
ADAMTS Proteins and Vascular Remodeling in Aortic Aneurysms


Abstract
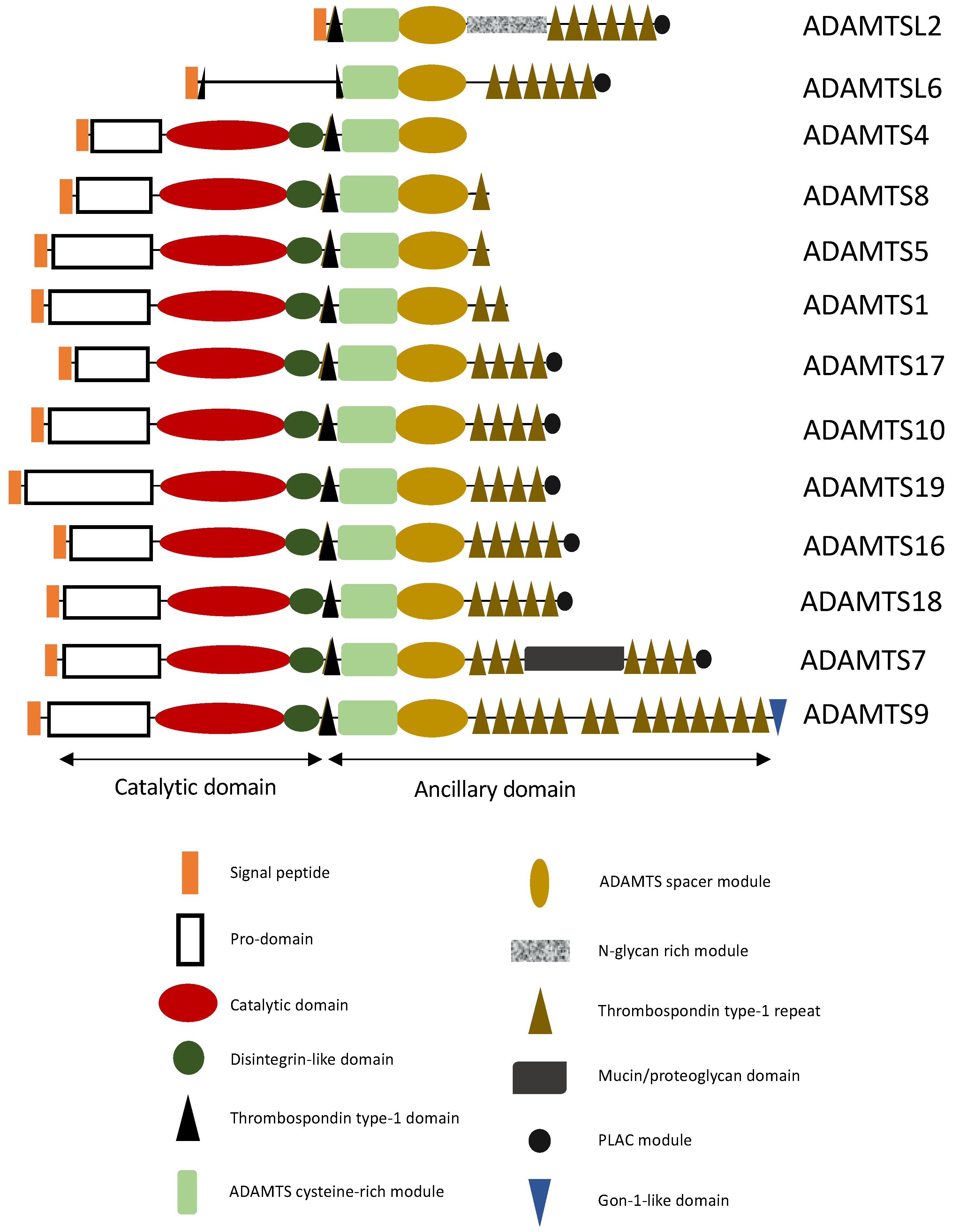
:1. Introduction
ADAMTS Proteins
2. ADAMTS/ADAMTSL and TAA
2.1. THSD4/ADAMTSL6 Mutations Responsible for TAA
2.2. Proteoglycanases Involved in TAA
2.2.1. ADAMTS1
2.2.2. ADAMTS4
2.2.3. ADAMTS5
2.2.4. ADAMTS16
3. ADAMTS Linked to AAA
3.1. ADAMTS8
3.2. ADAMTS9
3.3. ADAMTS7
4. ADAMTSL2, ADAMTS10 and 17 Possibly Involved in Aorta Pathology
5. ADAMTS18 Involved in Aorta Development
6. ADAMTS19 Implicated in Valve Development
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milewicz, D.M.; Ramirez, F. Therapies for Thoracic Aortic Aneurysms and Acute Aortic Dissections: Old Controversies and New Opportunities. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Dubail, J.; Apte, S.S. Insights on ADAMTS Proteases and ADAMTS-like Proteins from Mammalian Genetics. Matrix Biol. 2015, 44–46, 24–37. [Google Scholar] [CrossRef]
- Apte, S.S. A Disintegrin-like and Metalloprotease (Reprolysin-Type) with Thrombospondin Type 1 Motif (ADAMTS) Superfamily: Functions and Mechanisms. J. Biol. Chem. 2009, 284, 31493–31497. [Google Scholar] [CrossRef] [Green Version]
- Majerus, E.M.; Zheng, X.; Tuley, E.A.; Sadler, J.E. Cleavage of the ADAMTS13 Propeptide Is Not Required for Protease Activity. J. Biol. Chem. 2003, 278, 46643–46648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, B.-H.; Longpré, J.-M.; Somerville, R.P.T.; Alexander, J.P.; Leduc, R.; Apte, S.S. Regulation of ADAMTS9 Secretion and Enzymatic Activity by Its Propeptide. J. Biol. Chem. 2007, 282, 16146–16154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somerville, R.P.T.; Jungers, K.A.; Apte, S.S. Discovery and Characterization of a Novel, Widely Expressed Metalloprotease, ADAMTS10, and Its Proteolytic Activation. J. Biol. Chem. 2004, 279, 51208–51217. [Google Scholar] [CrossRef] [Green Version]
- Koo, B.-H.; Apte, S.S. Cell-Surface Processing of the Metalloprotease Pro-ADAMTS9 Is Influenced by the Chaperone GRP94/Gp96. J. Biol. Chem. 2010, 285, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-M.; Ge, G.; Lim, N.H.; Nagase, H.; Greenspan, D.S. TIMP-3 Inhibits the Procollagen N-Proteinase ADAMTS-2. Biochem. J. 2006, 398, 515–519. [Google Scholar] [CrossRef]
- Kashiwagi, M.; Tortorella, M.; Nagase, H.; Brew, K. TIMP-3 Is a Potent Inhibitor of Aggrecanase 1 (ADAM-TS4) and Aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 2001, 276, 12501–12504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortorella, M.D.; Arner, E.C.; Hills, R.; Easton, A.; Korte-Sarfaty, J.; Fok, K.; Wittwer, A.J.; Liu, R.-Q.; Malfait, A.-M. A2-Macroglobulin Is a Novel Substrate for ADAMTS-4 and ADAMTS-5 and Represents an Endogenous Inhibitor of These Enzymes. J. Biol. Chem. 2004, 279, 17554–17561. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Owen, K.; Parker, A.E.; Scilabra, S.D.; Dudhia, J.; Strickland, D.K.; Troeberg, L.; Nagase, H. Low Density Lipoprotein Receptor-Related Protein 1 (LRP1)-Mediated Endocytic Clearance of a Disintegrin and Metalloproteinase with Thrombospondin Motifs-4 (ADAMTS-4). J. Biol. Chem. 2014, 289, 6462–6474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huxley-Jones, J.; Apte, S.S.; Robertson, D.L.; Boot-Handford, R.P. The Characterisation of Six ADAMTS Proteases in the Basal Chordate Ciona Intestinalis Provides New Insights into the Vertebrate ADAMTS Family. Int. J. Biochem. Cell Biol. 2005, 37, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.R.; Goff, C.L.; Bhatt, S.; Dixon, L.J.; Sandy, J.D.; Apte, S.S. Adamts5, the Gene Encoding a Proteoglycan-Degrading Metalloprotease, Is Expressed by Specific Cell Lineages during Mouse Embryonic Development and in Adult Tissues. Gene Expr. Patterns 2009, 9, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Mead, T.J.; Apte, S.S. ADAMTS Proteins in Human Disorders. Matrix Biol. 2018, 71–72, 225–239. [Google Scholar] [CrossRef]
