
The Mucolipin TRPML2 Channel Enhances the Sensitivity of Multiple Myeloma Cell Lines to Ibrutinib and/or Bortezomib Treatment

 ,
,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. MTT Assay
2.3. Cell Cycle Analysis
2.4. Annexin-V/PI Assay
2.5. Gene Expression Analysis
2.6. Mitochondrial Transmembrane Potential (Δψm) Assay
2.7. TRPML2 Gene Silencing
2.8. Western Blot
2.9. Statistical Analysis
3. Results
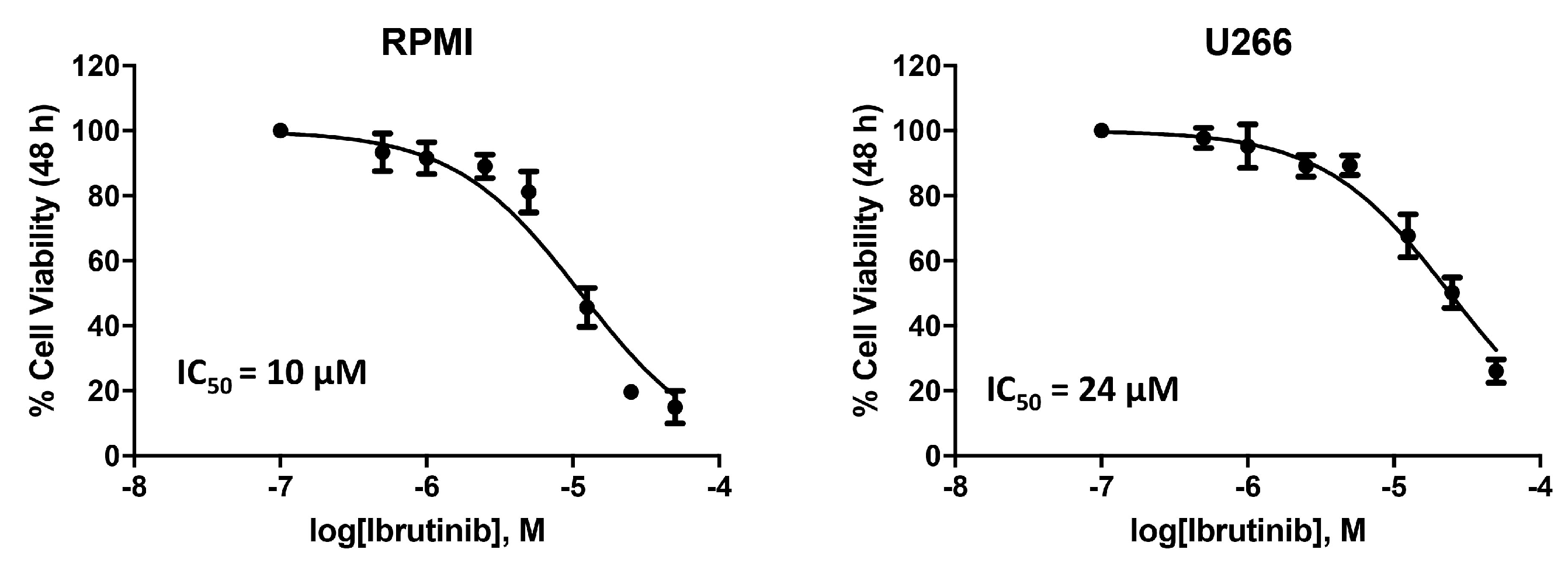
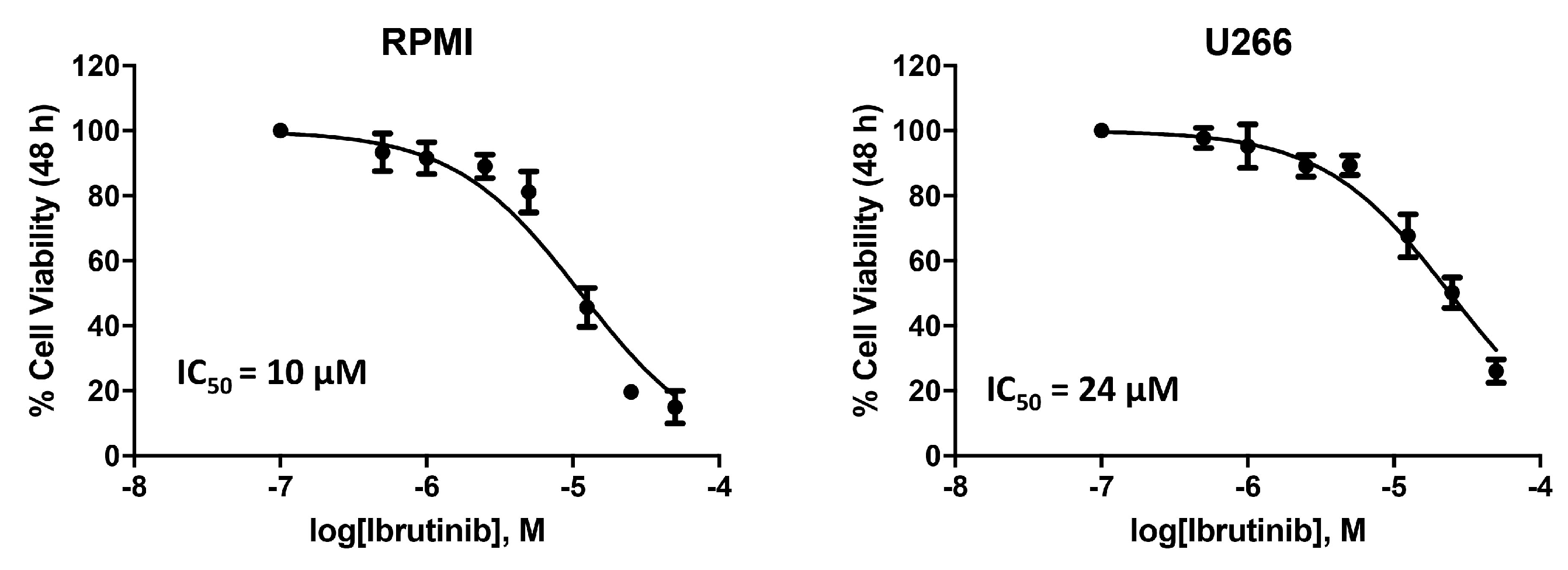
3.1. Cytotoxic Effect of Ibrutinib on MM Cell Viability
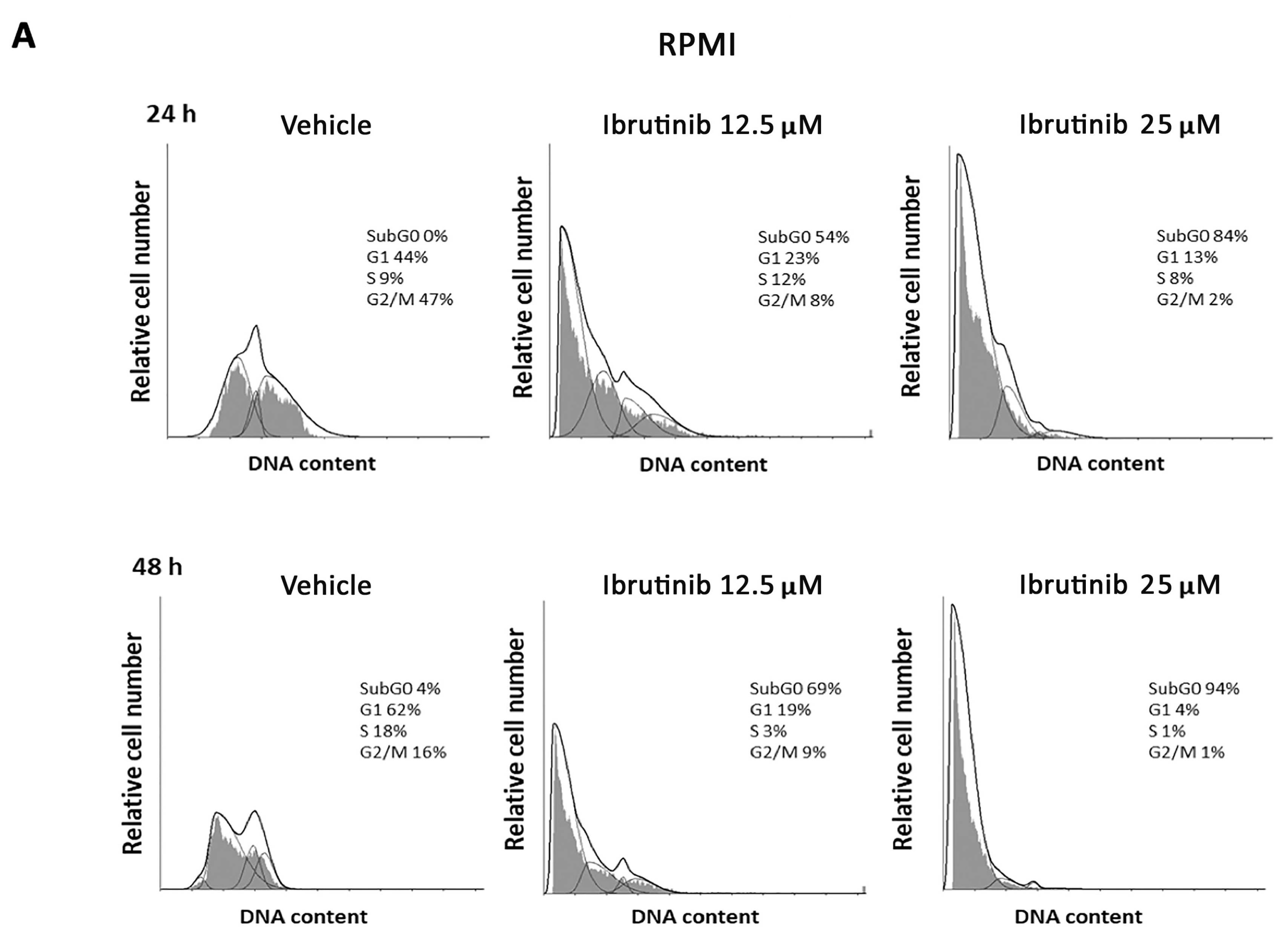
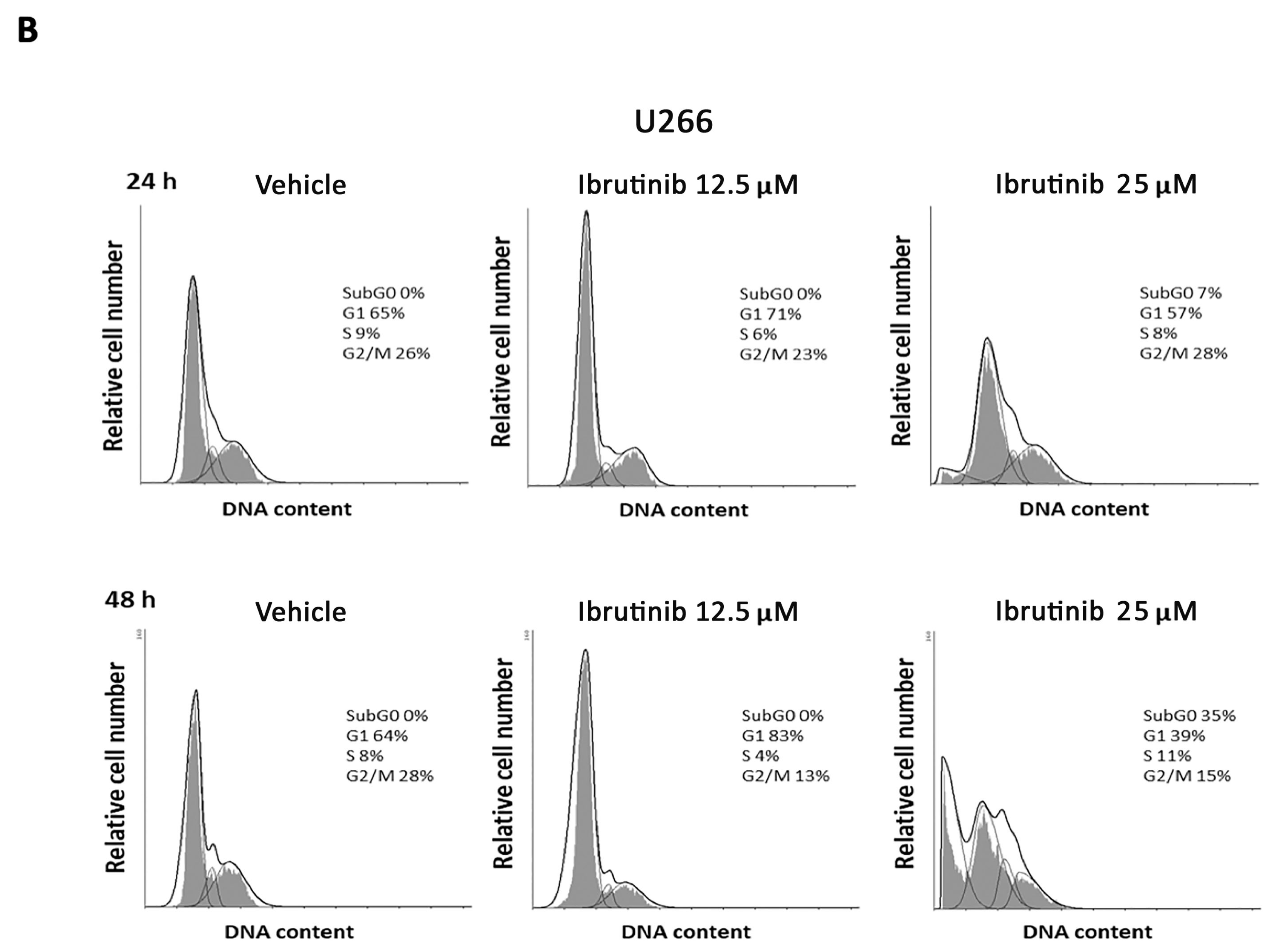
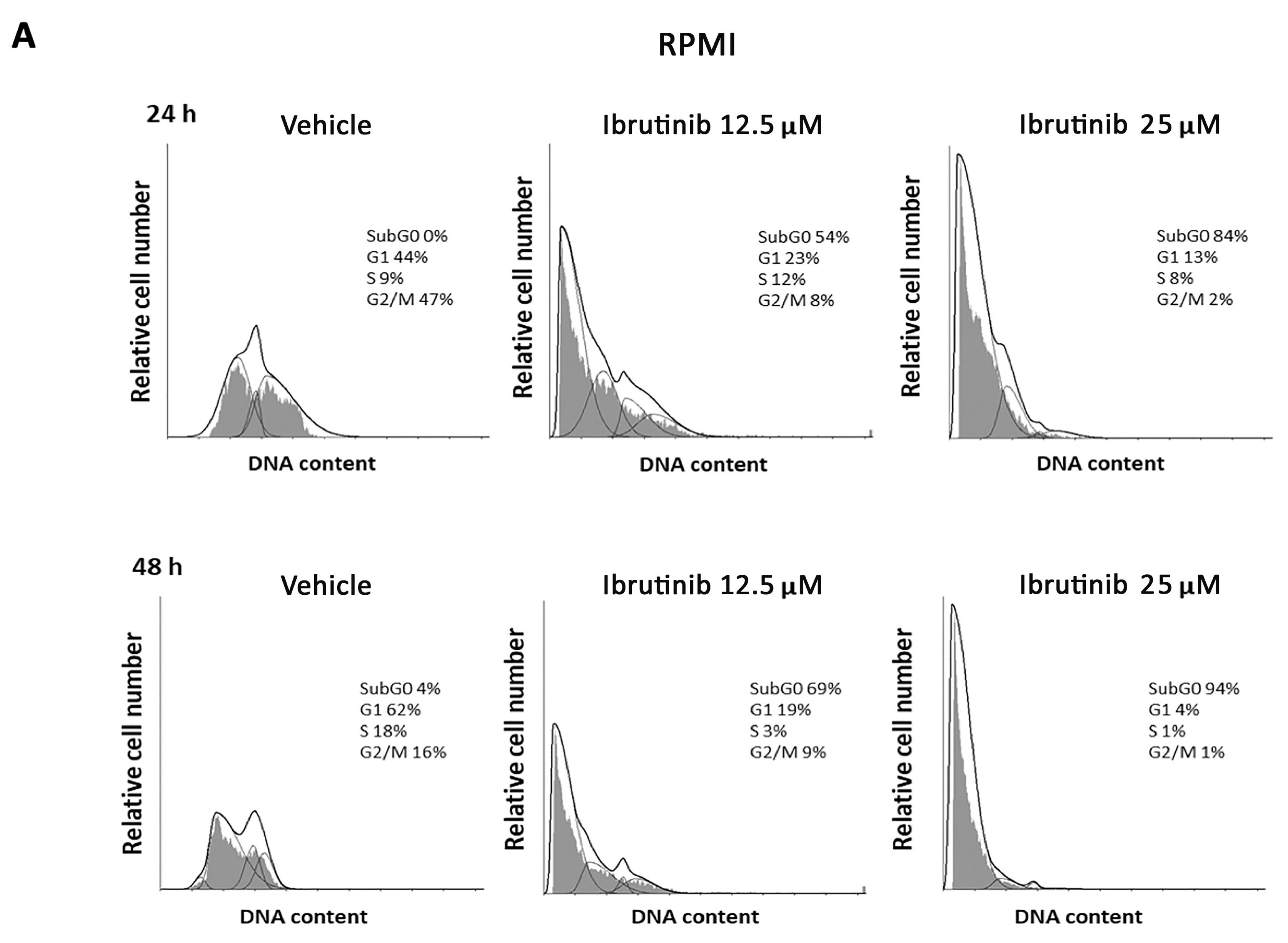
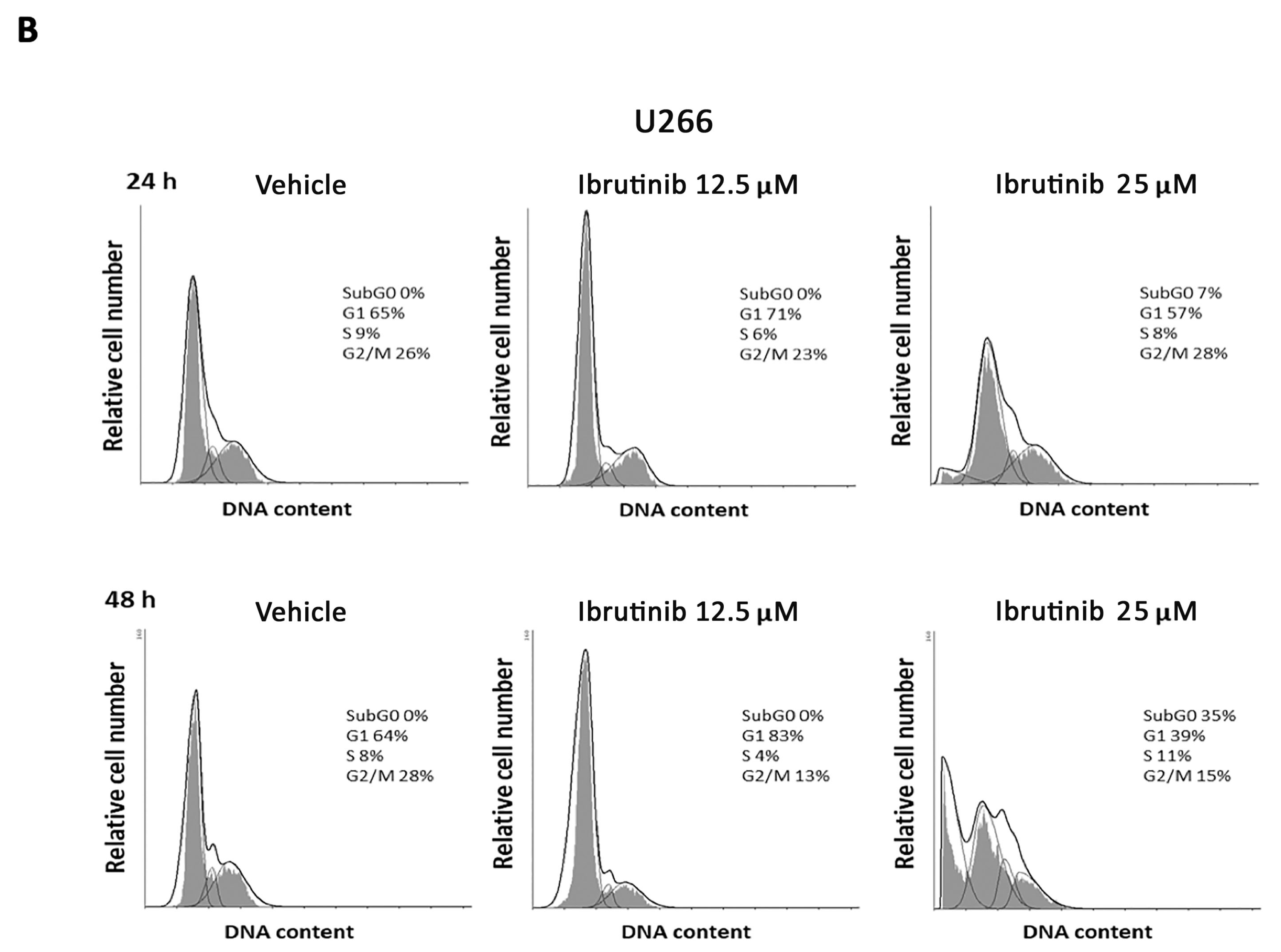
3.2. Ibrutinib Effects on Cell Cycle
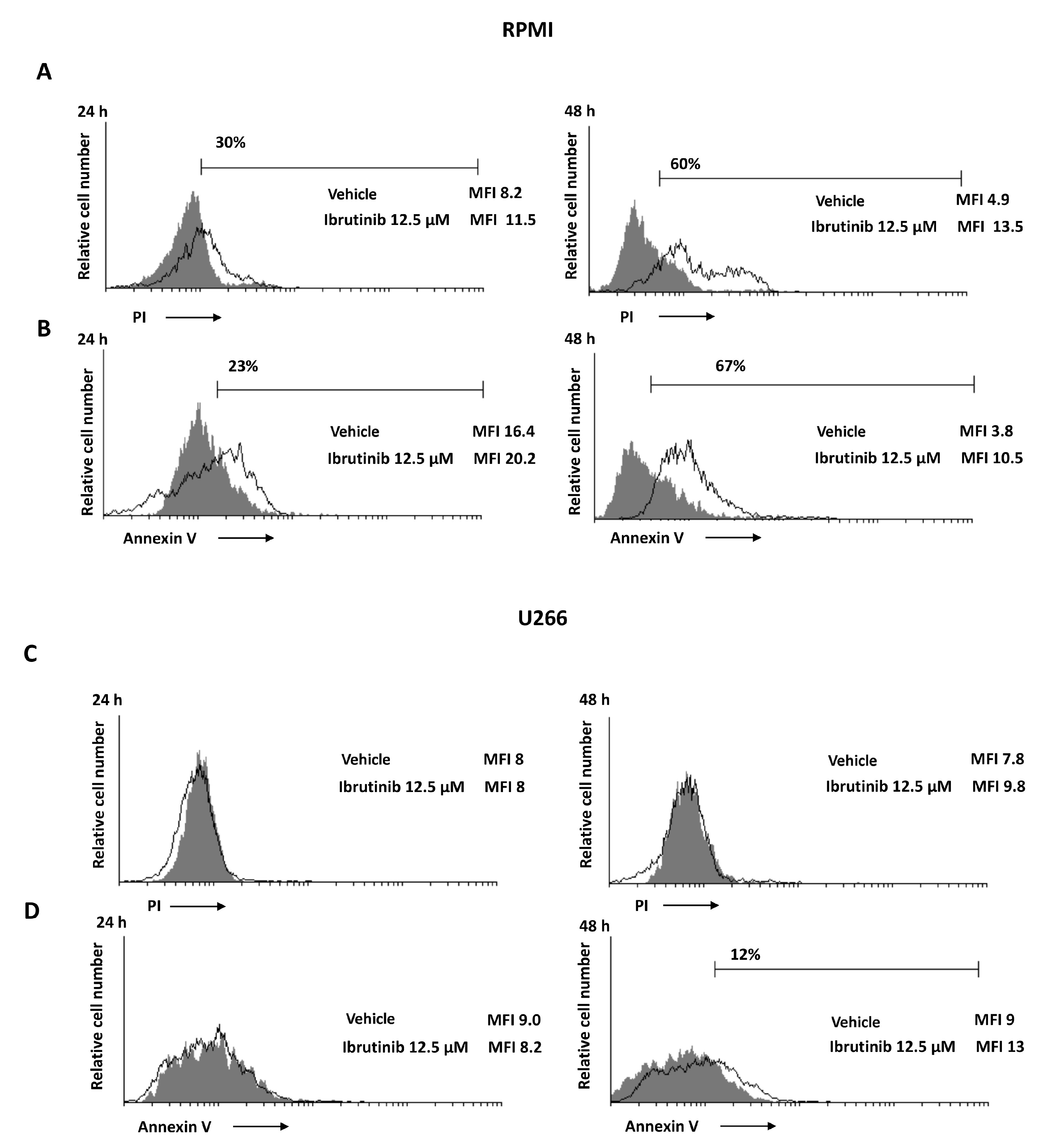
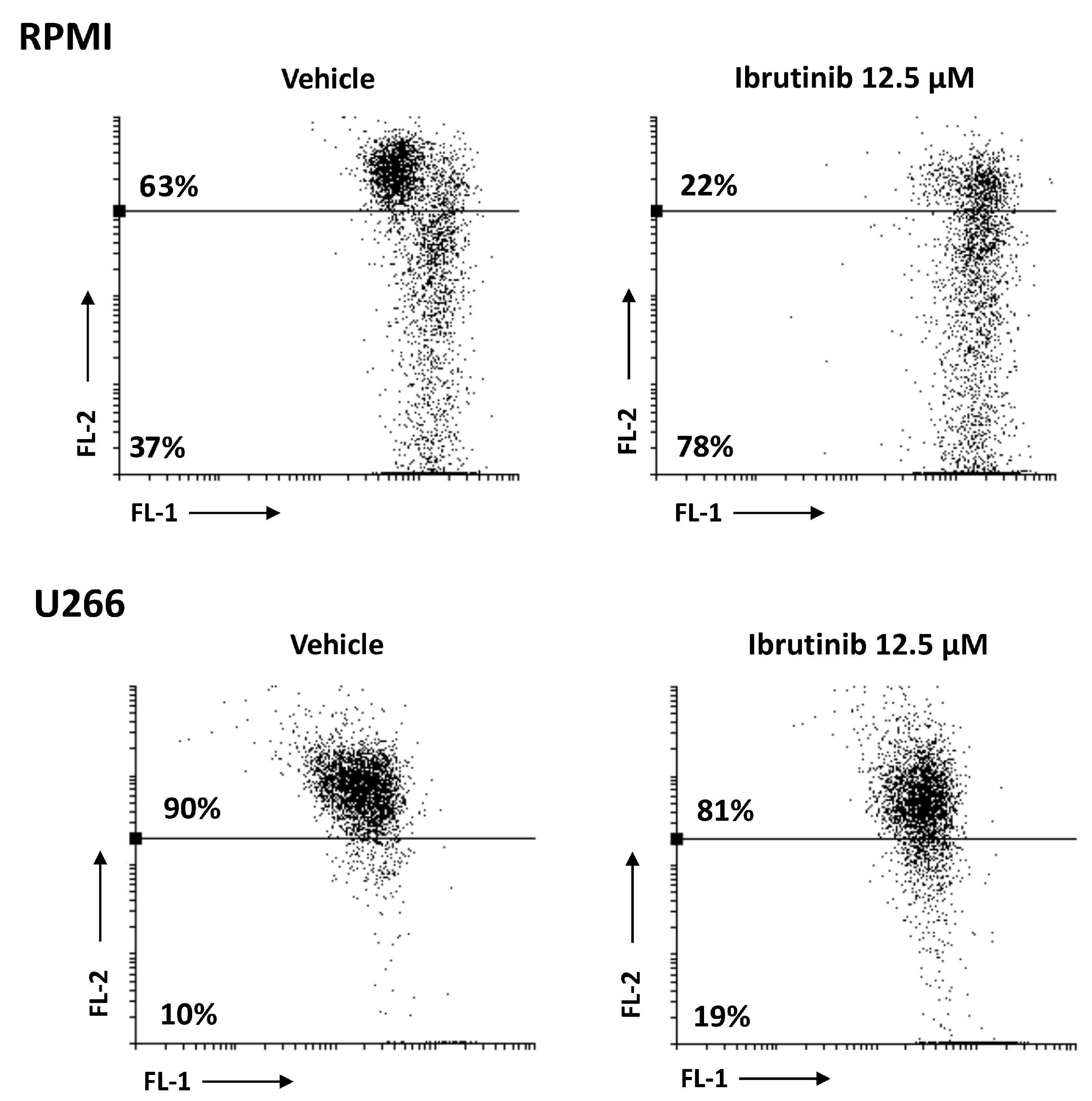
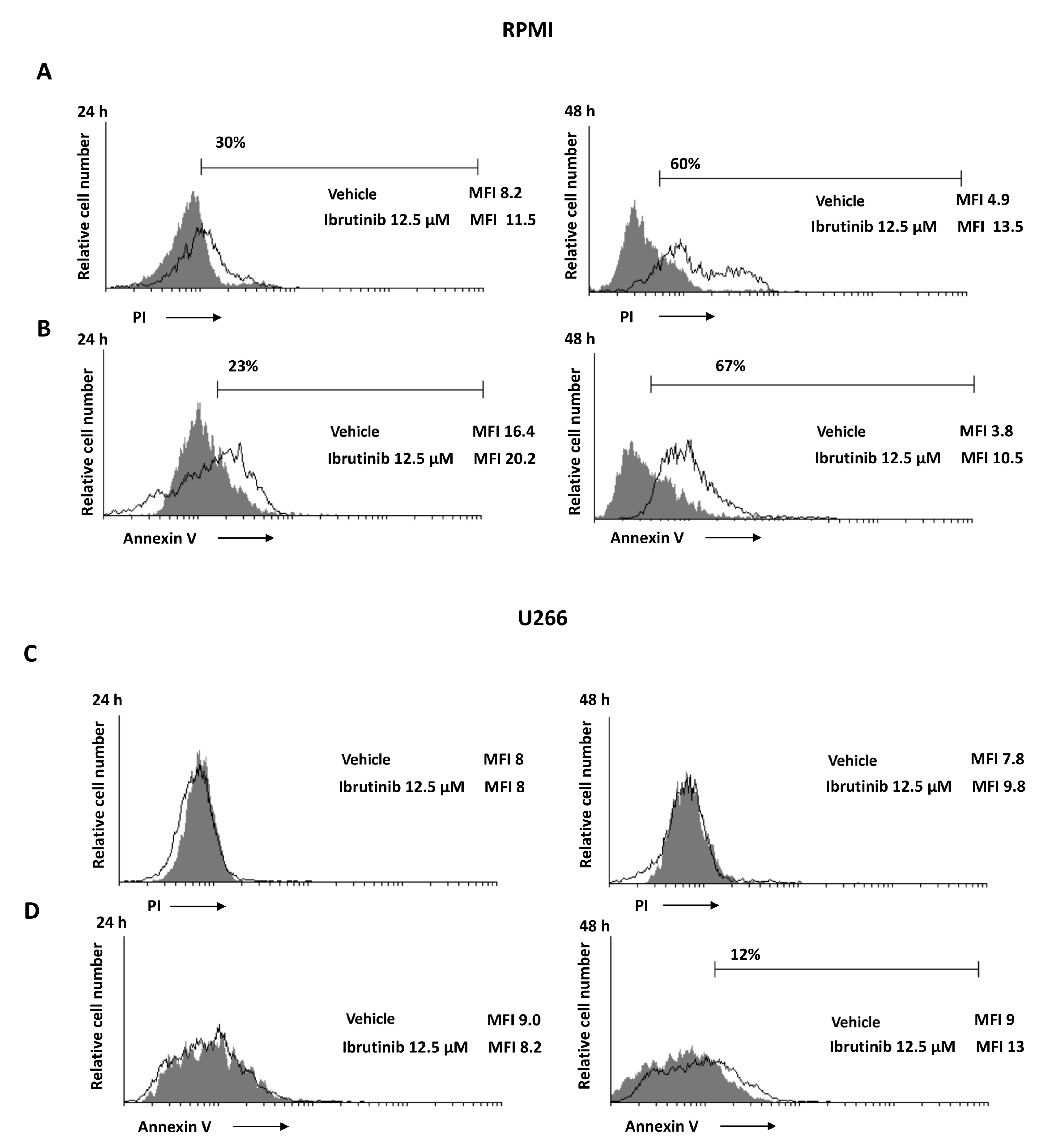
3.3. Ibrutinib Induces Apoptotic Cell Death in MM Cell Lines
3.4. Ibrutinib Induces Mitochondrial Depolarization, Mainly in RPMI Cells
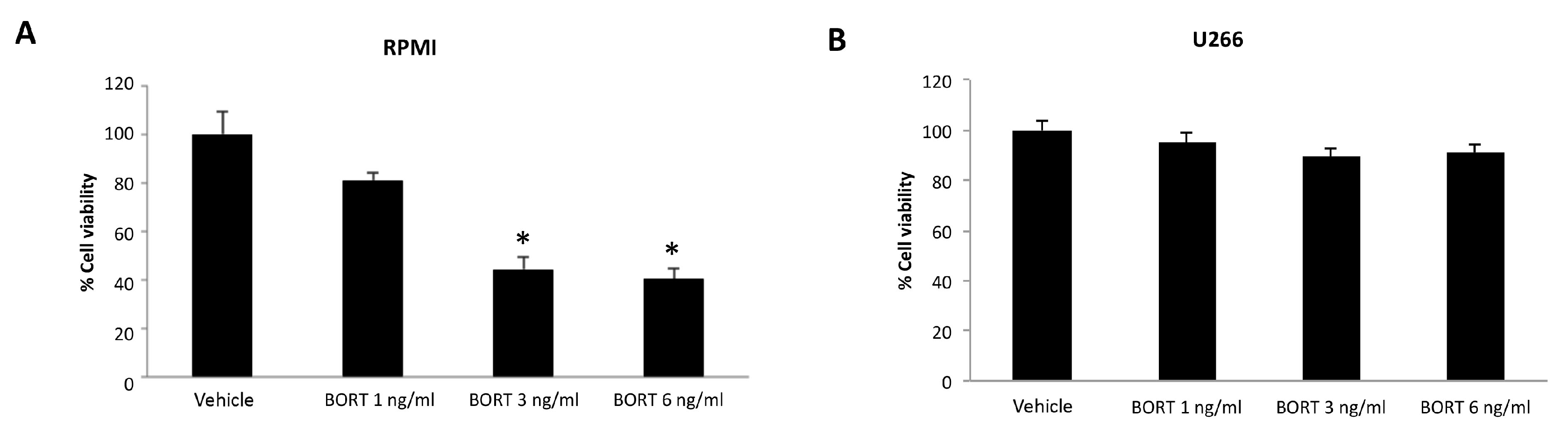
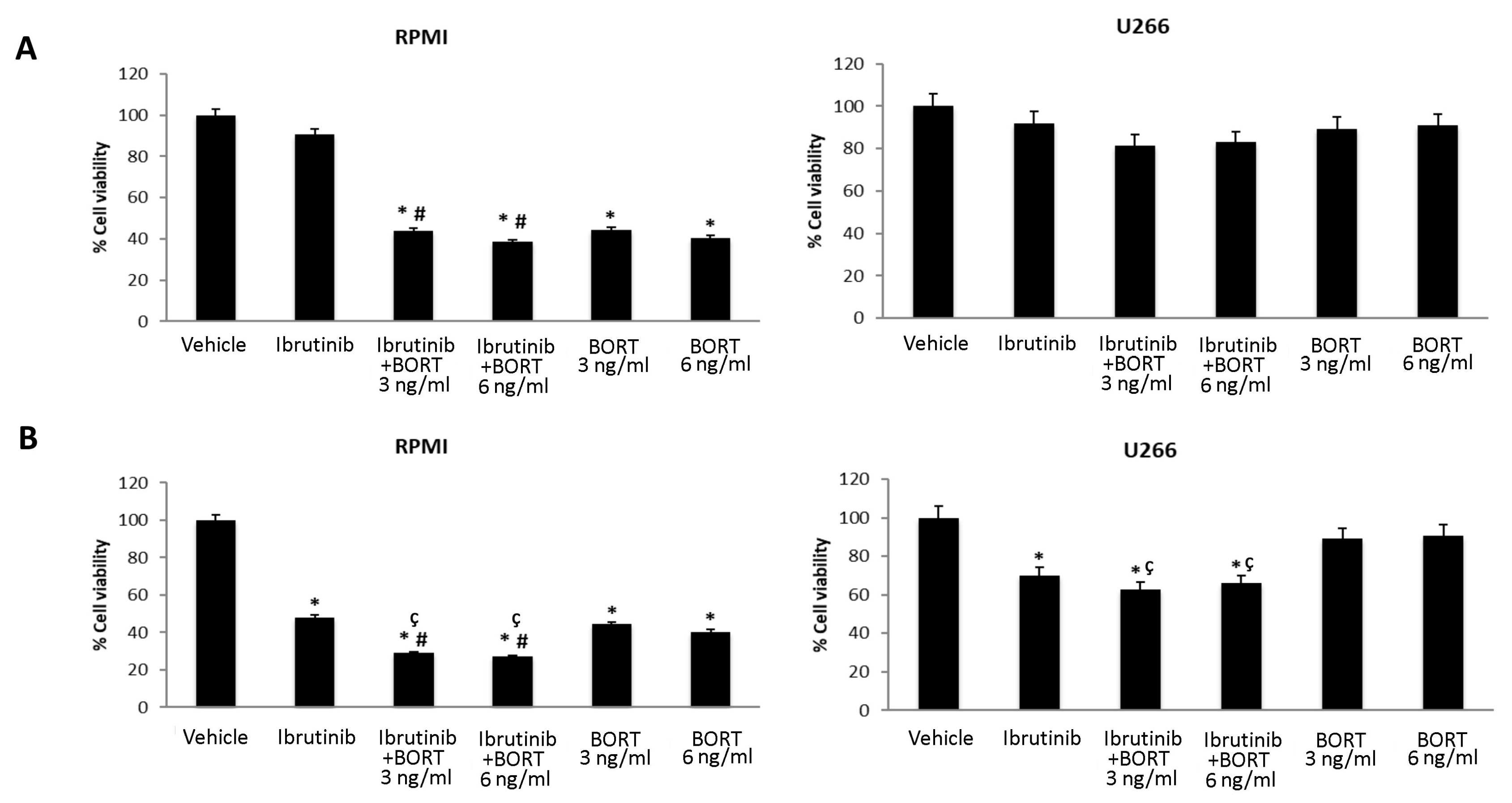
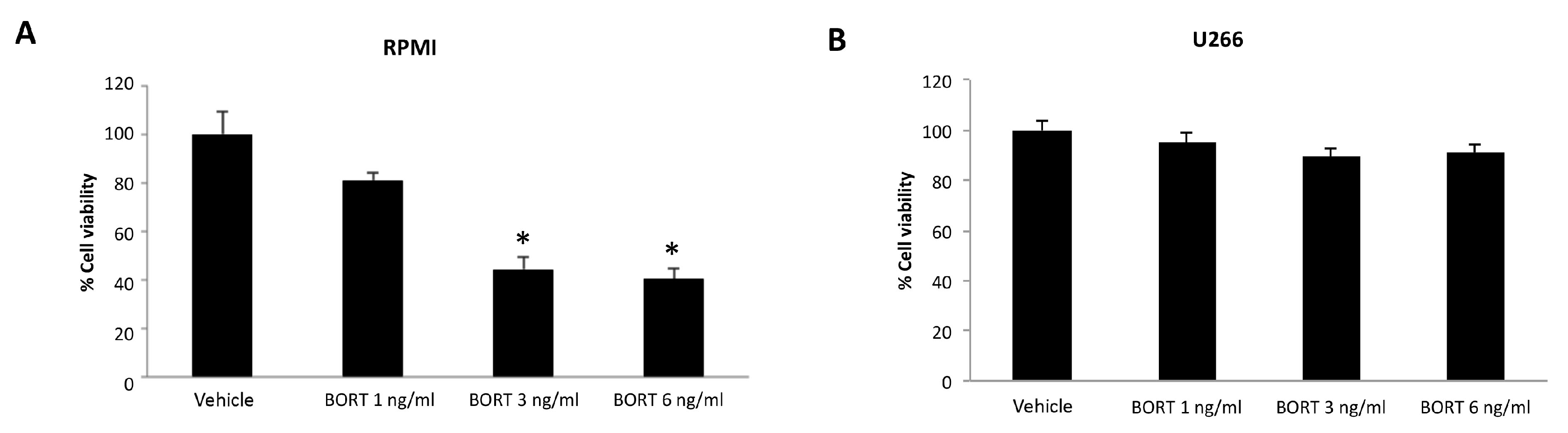
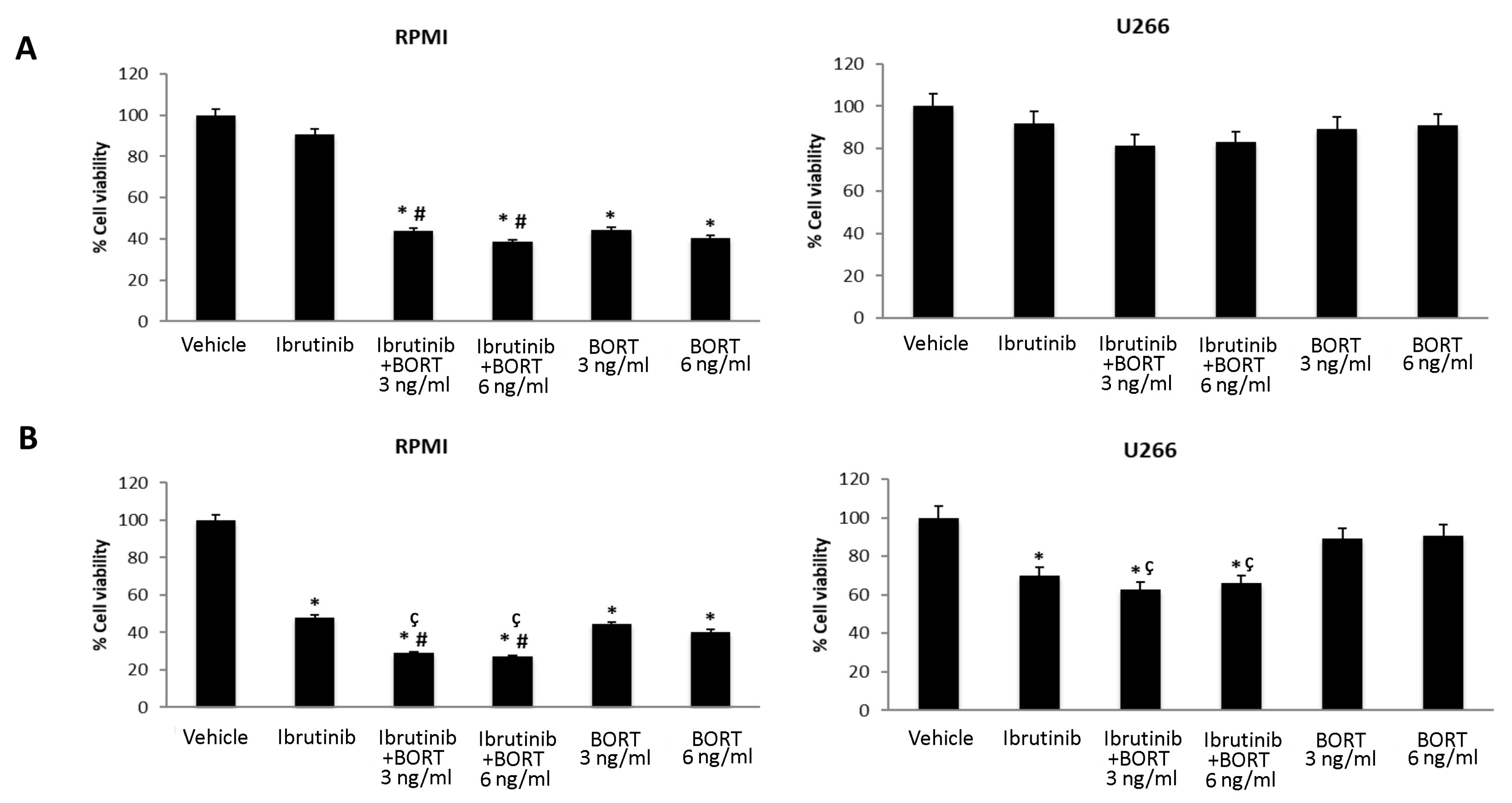
3.5. Ibrutinib and Bortezomib in Combination Increase the Cytotoxicity in RPMI But Not in U266 Cells
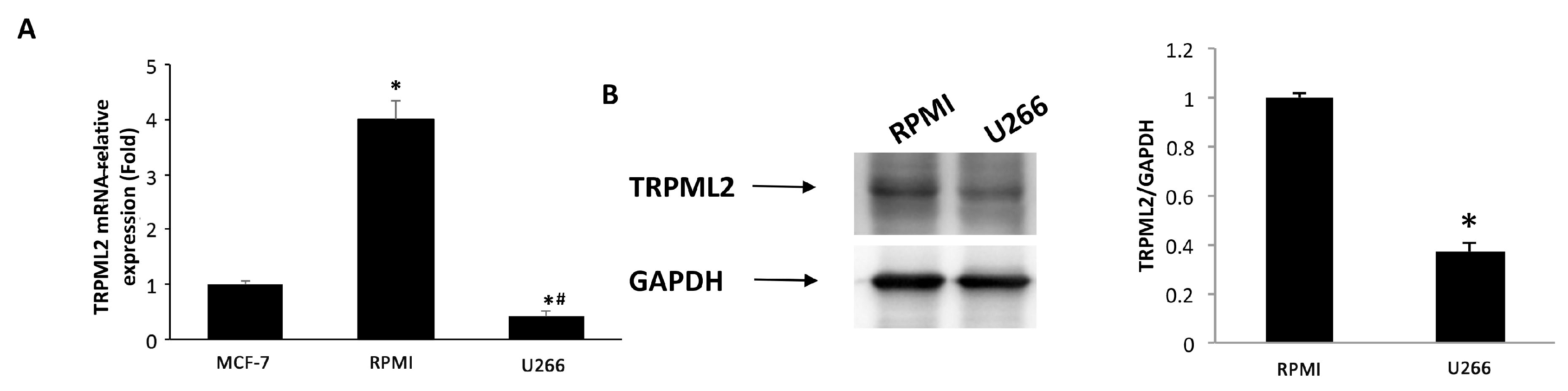
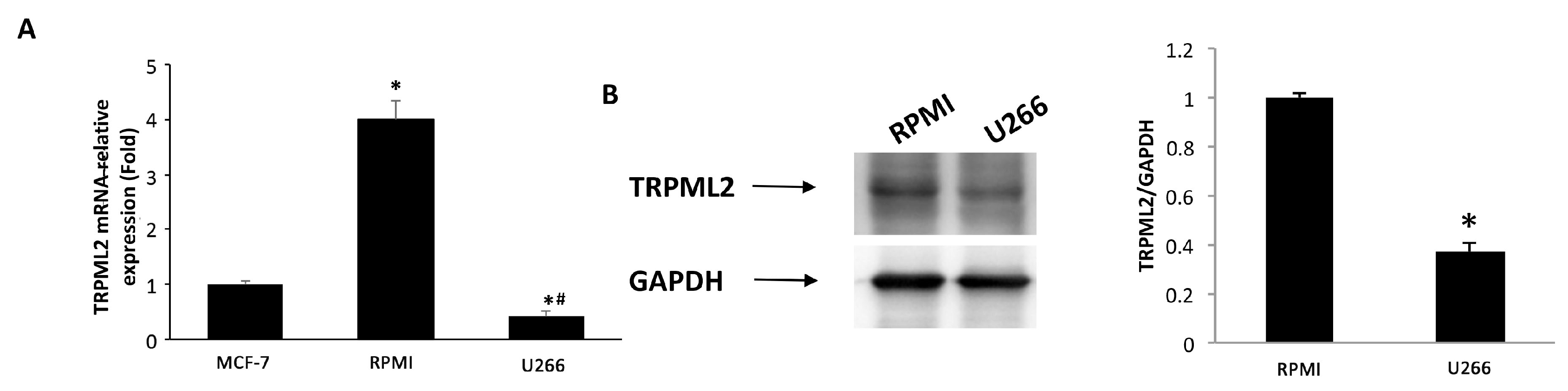
3.6. Expression of TRPML2 Channel in RPMI and U266 Cell Lines
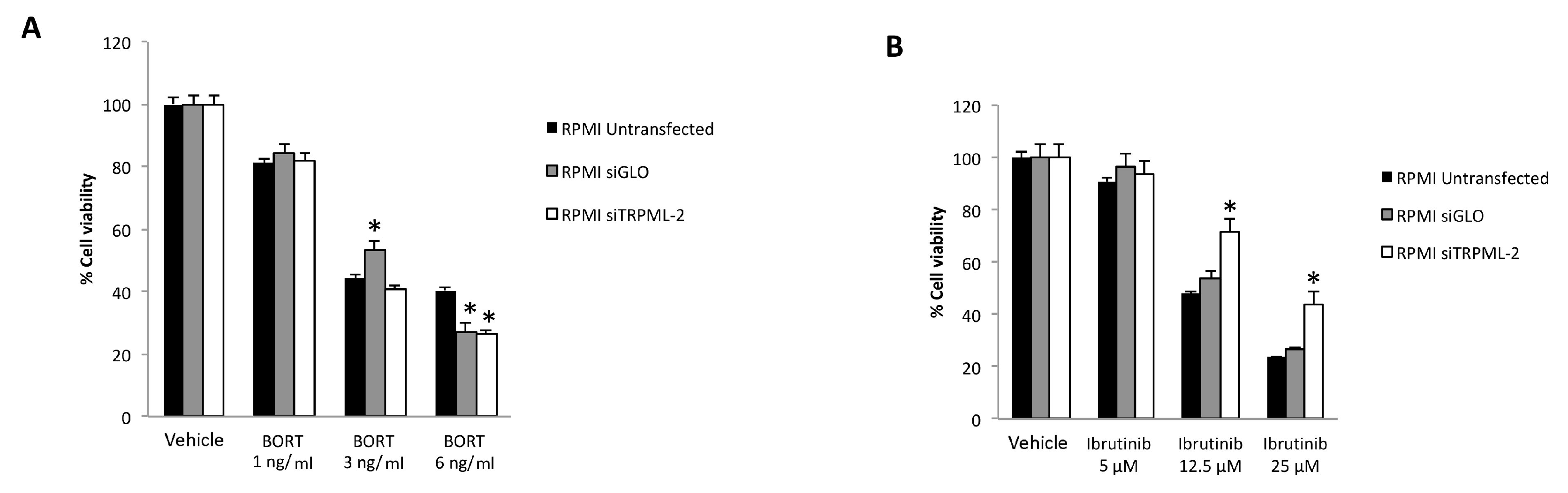
