S.C.; Cherkasov, A. The effect of insertions and deletions on wirings in protein-protein interaction networks: A large-scale study. J. Comput. Biol. 2009, 16, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Rockah-Shmuel, L.; Tóth-Petróczy, Á.; Sela, A.; Wurtzel, O.; Sorek, R.; Tawfik, D.S. Correlated occurrence and bypass of frame-shifting insertion-deletions (InDels) to give functional proteins. PLoS Genet. 2013, 9, e1003882. [Google Scholar] [CrossRef] [PubMed]
- Chuzhanova, N.A.; Anassis, E.J.; Ball, E.V.; Krawczak, M.; Cooper, D.N. Meta-analysis of indels causing human genetic disease: Mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum. Mutat. 2003, 21, 28–44. [Google Scholar] [CrossRef]
- Lin, M.; Whitmire, S.; Chen, J.; Farrel, A.; Shi, X.; Guo, J.T. Effects of short indels on protein structure and function in human genomes. Sci. Rep. 2017, 7, 9313. [Google Scholar] [CrossRef]
- Light, S.; Sagit, R.; Sachenkova, O.; Ekman, D.; Elofsson, A. Protein Expansion Is Primarily due to Indels in Intrinsically Disordered Regions. Mol. Biol. Evol. 2013, 30, 2645–2653. [Google Scholar] [CrossRef]
- Heinz, D.W.; Baase, W.A.; Dahlquist, F.W.; Matthews, B.W. How amino-acid insertions are allowed in an α-helix of T4 lysozyme. Nature 1993, 361, 561–564. [Google Scholar] [CrossRef]
- Cai, Z.W.; Liu, J.; Li, H.Y.; Sheppard, D.N. Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis. Acta Pharmacol. Sin. 2011, 32, 693–701. [Google Scholar] [CrossRef]
- Wolf, Y.; Madej, T.; Babenko, V.; Shoemaker, B.; Panchenko, A.R. Long-term trends in evolution of indels in protein sequences. BMC Evol. Biol. 2007, 7, 19. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Kim, R.; Guo, J.T. Systematic analysis of short internal indels and their impact on protein folding. BMC Struct. Biol. 2010, 10, 24. [Google Scholar] [CrossRef]
- Tsui, L.C.; Dorfman, R. The cystic fibrosis gene: A molecular genetic perspective. Cold Spring Harb. Perspect. Med. 2013, 3, a009472. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Iengar, P. An analysis of substitution, deletion and insertion mutations in cancer genes. Nucleic Acids Res. 2012, 40, 6401–6413. [Google Scholar] [CrossRef]
- Garry, R.F.; Gallaher, W.R. Naturally occurring indels in multiple coronavirus spikes. Image 2020, 1039, 136. [Google Scholar]
- Liu, Z.; Zheng, H.; Lin, H.; Li, M.; Yuan, R.; Peng, J.; Xiong, Q.; Sun, J.; Li, B.; Wu, J.; et al. Identification of common deletions in the spike protein of severe acute respiratory syndrome coronavirus 2. J. Virol. 2020, 94, e00790-20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, J.; Wang, Z.; Wang, L.; Gao, P. Impact of Indels on the Flanking Regions in Structural Domains. Mol. Biol. Evol. 2010, 28, 291–301. [Google Scholar] [CrossRef]
- Hsing, M.; Cherkasov, A. Indel PDB: A database of structural insertions and deletions derived from sequence alignments of closely related proteins. BMC Bioinform. 2008, 9, 293. [Google Scholar] [CrossRef]