- Furlan, M.; Robles, R.; Lämmle, B. Partial Purification and Characterization of a Protease from Human Plasma Cleaving von Willebrand Factor to Fragments Produced by in Vivo Proteolysis. Blood 1996, 87, 4223–4234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colige, A.; Sieron, A.L.; Li, S.-W.; Schwarze, U.; Petty, E.; Wertelecki, W.; Wilcox, W.; Krakow, D.; Cohn, D.H.; Reardon, W.; et al. Human Ehlers-Danlos Syndrome Type VII C and Bovine Dermatosparaxis Are Caused by Mutations in the Procollagen I N-Proteinase Gene. Am. J. Hum. Genet. 1999, 65, 308–317. [Google Scholar] [CrossRef] [Green Version]
- Janssen, L.; Dupont, L.; Bekhouche, M.; Noel, A.; Leduc, C.; Voz, M.; Peers, B.; Cataldo, D.; Apte, S.S.; Dubail, J.; et al. ADAMTS3 Activity Is Mandatory for Embryonic Lymphangiogenesis and Regulates Placental Angiogenesis. Angiogenesis 2016, 19, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Nardi, M.A.; Li, Y.-S.; Zhang, W.; Pan, R.; Dang, S.; Yee, H.; Quartermain, D.; Jonas, S.; Karpatkin, S. C-Terminal ADAMTS-18 Fragment Induces Oxidative Platelet Fragmentation, Dissolves Platelet Aggregates, and Protects against Carotid Artery Occlusion and Cerebral Stroke. Blood 2009, 113, 6051–6060. [Google Scholar] [CrossRef] [Green Version]
- Le Goff, C.; Morice-Picard, F.; Dagoneau, N.; Wang, L.W.; Perrot, C.; Crow, Y.J.; Bauer, F.; Flori, E.; Prost-Squarcioni, C.; Krakow, D.; et al. ADAMTSL2 Mutations in Geleophysic Dysplasia Demonstrate a Role for ADAMTS-like Proteins in TGF-β Bioavailability Regulation. Nat. Genet. 2008, 40, 1119–1123. [Google Scholar] [CrossRef] [Green Version]
- Bader, H.L.; Wang, L.W.; Ho, J.C.; Tran, T.; Holden, P.; Fitzgerald, J.; Atit, R.P.; Reinhardt, D.P.; Apte, S.S. A Disintegrin-like and Metalloprotease Domain Containing Thrombospondin Type 1 Motif-like 5 (ADAMTSL5) Is a Novel Fibrillin-1-, Fibrillin-2-, and Heparin-Binding Member of the ADAMTS Superfamily Containing a Netrin-like Module. Matrix Biol. 2012, 31, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, K.; Manabe, R.; Yamada, T.; Nakano, I.; Oguri, Y.; Keene, D.R.; Sengle, G.; Sakai, L.Y.; Sekiguchi, K. ADAMTSL-6 Is a Novel Extracellular Matrix Protein That Binds to Fibrillin-1 and Promotes Fibrillin-1 Fibril Formation. J. Biol. Chem. 2010, 285, 4870–4882. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, L.A.R.; Wang, L.W.; Bader, H.; Ho, J.C.; Majors, A.K.; Hollyfield, J.G.; Traboulsi, E.I.; Apte, S.S. ADAMTSL4, a Secreted Glycoprotein Widely Distributed in the Eye, Binds Fibrillin-1 Microfibrils and Accelerates Microfibril Biogenesis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 461. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.A.; Kawaguchi, N.; Fessler, L.I.; Nelson, R.E.; Chen, Y.; Kramerov, A.A.; Kusche-Gullberg, M.; Kramer, J.M.; Ackley, B.D.; Sieron, A.L.; et al. Papilin in Development; a Pericellular Protein with a Homology to the ADAMTS Metalloproteinases. Development 2000, 127, 5475–5485. [Google Scholar] [CrossRef]
- Elbitar, S.; Renard, M.; Arnaud, P.; Hanna, N.; Jacob, M.-P.; Guo, D.-C.; Tsutsui, K.; Gross, M.-S.; Kessler, K.; Tosolini, L.; et al. Pathogenic Variants in THSD4, Encoding the ADAMTS-like 6 Protein, Predispose to Inherited Thoracic Aortic Aneurysm. Genet. Med. 2021, 23, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, Z.; Chen, H.-N.; Qin, S.; Chen, Y.; Jiang, J.; Zhang, Z.; Luo, M.; Ye, Q.; Xie, N.; et al. ZNF37A Promotes Tumor Metastasis through Transcriptional Control of THSD4/TGF-β Axis in Colorectal Cancer. Oncogene 2021, 40, 3394–3407. [Google Scholar] [CrossRef]