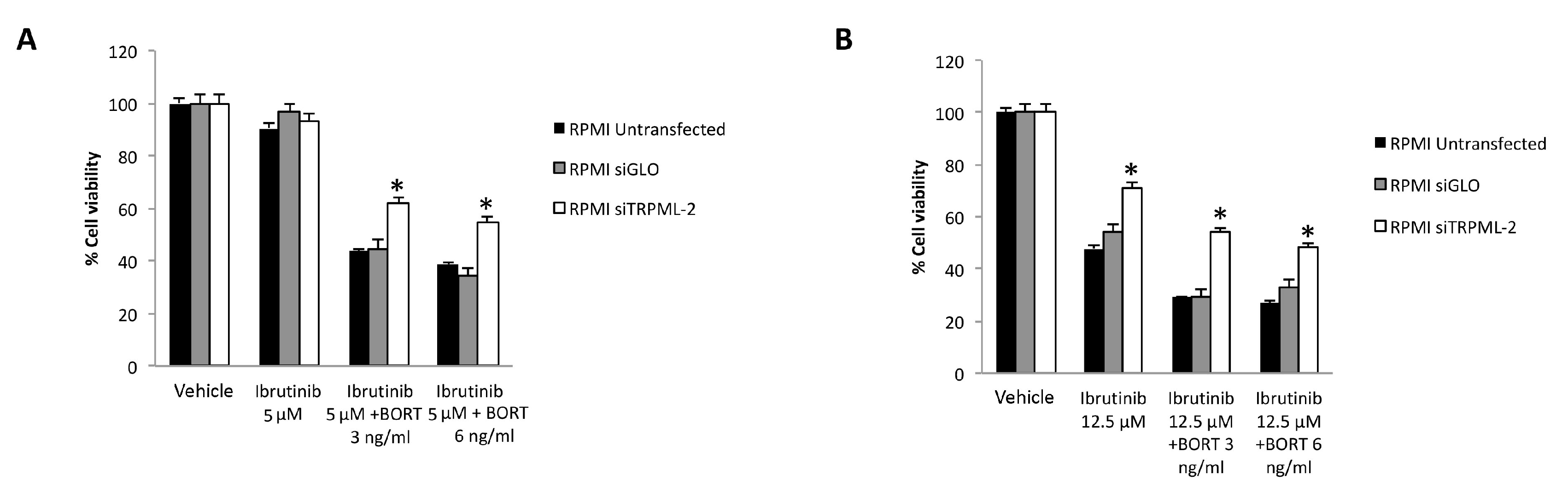
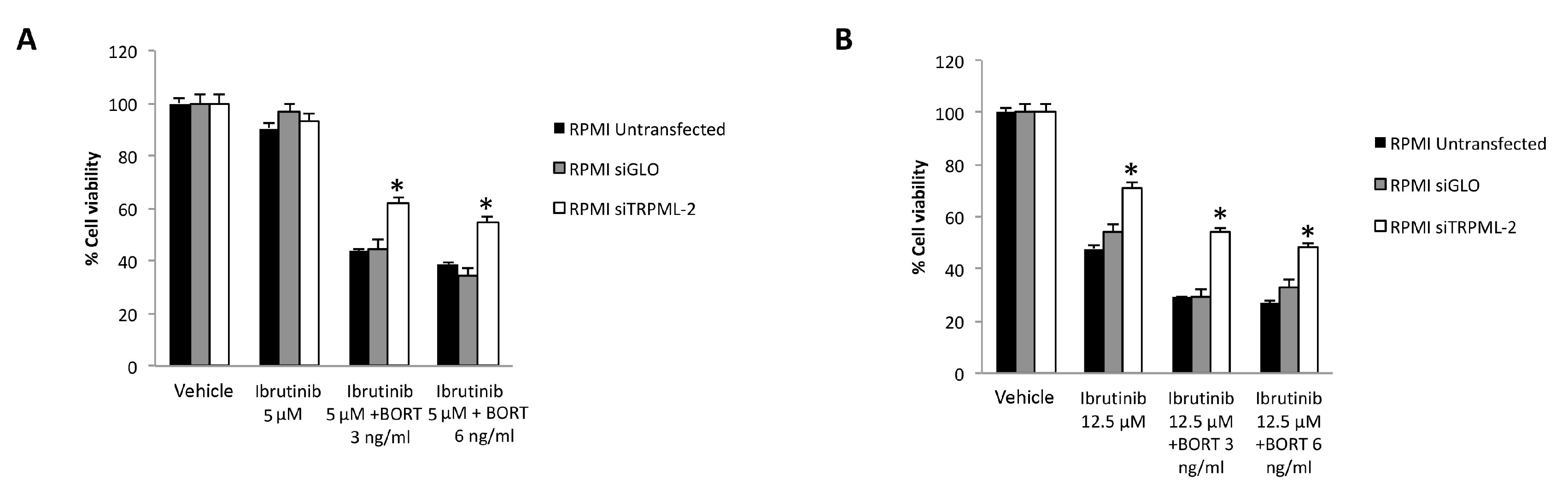
3.7. TRPML2 Silencing in RPMI Cells Reduces the Cytotoxic Effect of Ibrunitinb Alone or in Combination with Bortezomib
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kazandjian, D. Multiple Myeloma Epidemiology and Survival: A Unique Malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, A.; Bringhen, S.; Ludwig, H.; Dimopoulos, M.A.; Bladé, J.; Mateos, M.V.; Rosiñol, L.; Boccadoro, M.; Cavo, M.; Lokhorst, H.; et al. Personalized Therapy in Multiple Myeloma According to Patient Age and Vulnerability: A Report of the European Myeloma Network (EMN). Blood 2011, 118, 4519–4529. [Google Scholar] [CrossRef] [PubMed]
- Fairfield, H.; Falank, C.; Avery, L.; Reagan, M.R. Multiple Myeloma in the Marrow: Pathogenesis and Treatments. Ann. N. Y. Acad. Sci. 2016, 1364, 32–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple Myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the Initiation of Multiple Myeloma. Nat. Commun. 2020, 11, 1917. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef] [PubMed]
- Simeon, V.; Todoerti, K.; la Rocca, F.; Caivano, A.; Trino, S.; Lionetti, M.; Agnelli, L.; de Luca, L.; Laurenzana, I.; Neri, A.; et al. Molecular Classification and Pharmacogenetics of Primary Plasma Cell Leukemia: An Initial Approach toward Precision Medicine. Int. J. Mol. Sci. 2015, 16, 17514–17534. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, Z.; Gao, S. Key Regulators of Sensitivity to Immunomodulatory Drugs in Cancer Treatment. Biomark. Res. 2021, 9, 43. [Google Scholar] [CrossRef]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Nunes, A.T.; Annunziata, C.M. Proteasome Inhibitors: Structure and Function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, A.; Li, X.; Ling, W.; Khan, S.; Zangari, M.; Yaccoby, S. The Proteasome Inhibitor, Bortezomib Suppresses Primary Myeloma and Stimulates Bone Formation in Myelomatous and Nonmyelomatous Bones in Vivo. Am. J. Hematol. 2009, 84, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, M.Y.; Zaitseva, L.; Auger, M.J.; Craig, J.I.; MacEwan, D.J.; Rushworth, S.A.; Bowles, K.M. Ibrutinib Inhibits BTK-Driven NF-ΚB P65 Activity to Overcome Bortezomib-Resistance in Multiple Myeloma. Cell Cycle 2015, 14, 2367–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, A.; Akhtar, S.; Yousaf, H.; Manna, A.; Paulus, S.M.; Bashir, Y.; Caulfield, T.R.; Kuranz-Blake, M.; Chitta, K.; Wang, X.; et al. Waldenstrom Macroglobulinemia Cells Devoid of BTKC481S or CXCR4WHIM-like Mutations Acquire Resistance to Ibrutinib through Upregulation of Bcl-2 and AKT Resulting in Vulnerability towards Venetoclax or MK2206 Treatment. Blood Cancer J. 2017, 7, e565. [Google Scholar] [CrossRef]
- Gu, C.; Peng, H.; Lu, Y.; Yang, H.; Tian, Z.; Yin, G.; Zhang, W.; Lu, S.; Zhang, Y.; Yang, Y. BTK Suppresses Myeloma Cellular Senescence through Activating AKT/P27/Rb Signaling. Oncotarget 2017, 8, 56858–56867. [Google Scholar] [CrossRef] [Green Version]