- Ajawatanawong, P.; Atkinson, G.C.; Watson-Haigh, N.S.; MacKenzie, B.; Baldauf, S.L. SeqFIRE: A web application for automated extraction of indel regions and conserved blocks from protein multiple sequence alignments. Nucleic Acids Res. 2012, 40, W340–W347. [Google Scholar] [CrossRef]
- Jilani, M.; Turcan, A.; Haspel, N.; Jagodzinski, F. Assessing the Effects of Amino Acid Insertion and Deletion Mutations. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 2511–2518. [Google Scholar]
- Abola, E.E.; Bernstein, F.C.; Koetzle, T. The protein data bank. In Neutrons in Biology; Springer: Boston, MA, USA, 1984. [Google Scholar]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. Uniprotkb/swiss-prot. In Plant Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 89–112. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. What’s next for AlphaFold and the AI protein-folding revolution. Nature 2022, 604, 234–238. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.W.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar]
- Kolodny, R.; Guibas, L.; Levitt, M.; Koehl, P. Inverse kinematics in biology: The protein loop closure problem. Int. J. Robot. Res. 2005, 24, 151–163. [Google Scholar] [CrossRef]
- Cortés, J.; Siméon, T. Sampling-based motion planning under kinematic loop-closure constraints. In Algorithmic Foundations of Robotics VI; Springer: Berlin/Heidelberg, Germany, 2004; pp. 75–90. [Google Scholar]
- Org, R. KIC Tutorial Generalized Kinematic Closure 1. 2017. Available online: https://www.rosettacommons.org/ (accessed on 6 October 2021).
- Yao, P.; Dhanik, A.; Marz, N.; Propper, R.; Kou, C.; Liu, G.; Van Den Bedem, H.; Latombe, J.C.; Halperin-Landsberg, I.; Altman, R.B. Efficient algorithms to explore conformation spaces of flexible protein loops. IEEE/ACM Trans. Comput. Biol. Bioinform. 2008, 5, 534–545. [Google Scholar] [CrossRef]
- Huang, P.S.; Ban, Y.E.A.; Richter, F.; Andre, I.; Vernon, R.; Schief, W.R.; Baker, D. RosettaRemodel: A generalized framework for flexible backbone protein design. PLoS ONE 2011, 6, e24109. [Google Scholar] [CrossRef]
- Mandell, D.J.; Coutsias, E.A.; Kortemme, T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 2009, 6, 551–552. [Google Scholar] [CrossRef]
- Stein, A.; Kortemme, T. Improvements to robotics-inspired conformational sampling in rosetta. PLoS ONE 2013, 8, e63090. [Google Scholar] [CrossRef]
- Nivón, L.G.; Moretti, R.; Baker, D. A Pareto-optimal refinement method for protein design scaffolds. PLoS ONE 2013, 8, e59004. [Google Scholar] [CrossRef]
- Popot, J.L.; Engelman, D.M. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 2000, 69, 881–922. [Google Scholar] [CrossRef] [PubMed]
- Karshikoff, A.; Nilsson, L.; Ladenstein, R. Rigidity versus flexibility: The dilemma of understanding protein thermal stability. FEBS J. 2015, 282, 3899–3917. [Google Scholar] [CrossRef] [PubMed]