- Saito, T.; Nishida, K.; Furumatsu, T.; Yoshida, A.; Ozawa, M.; Ozaki, T. Histone Deacetylase Inhibitors Suppress Mechanical Stress-Induced Expression of RUNX-2 and ADAMTS-5 through the Inhibition of the MAPK Signaling Pathway in Cultured Human Chondrocytes. Osteoarthr. Cartil. 2013, 21, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cikach, F.S.; Koch, C.D.; Mead, T.J.; Galatioto, J.; Willard, B.B.; Emerton, K.B.; Eagleton, M.J.; Blackstone, E.H.; Ramirez, F.; Roselli, E.E.; et al. Massive Aggrecan and Versican Accumulation in Thoracic Aortic Aneurysm and Dissection. JCI Insight 2018, 3, e97167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankunas, K.; Hang, C.T.; Tsun, Z.-Y.; Chen, H.; Lee, N.V.; Wu, J.I.; Shang, C.; Bayle, J.H.; Shou, W.; Iruela-Arispe, M.L.; et al. Endocardial Brg1 Represses ADAMTS1 to Maintain the Microenvironment for Myocardial Morphogenesis. Dev. Cell 2008, 14, 298–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooley, M.A.; Fresco, V.M.; Dorlon, M.E.; Twal, W.O.; Lee, N.V.; Barth, J.L.; Kern, C.B.; Iruela-Arispe, M.L.; Argraves, W.S. Fibulin-1 Is Required during Cardiac Ventricular Morphogenesis for Versican Cleavage, Suppression of ErbB2 and Erk1/2 Activation, and to Attenuate Trabecular Cardiomyocyte Proliferation. Dev. Dyn. 2012, 241, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Manzaneque, J.C.; Fernández-Rodríguez, R.; Rodríguez-Baena, F.J.; Iruela-Arispe, M.L. ADAMTS Proteases in Vascular Biology. Matrix Biol. 2015, 44–46, 38–45. [Google Scholar] [CrossRef]
- Luque, A.; Carpizo, D.R.; Iruela-Arispe, M.L. ADAMTS1/METH1 Inhibits Endothelial Cell Proliferation by Direct Binding and Sequestration of VEGF165. J. Biol. Chem. 2003, 278, 23656–23665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wu, W.; Yu, C.; Zhong, F.; Li, G.; Kong, W.; Zheng, J. A Disintegrin and Metalloproteinase with Thrombospondin Motif 1 (ADAMTS1) Expression Increases in Acute Aortic Dissection. Sci. China Life Sci. 2016, 59, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Y.; Zhao, G.; He, L.; Fu, Y.; Yu, C.; Wang, Z.; Zhao, T.; Cao, F.; Gao, Y.; et al. Postnatal Deficiency of ADAMTS1 Ameliorates Thoracic Aortic Aneurysm and Dissection in Mice. Exp. Physiol. 2018, 103, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oller, J.; Alfranca, A.; Méndez-Barbero, N.; Villahoz, S.; Lozano-Vidal, N.; Martín-Alonso, M.; Arroyo, A.G.; Escolano, A.; Armesilla, A.L.; Campanero, M.R.; et al. C/EBPβ and Nuclear Factor of Activated T Cells Differentially Regulate Adamts-1 Induction by Stimuli Associated with Vascular Remodeling. Mol. Cell. Biol. 2015, 35, 3409–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oller, J.; Méndez-Barbero, N.; Ruiz, E.J.; Villahoz, S.; Renard, M.; Canelas, L.I.; Briones, A.M.; Alberca, R.; Lozano-Vidal, N.; Hurlé, M.A.; et al. Nitric Oxide Mediates Aortic Disease in Mice Deficient in the Metalloprotease Adamts1 and in a Mouse Model of Marfan Syndrome. Nat. Med. 2017, 23, 200–212. [Google Scholar] [CrossRef]
- Ren, P.; Zhang, L.; Xu, G.; Palmero, L.C.; Albini, P.T.; Coselli, J.S.; Shen, Y.H.; LeMaire, S.A. ADAMTS-1 and ADAMTS-4 Levels Are Elevated in Thoracic Aortic Aneurysms and Dissections. Ann. Thorac. Surg. 2013, 95, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Güneş, M.F.; Akpinar, M.B.; Cömertoğlu, I.; Akyol, S.; Demirçelik, B.; Gürel, Ö.M.; Aynekin, B.; Erdemli, H.K.; Ateş, M.; Eryonucu, B.; et al. The Investigation of a Disintegrin and Metalloproteinase with ThromboSpondin Motifs (ADAMTS) 1, 5 and 16 in Thoracic Aortic Aneurysms and Dissections. Clin. Lab. 2016, 62, 425–433. [Google Scholar] [CrossRef]