- Niemann, C.U.; Wiestner, A. B-Cell Receptor Signaling as a Driver of Lymphoma Development and Evolution. Semin. Cancer Biol. 2013, 23, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Bowles, K.M.; Barrera, L.N.; Murray, M.Y.; Zaitseva, L.; MacEwan, D.J. BTK Inhibitor Ibrutinib Is Cytotoxic to Myeloma and Potently Enhances Bortezomib and Lenalidomide Activities through NF-ΚB. Cell. Signal. 2013, 25, 106–112. [Google Scholar] [CrossRef]
- Singh, S.P.; Pillai, S.Y.; de Bruijn, M.J.W.; Stadhouders, R.; Corneth, O.B.J.; van den Ham, H.J.; Muggen, A.; van IJcken, W.; Slinger, E.; Kuil, A.; et al. Cell Lines Generated from a Chronic Lymphocytic Leukemia Mouse Model Exhibit Constitutive Btk and Akt Signaling. Oncotarget 2017, 8, 71981–71995. [Google Scholar] [CrossRef]
- Lindvall, J.M.; Blomberg, K.E.M.; Wennborg, A.; Smith, C.I.E. Differential Expression and Molecular Characterisation of Lmo7, Myo1e, Sash1, and Mcoln2 Genes in Btk-Defective B-Cells. Cell. Immunol. 2005, 235, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Samie, M.A.; Grimm, C.; Evans, J.A.; Curcio-Morelli, C.; Heller, S.; Slaugenhaupt, S.A.; Cuajungco, M.P. The Tissue-Specific Expression of TRPML2 (MCOLN-2) Gene Is Influenced by the Presence of TRPML1. Pflügers Arch.-Eur. J. Physiol. 2009, 459, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Flajnik, M.F.; Kasahara, M. Origin and Evolution of the Adaptive Immune System: Genetic Events and Selective Pressures. Nat. Rev. Genet. 