- Jagodzinski, F.; Hardy, J.; Streinu, I. Using rigidity analysis to probe mutation-induced structural changes in proteins. J. Bioinform. Comput. Biol. 2012, 10, 1242010. [Google Scholar] [CrossRef]
- Dehghanpoor, R.; Ricks, E.; Hursh, K.; Gunderson, S.; Farhoodi, R.; Haspel, N.; Hutchinson, B.; Jagodzinski, F. Predicting the effect of single and multiple mutations on protein structural stability. Molecules 2018, 23, 251. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.; Jagodzinski, F.; Streinu, I. KINARI-Lib: A C++ library for mechanical modeling and pebble game rigidity analysis. In Proceedings of the Minisymposium on Publicly Available Geometric/-Topological Software, Chapel Hill, NC, USA, 17–20 June 2012; pp. 29–32. [Google Scholar]
- Andersson, E.; Hsieh, R.; Szeto, H.; Farhoodi, R.; Jagodzinski, F.; Haspel, N. Assessing How Multiple Mutations Affect Protein Stability Using Rigid Cluster Size Distributions. In Proceedings of the IEEE-ICCABS (International Conference on Computational Advances in Bio and Medical Sciences), Atlanta, GA, USA, 13–15 October 2016. [Google Scholar] [CrossRef]
- Schenkmayerova, A.; Pinto, G.P.; Toul, M.; Marek, M.; Hernychova, L.; Planas-Iglesias, J.; Daniel Liskova, V.; Pluskal, D.; Vasina, M.; Emond, S.; et al. Engineering the protein dynamics of an ancestral luciferase. Nat. Commun. 2021, 12, 3616. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.; Spielman, S.; Wilke, C. Computational prediction of the tolerance to amino-acid deletion in green-fluorescent protein. PLoS ONE 2017, 12, e0164905. [Google Scholar] [CrossRef] [PubMed]
- Turcan, A.; Zivkovic, A.; Thompson, D.; Wong, L.; Johnson, L.; Jagodzinski, F. CGRAP: A Web Server for Coarse-Grained Rigidity Analysis of Proteins. Symmetry 2021, 13, 2401. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Wissler, C. The Spearman correlation formula. Science 1905, 22, 309–311. [Google Scholar] [CrossRef]
- Figueiredo Filho, D.B.; Paranhos, R.; Rocha, E.C.d.; Batista, M.; Silva Jr, J.A.d.; Santos, M.L.W.D.; Marino, J.G. When is statistical significance not significant? Braz. Political Sci. Rev. 2013, 7, 31–55. [Google Scholar] [CrossRef]
- Mendelman, P.M.; Chaffin, D.; Stull, T.; Rubens, C.; Mack, K.; Smith, A. Characterization of non-beta-lactamase-mediated ampicillin resistance in Haemophilus influenzae. Antimicrob. Agents Chemother. 1984, 26, 235–244. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wildtype | Mutant | InDel Type | Global RMSD | Local RMSD | TLCCS |
|---|---|---|---|---|---|
| 5GQL | 5GQI | d:1, l | 0.138 | 0.126 | 0.848 |
| 5GQL | 5GQJ | d:2, l | 0.767 | 0.708 | 0.665 |
| 5GQL | 5GQN | d:3, l | 0.836 | 0.678 | 0.774 |
| 6J6C | 6AIS | d:2,l | 0.96 | 0.509 | 0.698 |
| 6J6C | 6ICS | d:4, l | 0.135 | 0.34 | 0.691 |
| 2VJI | 4XQF | d:2, l | 0.16 | 0.135 | 0.765 |
| 2BBO | 1XMJ | d:1, l | 0.652 | 0.768 | 0.023 |
| 2IQ1 | 6AK7 | d:3, l | 0.79 | 0.392 | 0.693 |
| 2VJJ | 6GVP | d:2, l | 0.186 | 0.376 | 0.789 |