- Rao, N.; Ke, Z.; Liu, H.; Ho, C.-J.; Kumar, S.; Xiang, W.; Zhu, Y.; Ge, R. ADAMTS4 and Its Proteolytic Fragments Differentially Affect Melanoma Growth and Angiogenesis in Mice: ADAMTS4 in Cancer. Int. J. Cancer 2013, 133, 294–306. [Google Scholar] [CrossRef]
- Kahn, J.; Mehraban, F.; Ingle, G.; Xin, X.; Bryant, J.E.; Vehar, G.; Schoenfeld, J.; Grimaldi, C.J.; Peale, F.; Draksharapu, A.; et al. Gene Expression Profiling in an in Vitro Model of Angiogenesis. Am. J. Pathol. 2000, 156, 1887–1900. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.; Hughes, M.; Krishnamoorthy, S.; Zou, S.; Zhang, L.; Wu, D.; Zhang, C.; Curci, J.A.; Coselli, J.S.; Milewicz, D.M.; et al. Critical Role of ADAMTS-4 in the Development of Sporadic Aortic Aneurysm and Dissection in Mice. Sci. Rep. 2017, 7, 12351. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ma, W.; Pan, S.; Li, Y.; Wang, H.; Wang, B.; Khalil, R.A. MiR-126a-5p Limits the Formation of Abdominal Aortic Aneurysm in Mice and Decreases ADAMTS-4 Expression. J. Cell. Mol. Med. 2020, 24, 7896–7906. [Google Scholar] [CrossRef]
- Zeng, T.; Gan, J.; Liu, Y.; Shi, L.; Lu, Z.; Xue, Y.; Xiong, R.; Liu, L.; Yang, Z.; Lin, Y.; et al. ADAMTS-5 Decreases in Aortas and Plasma From Aortic Dissection Patients and Alleviates Angiotensin II-Induced Smooth Muscle-Cell Apoptosis. Front. Cardiovasc. Med. 2020, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Fava, M.; Barallobre-Barreiro, J.; Mayr, U.; Lu, R.; Didangelos, A.; Baig, F.; Lynch, M.; Catibog, N.; Joshi, A.; Barwari, T.; et al. Role of ADAMTS-5 in Aortic Dilatation and Extracellular Matrix Remodeling. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1537–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.M.; Rateri, D.L.; Balakrishnan, A.; Howatt, D.A.; Strickland, D.K.; Muratoglu, S.C.; Haggerty, C.M.; Fornwalt, B.K.; Cassis, L.A.; Daugherty, A. Smooth Muscle Cell Deletion of Low-Density Lipoprotein Receptor–Related Protein 1 Augments Angiotensin II–Induced Superior Mesenteric Arterial and Ascending Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, S.; de Groot, R. ADAMTS Proteases in Cardiovascular Physiology and Disease. Open Biol. 2020, 10, 200333. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.E.; Nelson, E.L.; Hozik, B.; Porto, S.C.; Rogers-DeCotes, A.; Fosang, A.; Kern, C.B. Adamts5−/− Mice Exhibit Altered Aggrecan Proteolytic Profiles That Correlate With Ascending Aortic Anomalies. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2067–2081. [Google Scholar] [CrossRef] [PubMed]
- Schnellmann, R.; Sack, R.; Hess, D.; Annis, D.S.; Mosher, D.F.; Apte, S.S.; Chiquet-Ehrismann, R. A Selective Extracellular Matrix Proteomics Approach Identifies Fibronectin Proteolysis by A Disintegrin-like and Metalloprotease Domain with Thrombospondin Type 1 Motifs (ADAMTS16) and Its Impact on Spheroid Morphogenesis. Mol. Cell. Proteom. 2018, 17, 1410–1425. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, K.; Kumarasamy, S.; Abdul-Majeed, S.; Kalinoski, A.L.; Morgan, E.E.; Gohara, A.F.; Nauli, S.M.; Filipiak, W.E.; Saunders, T.L.; Joe, B. Targeted Disruption of Adamts16 Gene in a Rat Genetic Model of Hypertension. Proc. Natl. Acad. Sci. USA 2012, 109, 20555–20559. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, S.; Martin, D.R.; Dong, X.; Yamamoto, K.; Apte, S.S.; Ahnström, J. Post-Translational Regulation And Proteolytic Activity Of The Metalloproteinase Adamts8. J. Biol. Chem. 2021, 297, 101323. [Google Scholar] [CrossRef] [PubMed]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Ohtsuki, T.; Al-Mamun, M.E.; Siddique, M.A.H.; Yaoita, N.; et al. ADAMTS8 Promotes the Development of Pulmonary Arterial Hypertension and Right Ventricular Failure: A Possible Novel Therapeutic Target. Circ. Res. 2019, 125, 884–906. [Google Scholar] [CrossRef]