2010, 11, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Spix, B.; Chao, Y.K.; Abrahamian, C.; Chen, C.-C.; Grimm, C. TRPML cation channels in inflammation and immunity. Front. Immunol. 2020, 11, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutt, S.L.; Heavey, B.; Rolink, A.G.; Busslinger, M. Commitment to the B-Lymphoid Lineage Depends on the Transcription Factor Pax5. Nature 1999, 401, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Valadez, J.A.; Cuajungco, M.P. PAX5 Is the Transcriptional Activator of Mucolipin-2 (MCOLN2) Gene. Gene 2015, 555, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Almamun, M.; Levinson, B.T.; van Swaay, A.C.; Johnson, N.T.; McKay, S.D.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. Integrated Methylome and Transcriptome Analysis Reveals Novel Regulatory Elements in Pediatric Acute Lymphoblastic Leukemia. Epigenetics 2015, 10, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Santoni, G.; Maggi, F.; Morelli, M.B.; Santoni, M.; Marinelli, O. Transient Receptor Potential Cation Channels in Cancer Therapy. Med. Sci. 2019, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Dong, X.-P. Endolysosomal TRPMLs in cancer. Biomolecules 2021, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Farfariello, V. TRP Channels and Cancer: New Targets for Diagnosis and Chemotherapy. Endocr. Metab. Immune Disord. Drug Targets 2011, 11, 54–67. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 Channel by Cannabidiol Sensitizes Glioblastoma Cells to Cytotoxic Chemotherapeutic Agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol Stimulates Aml-1a-Dependent Glial Differentiation and Inhibits Glioma Stem-like Cells Proliferation by Inducing Autophagy in a TRPV2-Dependent Manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The Effects of Cannabidiol and Its Synergism with Bortezomib in Multiple Myeloma Cell Lines. A Role for Transient Receptor Potential Vanilloid Type-2. Int. J. Cancer 2014, 134, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of Transient Receptor Potential Mucolipin-2 Ion Channels in Gliomas: Role in Tumor Growth and Progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, G.; Anderson, K.C. Understanding Biology to Tackle the Disease: Multiple Myeloma from Bench to Bedside, and Back. CA A Cancer J. Clin. 2014, 64, 422–444. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Larson, S.; Holkova, B.; Cornell, R.F.; Gasparetto, C.; Karanes, C.; Matous, J.V.; Niesvizky, R.; Valent, J.; Lunning, M.; et al. Phase 1 Trial of Ibrutinib and Carfilzomib Combination Therapy for Relapsed or Relapsed and Refractory Multiple Myeloma. Leuk. Lymphoma 2018, 59, 2588–2594. [Google Scholar] [CrossRef] [Green Version]