| 1A7N | 1A7O | d:2, l | 0.283 | 0.251 | 0.706 |
| 1STN | 1STA | i:2, l | 0.506 | 0.47 | 0.629 |
| 2VJJ | 6GVR | d:2, l | 0.168 | 0.329 | 0.790 |
| 5GQM | 5GQK | d:3, l | 0.796 | 0.269 | 0.899 |
| 1JWR | 1DI4 | d:2, l | 0.563 | 0.498 | 0.54 |
| 1JWR | 1DI5 | d:1, l | 0.283 | 0.251 | 0.81 |
| 2Y0G | 4KA9 | i:1, d:1, l | 0.72 | 0.58 | 0.656 |
| 4KJK | 4KJL | i:1, l | 0.378 | 0.574 | 0.717 |
| 2NIP | 1RW4 | d:1, l | 0.557 | 0.53 | 0.189 |
| 4EUL | 6FLL | d:2, l | 0.206 | 0.36 | 0.513 |
| 1ANF | 1MDQ | i:1, l | 0.701 | 0.607 | 0.312 |
| 1OMF | 1GFN | d:6, l | 0.245 | 0.342 | 0.554 |
| 1F21 | 1GOA | i:1, l | 0.97 | 0.85 | 0.498 |
| 2Y0G | 4KAG | i:1, d:1, l | 0.69 | 0.74 | 0.587 |
| 5YHA | 5YHB | d:3, l | 0.623 | 0.807 | 0.460 |
| 2LZM | 103L | i:3,hs | 0.221 | 0.125 | 0.313 |
| 2LZM | 102L | i:1, h | 0.638 | 0.23 | 0.534 |
| 2EY6 | 6XSF | d:6, h | 0.735 | 0.43 | 0.467 |
| 1EY0 | 5IIF | d:6, s | 0.7 | 0.36 | 0.447 |
| 1SNC | 1STY | i:1, h | 0.709 | 0.385 | 0.671 |
| 1C7C | 1C7D | i:1, h | 0.745 | 0.35 | 0.775 |
| 1TXX | 1THO | i:1, s | 0.685 | 0.221 | 0.654 |
| 4EUL | 4KEX | d:1, l | 0.263 | 0.437 | 0.616 |
| 2LZM | 210L | d:1, h | 0.961 | 0.31 | 0.625 |
| 1C7D | 1O1p | d:1, s | 0.708 | 0.224 | 0.762 |
| 2LZM | 215L | i:1, h | 0.657 | 0.3 | 0.686 |
| Insertion | Fitness | TLCCS | RDSM1 | RDSM2 | RDSM3 | RDSM5 |
|---|---|---|---|---|---|---|
| 37, Q | 0.164355 | 0.00051 | 0.655974 | 0.000121 | 0.000036 | 2.01 |
| 45, P | 0.000911 | 0.00062 | 0.662656 | 0.000146 | 0.000047 | 2.72 |
| 89, V | 0.022674 | 0.00046 | 0.651466 | 0.000086 | 0.000022 | 1.10 |
| 130, C | 0.001328 | 0.00033 | 0.672744 | 0.000109 | 0.000031 | 1.67 |
| 148, K | 0.237953 | 0.00008 | 0.669186 | 0.000097 | 0.000027 | 1.50 |
| 150, V | 0.305583 | 0.00011 | 0.697794 | 0.000062 | 0.000016 | 8.42 |
| 172, M | 0.435899 | 0.00043 | 0.676728 | 0.000128 | 0.000039 | 2.22 |
| 191, G | 0.005969 | 0.00041 | 0.676746 | 0.000117 | 0.000034 | 1.89 |
| 210, G | 0.028093 | 0.00036 | 0.698456 | 0.000099 | 0.000028 | 1.55 |
| 214, I | 0.083574 | 0.00056 | 0.673495 | 0.000123 | 0.000037 | 2.08 |
| R | 0.239 | −0.423 | −0.191 | −0.153 | −0.254 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jilani, M.; Turcan, A.; Haspel, N.; Jagodzinski, F. Elucidating the Structural Impacts of Protein InDels. Biomolecules 2022, 12, 1435. https://doi.org/10.3390/biom12101435
Jilani M, Turcan A, Haspel N, Jagodzinski F. Elucidating the Structural Impacts of Protein InDels. Biomolecules. 2022; 12(10):1435. https://doi.org/10.3390/biom12101435
Chicago/Turabian StyleJilani, Muneeba, Alistair Turcan, Nurit Haspel, and Filip Jagodzinski. 2022. "Elucidating the Structural Impacts of Protein InDels" Biomolecules 12, no. 10: 1435. https://doi.org/10.3390/biom12101435
APA StyleJilani, M., Turcan, A., Haspel, N., & Jagodzinski, F. (2022). Elucidating the Structural Impacts of Protein InDels. Biomolecules, 12(10), 1435. https://doi.org/10.3390/biom12101435