- Farrell, K.; Simmers, P.; Mahajan, G.; Boytard, L.; Camardo, A.; Joshi, J.; Ramamurthi, A.; Pinet, F.; Kothapalli, C.R. Alterations in Phenotype and Gene Expression of Adult Human Aneurysmal Smooth Muscle Cells by Exogenous Nitric Oxide. Exp. Cell Res. 2019, 384, 111589. [Google Scholar] [CrossRef] [Green Version]
- Ashvetiya, T.; Fan, S.X.; Chen, Y.-J.; Williams, C.H.; O’Connell, J.R.; Perry, J.A.; Hong, C.C. Identification of Novel Genetic Susceptibility Loci for Thoracic and Abdominal Aortic Aneurysms via Genome-Wide Association Study Using the UK Biobank Cohort. PLoS ONE 2021, 16, e0247287. [Google Scholar] [CrossRef] [PubMed]
- Nandadasa, S.; Kraft, C.M.; Wang, L.W.; O’Donnell, A.; Patel, R.; Gee, H.Y.; Grobe, K.; Cox, T.C.; Hildebrandt, F.; Apte, S.S. Secreted Metalloproteases ADAMTS9 and ADAMTS20 Have a Non-Canonical Role in Ciliary Vesicle Growth during Ciliogenesis. Nat. Commun. 2019, 10, 953. [Google Scholar] [CrossRef] [Green Version]
- Kern, C.B.; Wessels, A.; McGarity, J.; Dixon, L.J.; Alston, E.; Argraves, W.S.; Geeting, D.; Nelson, C.M.; Menick, D.R.; Apte, S.S. Reduced Versican Cleavage Due to Adamts9 Haploinsufficiency Is Associated with Cardiac and Aortic Anomalies. Matrix Biol. 2010, 29, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Gäbel, G.; Northoff, B.H.; Weinzierl, I.; Ludwig, S.; Hinterseher, I.; Wilfert, W.; Teupser, D.; Doderer, S.A.; Bergert, H.; Schönleben, F.; et al. Molecular Fingerprint for Terminal Abdominal Aortic Aneurysm Disease. J. Am. Heart Assoc. 2017, 6, e006798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Kong, W.; Ilalov, K.; Yu, S.; Xu, K.; Prazak, L.; Fajardo, M.; Sehgal, B.; Di Cesare, P.E.; Liu, C.; et al. ADAMTS-7: A Metalloproteinase That Directly Binds to and Degrades Cartilage Oligomeric Matrix Protein. FASEB J. 2006, 20, 988–990. [Google Scholar] [CrossRef]
- Riessen, R.; Fenchel, M.; Chen, H.; Axel, D.I.; Karsch, K.R.; Lawler, J. Cartilage Oligomeric Matrix Protein (Thrombospondin-5) Is Expressed by Human Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zheng, J.; Bai, X.; Liu, B.; Liu, C.; Xu, Q.; Zhu, Y.; Wang, N.; Kong, W.; Wang, X. ADAMTS-7 Mediates Vascular Smooth Muscle Cell Migration and Neointima Formation in Balloon-Injured Rat Arteries. Circ. Res. 2009, 104, 688–698. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.C.; Tohyama, J.; Cui, J.; Cheng, L.; Yang, J.; Zhang, X.; Ou, K.; Paschos, G.K.; Zheng, X.L.; Parmacek, M.S.; et al. Knockout of Adamts7, a Novel Coronary Artery Disease Locus in Humans, Reduces Atherosclerosis in Mice. Circulation 2015, 131, 1202–1213. [Google Scholar] [CrossRef] [Green Version]
- Kessler, T.; Zhang, L.; Liu, Z.; Yin, X.; Huang, Y.; Wang, Y.; Fu, Y.; Mayr, M.; Ge, Q.; Xu, Q.; et al. ADAMTS-7 Inhibits Re-Endothelialization of Injured Arteries and Promotes Vascular Remodeling Through Cleavage of Thrombospondin-1. Circulation 2015, 131, 1191–1201. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Cao, Y.; Li, L.; Chen, W.; Chen, X. Upregulation of ADAMTS-7 and Downregulation of COMP Are Associated with Aortic Aneurysm. Mol. Med. Rep. 2017, 16, 5459–5463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; Keshishian, H.; Mundorff, C.C.; Arduini, A.; Lai, D.; Bendinelli, K.; Popp, N.R.; Bhandary, B.; Clauser, K.R.; Specht, H.; et al. TAILS Identifies Candidate Substrates and Biomarkers of ADAMTS7, a Therapeutic Protease Target in Coronary Artery Disease. bioRxiv 2021. [Google Scholar] [CrossRef]