- Hajek, R.; Pour, L.; Ozcan, M.; Martin Sánchez, J.; García Sanz, R.; Anagnostopoulos, A.; Oriol, A.; Cascavilla, N.; Terjung, A.; Lee, Y.; et al. A Phase 2 Study of Ibrutinib in Combination with Bortezomib and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Eur. J. Haematol. 2020, 104, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Cornell, R.F.; Gasparetto, C.; Karanes, C.; Matous, J.V.; Niesvizky, R.; Lunning, M.; Usmani, S.Z.; Anderson, L.D.; Chhabra, S.; et al. Final Analysis of a Phase 1/2b Study of Ibrutinib Combined with Carfilzomib/Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Hematol. Oncol. 2020, 38, 353–362. [Google Scholar] [CrossRef]
- Wei, L.; Su, Y.-K.; Lin, C.-M.; Chao, T.-Y.; Huang, S.-P.; Huynh, T.-T.; Jan, H.-J.; Whang-Peng, J.; Chiou, J.-F.; Wu, A.T.H.; et al. Preclinical Investigation of Ibrutinib, a Bruton’s Kinase Tyrosine (Btk) Inhibitor, in Suppressing Glioma Tumorigenesis and Stem Cell Phenotypes. Oncotarget 2016, 7, 69961–69975. [Google Scholar] [CrossRef] [Green Version]
- Sala, L.; Cirillo, G.; Riva, G.; Romano, G.; Giussani, C.; Cialdella, A.; Todisco, A.; Virtuoso, A.; Cerrito, M.G.; Bentivegna, A.; et al. Specific Expression of a New Bruton Tyrosine Kinase Isoform (P65BTK) in the Glioblastoma Gemistocytic Histotype. Front. Mol. Neurosci. 2019, 12, 2. [Google Scholar] [CrossRef]
- Yue, C.; Niu, M.; Shan, Q.Q.; Zhou, T.; Tu, Y.; Xie, P.; Hua, L.; Yu, R.; Liu, X. High Expression of Bruton’s Tyrosine Kinase (BTK) Is Required for EGFR-Induced NF-ΚB Activation and Predicts Poor Prognosis in Human Glioma. J. Exp. Clin. Cancer Res. 2017, 36, 132. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Hong, Y.; Wang, S.; Chen, P.; Gu, A.; Guo, X.; Zhao, P. Ibrutinib, a Bruton’s Tyrosine Kinase Inhibitor, Exhibits Antitumoral Activity and Induces Autophagy in Glioblastoma. J. Exp. Clin. Cancer Res. 2017, 36, 96. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoni, G.; Amantini, C.; Maggi, F.; Marinelli, O.; Santoni, M.; Morelli, M.B. The Mucolipin TRPML2 Channel Enhances the Sensitivity of Multiple Myeloma Cell Lines to Ibrutinib and/or Bortezomib Treatment. Biomolecules 2022, 12, 107. https://doi.org/10.3390/biom12010107
Santoni G, Amantini C, Maggi F, Marinelli O, Santoni M, Morelli MB. The Mucolipin TRPML2 Channel Enhances the Sensitivity of Multiple Myeloma Cell Lines to Ibrutinib and/or Bortezomib Treatment. Biomolecules. 2022; 12(1):107. https://doi.org/10.3390/biom12010107
Chicago/Turabian StyleSantoni, Giorgio, Consuelo Amantini, Federica Maggi, Oliviero Marinelli, Matteo Santoni, and Maria Beatrice Morelli. 2022. "The Mucolipin TRPML2 Channel Enhances the Sensitivity of Multiple Myeloma Cell Lines to Ibrutinib and/or Bortezomib Treatment" Biomolecules 12, no. 1: 107. https://doi.org/10.3390/biom12010107
APA StyleSantoni, G., Amantini, C., Maggi, F., Marinelli, O., Santoni, M., & Morelli, M. B. (2022). The Mucolipin TRPML2 Channel Enhances the Sensitivity of Multiple Myeloma Cell Lines to Ibrutinib and/or Bortezomib Treatment. Biomolecules, 12(1), 107. https://doi.org/10.3390/biom12010107