- Colige, A.; Monseur, C.; Crawley, J.T.B.; Santamaria, S.; de Groot, R. Proteomic Discovery of Substrates of the Cardiovascular Protease ADAMTS7. J. Biol. Chem. 2019, 294, 8037–8045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; LeMaire, S.A.; Chen, L.; Carter, S.A.; Shen, Y.H.; Gan, Y.; Bartsch, H.; Wilks, J.A.; Utama, B.; Ou, H.; et al. Decreased Expression of Fibulin-5 Correlates with Reduced Elastin in Thoracic Aortic Dissection. Surgery 2005, 138, 352–359. [Google Scholar] [CrossRef]
- Le Goff, C.; Cormier-Daire, V. The ADAMTS(L) Family and Human Genetic Disorders. Hum. Mol. Genet. 2011, 20, R163–R167. [Google Scholar] [CrossRef] [Green Version]
- Spranger, J.; Gilbert, E.; Tuffli, G.; Rossiter, F.; Opitz, J. Geleophysic Dwarfism—A “Focal” Mucopolysaccharidosis ? Lancet 1971, 298, 97–98. [Google Scholar] [CrossRef]
- Allali, S.; Le Goff, C.; Pressac-Diebold, I.; Pfennig, G.; Mahaut, C.; Dagoneau, N.; Alanay, Y.; Brady, A.F.; Crow, Y.J.; Devriendt, K.; et al. Molecular Screening of ADAMTSL2 Gene in 33 Patients Reveals the Genetic Heterogeneity of Geleophysic Dysplasia. J. Med. Genet. 2011, 48, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Ben-Salem, S.; Hertecant, J.; Al-Shamsi, A.M.; Ali, B.R.; Al-Gazali, L. Novel Mutations in ADAMTSL2 Gene Underlying Geleophysic Dysplasia in Families from United Arab Emirates: Birth Defects and Prenatal Alcohol Exposure. Birth Defects Res. Part A Clin. Mol. Teratol. 2013, 97, 764–769. [Google Scholar] [CrossRef]
- García-Ortiz, L.; Gutiérrez-Salinas, J.; del Carmen Chima Galán, M.; García, R.L.R.; de la Concepción, A.; Yerena, M. Geleophysic Dysplasia: A Novel in-Frame Deletion of a Tandem Repeat in the ADAMTSL2 Gene. Am. J. Med. Genet. 2015, 167, 1949–1951. [Google Scholar] [CrossRef]
- Mackenroth, L.; Rump, A.; Lorenz, P.; Schröck, E.; Tzschach, A. Novel ADAMTSL2-Mutations in a Patient with Geleophysic Dysplasia Type I. Clin. Dysmorphol. 2016, 25, 106–109. [Google Scholar] [CrossRef]
- Legare, J.M.; Modaff, P.; Strom, S.P.; Pauli, R.M.; Bartlett, H.L. Geleophysic Dysplasia: 48 Year Clinical Update with Emphasis on Cardiac Care. Am. J. Med. Genet. 2018, 176, 2237–2242. [Google Scholar] [CrossRef]
- Trenson, S.; Hermans, H.; Craps, S.; Pokreisz, P.; de Zeeuw, P.; Van Wauwe, J.; Gillijns, H.; Veltman, D.; Wei, F.; Caluwé, E.; et al. Cardiac Microvascular Endothelial Cells in Pressure Overload–Induced Heart Disease. Circ. Heart Fail. 2021, 14, e006979. [Google Scholar] [CrossRef]
- Xie, T.; Guo, J.; Jiang, Y.; Li, L.; Jiang, L.; Wei, Y. Screening Differentially Expressed Proteins of Coronary Heart Disease with Congenital Cold Syndrome Based on Tandem Mass Tag (TMT) Technology. Bioengineered 2021, 12, 1338–1350. [Google Scholar] [CrossRef]
- Hubmacher, D.; Wang, L.W.; Mecham, R.P.; Reinhardt, D.P.; Apte, S.S. Adamtsl2 Deletion Results in Bronchial Fibrillin Microfibril Accumulation and Bronchial Epithelial Dysplasia—A Novel Mouse Model Providing Insights into Geleophysic Dysplasia. Dis. Models Mech. 2015, 8, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhon, L.; Mahaut, C.; Goudin, N.; Gaudas, E.; Piquand, K.; Le Goff, W.; Cormier-Daire, V.; Le Goff, C. Impairment of Chondrogenesis and Microfibrillar Network in Adamtsl2 Deficiency. FASEB J. 2019, 33, 2707–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rypdal, K.B.; Erusappan, P.M.; Melleby, A.O.; Seifert, D.E.; Palmero, S.; Strand, M.E.; Tønnessen, T.; Dahl, C.P.; Almaas, V.; Hubmacher, D.; et al. The Extracellular Matrix Glycoprotein ADAMTSL2 Is Increased in Heart Failure and Inhibits TGFβ Signalling in Cardiac Fibroblasts. Sci. Rep. 2021, 11, 19757. [Google Scholar] [CrossRef]
- Weill, G. Ectopie Du Cristallin et Malformations Générales. Ann. Ocul. 1932, 169, 21–44. [Google Scholar]
- Marchesani, O. Brachydaktylie Und Angeborene Kugellinse Als Systemerkrankung. Klin. Monatsbl. Augenheilkd 1939, 103, 392–406. [Google Scholar]
- Faivre, L.; Mégarbané, A.; Alswaid, A.; Zylberberg, L.; Aldohayan, N.; Campos-Xavier, A.; Bacq, D.; Legeai-Mallet, L.; Bonaventure, J.; Munnich, A.; et al. Homozygosity Mapping of a Weill-Marchesani Syndrome Locus to Chromosome 19p13.3-P13.2. Hum. Genet. 2002, 110, 366–370. [Google Scholar] [CrossRef]
- Kutz, W.E.; Wang, L.W.; Dagoneau, N.; Odrcic, K.J.; Cormier-Daire, V.; Traboulsi, E.I.; Apte, S.S. Functional Analysis of an ADAMTS10 Signal Peptide Mutation in Weill-Marchesani Syndrome Demonstrates a Long-Range Effect on Secretion of the Full-Length Enzyme. Hum. Mutat. 2008, 29, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Al-Sharif, L.; Khalil, D.S.; Shinwari, J.M.A.; Bavi, P.; Al-Mahrouqi, R.A.; Al-Rajhi, A.; Alkuraya, F.S.; Meyer, B.F.; Al Tassan, N. Homozygous Mutations in ADAMTS10 and ADAMTS17 Cause Lenticular Myopia, Ectopia Lentis, Glaucoma, Spherophakia, and Short Stature. Am. J. Hum. Genet. 2009, 85, 558–568. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O.; Aldahmesh, M.A.; Al-Ghadeer, H.; Mohamed, J.Y.; Alkuraya, F.S. Familial Spherophakia with Short Stature Caused by a Novel Homozygous ADAMTS17 Mutation. Ophthalmic Genet. 2012, 33, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Kojuri, J.; Razeghinejad, M.R.; Aslani, A. Cardiac Findings in Weill–Marchesani Syndrome. Am. J. Med. Genet. 2007, 143A, 2062–2064. [Google Scholar] [CrossRef]
- Dagoneau, N.; Benoist-Lasselin, C.; Huber, C.; Faivre, L.; Mégarbané, A.; Alswaid, A.; Dollfus, H.; Alembik, Y.; Munnich, A.; Legeai-Mallet, L.; et al. ADAMTS10 Mutations in Autosomal Recessive Weill-Marchesani Syndrome. Am. J. Hum. Genet. 2004, 75, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.R.; Care4Rare Canada Consortium; Green, J.S.; Fahiminiya, S.; Majewski, J.; Fernandez, B.A.; Deardorff, M.A.; Johnson, G.J.; Whelan, J.H.; Hubmacher, D.; et al. A Novel Pathogenic Missense ADAMTS17 Variant That Impairs Secretion Causes Weill-Marchesani Syndrome with Variably Dysmorphic Hand Features. Sci. Rep. 2020, 10, 10827. [Google Scholar] [CrossRef] [PubMed]
- Steinkellner, H.; Etzler, J.; Gogoll, L.; Neesen, J.; Stifter, E.; Brandau, O.; Laccone, F. Identification and Molecular Characterisation of a Homozygous Missense Mutation in the ADAMTS10 Gene in a Patient with Weill–Marchesani Syndrome. Eur. J. Hum. Genet. 2015, 23, 1186–1191. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.W.; Kutz, W.E.; Mead, T.J.; Beene, L.C.; Singh, S.; Jenkins, M.W.; Reinhardt, D.P.; Apte, S.S. Adamts10 Inactivation in Mice Leads to Persistence of Ocular Microfibrils Subsequent to Reduced Fibrillin-2 Cleavage. Matrix Biol. 2019, 77, 117–128. [Google Scholar] [CrossRef]
- Ye, S.; Yang, N.; Lu, T.; Wu, T.; Wang, L.; Pan, Y.-H.; Cao, X.; Yuan, X.; Wisniewski, T.; Dang, S.; et al. Adamts18 Modulates the Development of the Aortic Arch and Common Carotid Artery. iScience 2021, 24, 102672. [Google Scholar] [CrossRef]
- Lu, T.; Lin, X.; Pan, Y.-H.; Yang, N.; Ye, S.; Zhang, Q.; Wang, C.; Zhu, R.; Zhang, T.; Wisniewski, T.M.; et al. ADAMTS18 Deficiency Leads to Pulmonary Hypoplasia and Bronchial Microfibril Accumulation. iScience 2020, 23, 101472. [Google Scholar] [CrossRef] [PubMed]
- Wünnemann, F.; MIBAVA Leducq Consortium Principal Investigators; Ta-Shma, A.; Preuss, C.; Leclerc, S.; van Vliet, P.P.; Oneglia, A.; Thibeault, M.; Nordquist, E.; Lincoln, J.; et al. Loss of ADAMTS19 Causes Progressive Non-Syndromic Heart Valve Disease. Nat. Genet. 2020, 52, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, T.; MacDonald, B.T.; Bhandary, B.; Popp, N.R.; Laprise, D.; Arduini, A.; Lai, D.; Zhu, Q.M.; Xing, Y.; Kaushik, V.K.; et al. Coronary Disease Association With ADAMTS7 Is Due to Protease Activity. Circ. Res. 2021, 129, 458–470. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Genotype | Treatment | Phenotype | Observations | References |
|---|---|---|---|---|
| Tshd4+/− | None | Thoracic aortic dilatation | Elastic fibers disruption, increased SMC apoptosis | [24] |
| Adamts1+/+ | Ang II | TAD | High ADAMTS1 expression | [32] |
| Adamts1−/− | BAPN | Decreased susceptibility to TAAD | No inflammatory cell infiltration | [33] |
| Adamts1+/− | Ang II and none | TAA | Medial degeneration, elastic fibers breaks, collagen and proteoglycans accumulation, increased TGFβ activation, elevated aortic NO and Nos2 | [35] |
| Adamts4+/+ | Ang II and high-fat diet | AAD | High ADAMTS4 expression in aortic SMC and macrophages | [40] |
| Adamts4−/− | Ang II and high-fat diet | Decreased susceptibility to AAD | Reduced elastic fibers destruction and versican degradation | [40] |
| Apoe−/− | Ang II and miR-126a-5p | Reduction AAA | ADAMTS4 downregulation, improvement elastic fibers fragmentation and ECM degradation | [41] |
| Adamts5Δcat | AngII | TAAD | Ascending aorta enlargement, ECM versican upregulation, and versikin reduction, low LRP1 protein levels, high ADAMTS1 protein levels | [43] |
| Adamts5−/− | AngII | Aortic dilatation | Versican accumulation, aortic SMC loss, elastin degradation, abnormal adventitia | [46] |
| Adamts16+/− Adamts16−/− | None | Hypotension | Low blood pressure, media layer thickness decrease, pulse wave velocity decrease | [48] |
| Adamts8ΔSM22 | Hypoxia | PAH reduction | PASMC proliferation decrease, AMPK upregulation, endothelial dysfunction, matrix metalloprotease activation | [50] |
| Adamts9+/− | None | Aortic wall defects, valve sinus and valve leaflets, abnormal myocardial projections, spongy myocardium | Abnormal versican accumulation, decrease in versican cleavage | [54] |
| Adamts7−/− | None | Excessive re-reendothelialization and reduced atherosclerosis | EC proliferation enhanced | [59,60,91] |
| Adamtsl2−/− | None | Post-natal death | Lung abnormalities associated with bronchial fibrillin microfibril accumulation, cardiac developmental defects | [74,75] |
| Adamts18−/− | None | Malformations in the aortic arch and carotid artery system, thymus hypoplasia, carotid body absence | Disordered elastic fibers, increased Fbn1 level, low blood pressure. | [88] |
| Adamts19−/− | None | Non-syndromic heart valve disease | ECM disorganization, high proteoglycan content, thin collagen fibers | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mougin, Z.; Huguet Herrero, J.; Boileau, C.; Le Goff, C. ADAMTS Proteins and Vascular Remodeling in Aortic Aneurysms. Biomolecules 2022, 12, 12. https://doi.org/10.3390/biom12010012
Mougin Z, Huguet Herrero J, Boileau C, Le Goff C. ADAMTS Proteins and Vascular Remodeling in Aortic Aneurysms. Biomolecules. 2022; 12(1):12. https://doi.org/10.3390/biom12010012
Chicago/Turabian StyleMougin, Zakaria, Julia Huguet Herrero, Catherine Boileau, and Carine Le Goff. 2022. "ADAMTS Proteins and Vascular Remodeling in Aortic Aneurysms" Biomolecules 12, no. 1: 12. https://doi.org/10.3390/biom12010012
APA StyleMougin, Z., Huguet Herrero, J., Boileau, C., & Le Goff, C. (2022). ADAMTS Proteins and Vascular Remodeling in Aortic Aneurysms. Biomolecules, 12(1), 12. https://doi.org/10.3390/biom12010012