
Progress in Gene-Editing Technology of Zebrafish

Abstract
:1. Introduction
2. Transgenic Technology
3. Targeting Induced Local Lesions in Genomes (TILLING)
4. Discovery and Application of Fixed-Point Shear Enzymes
5. Knockout Gene Editing

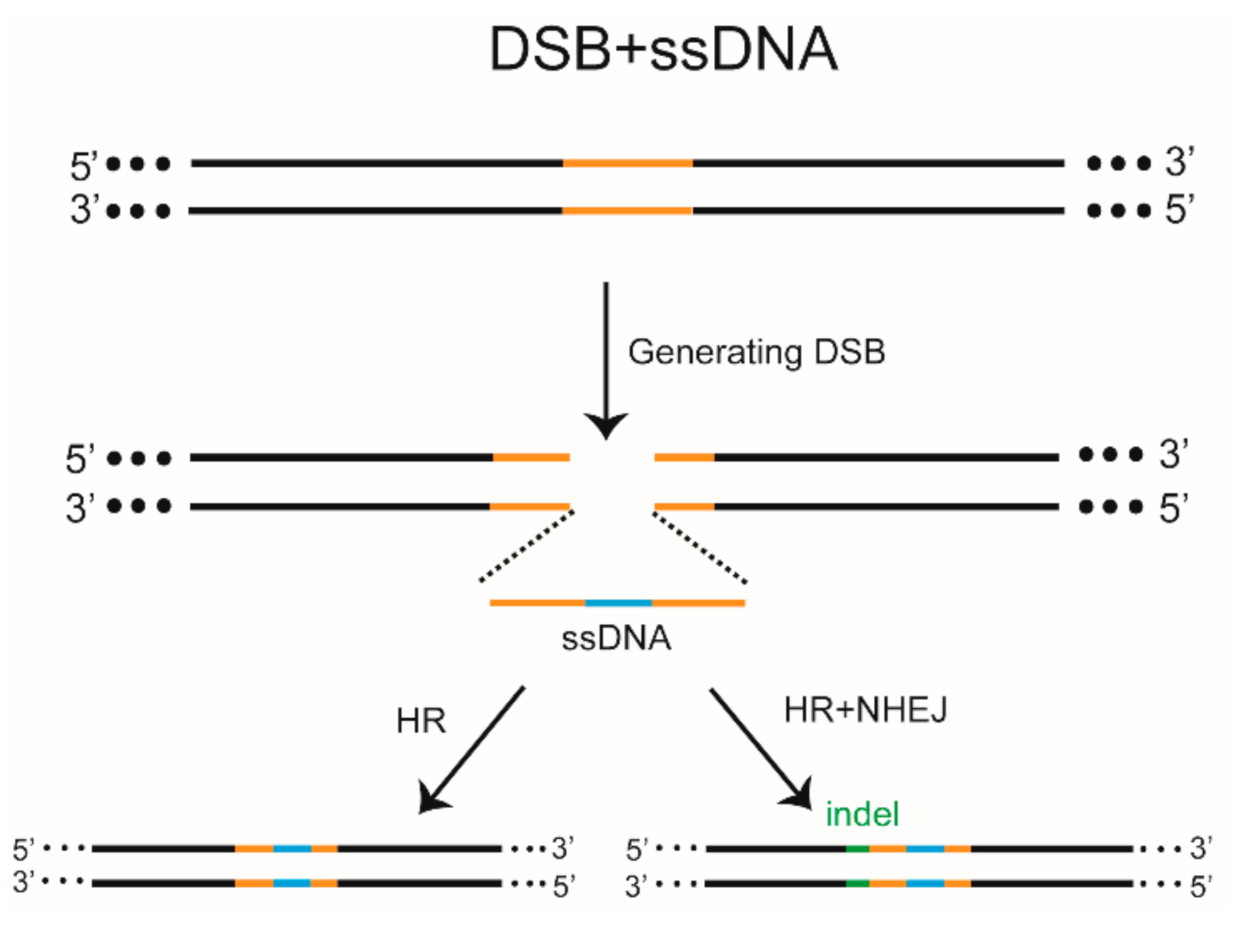
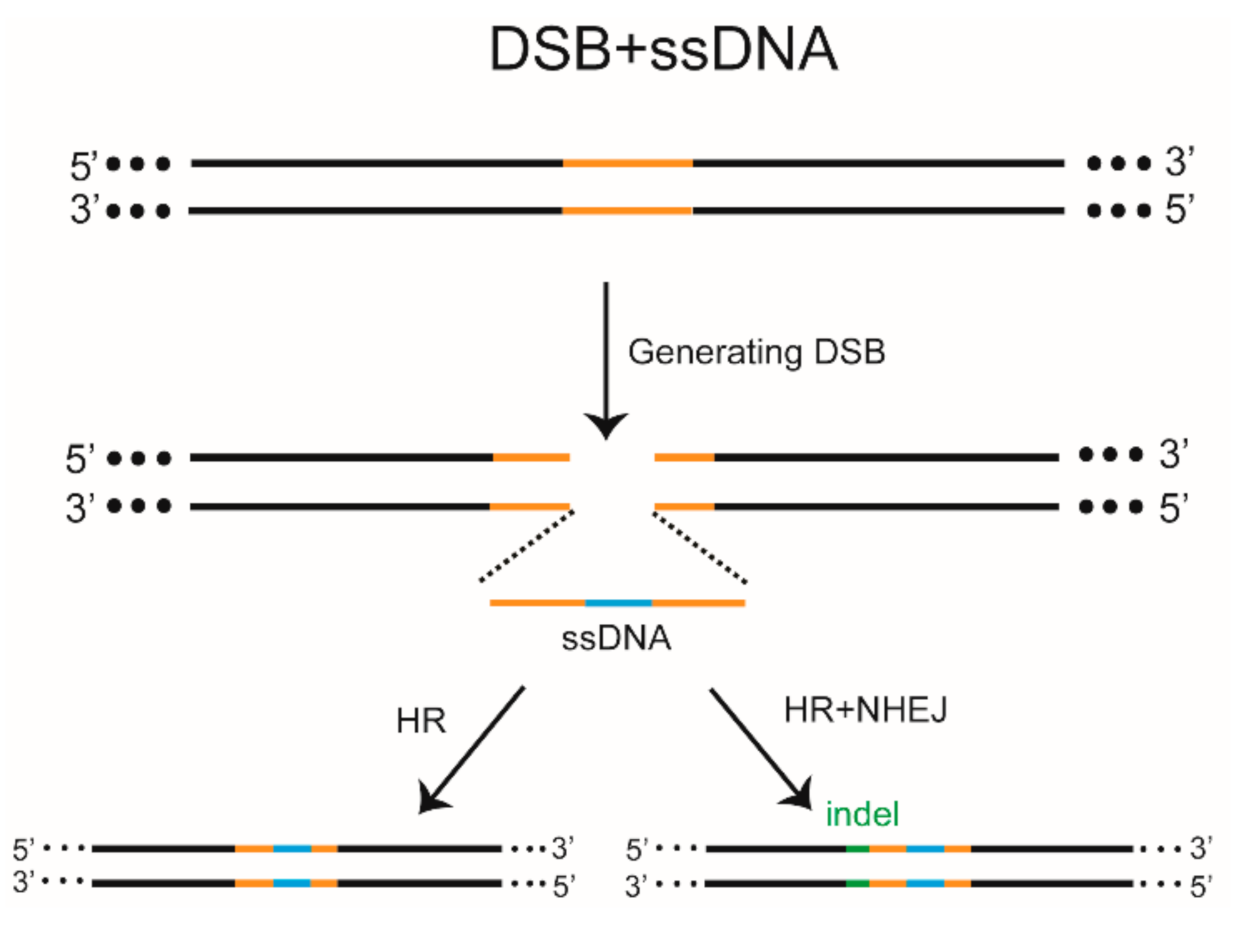{kind=link}
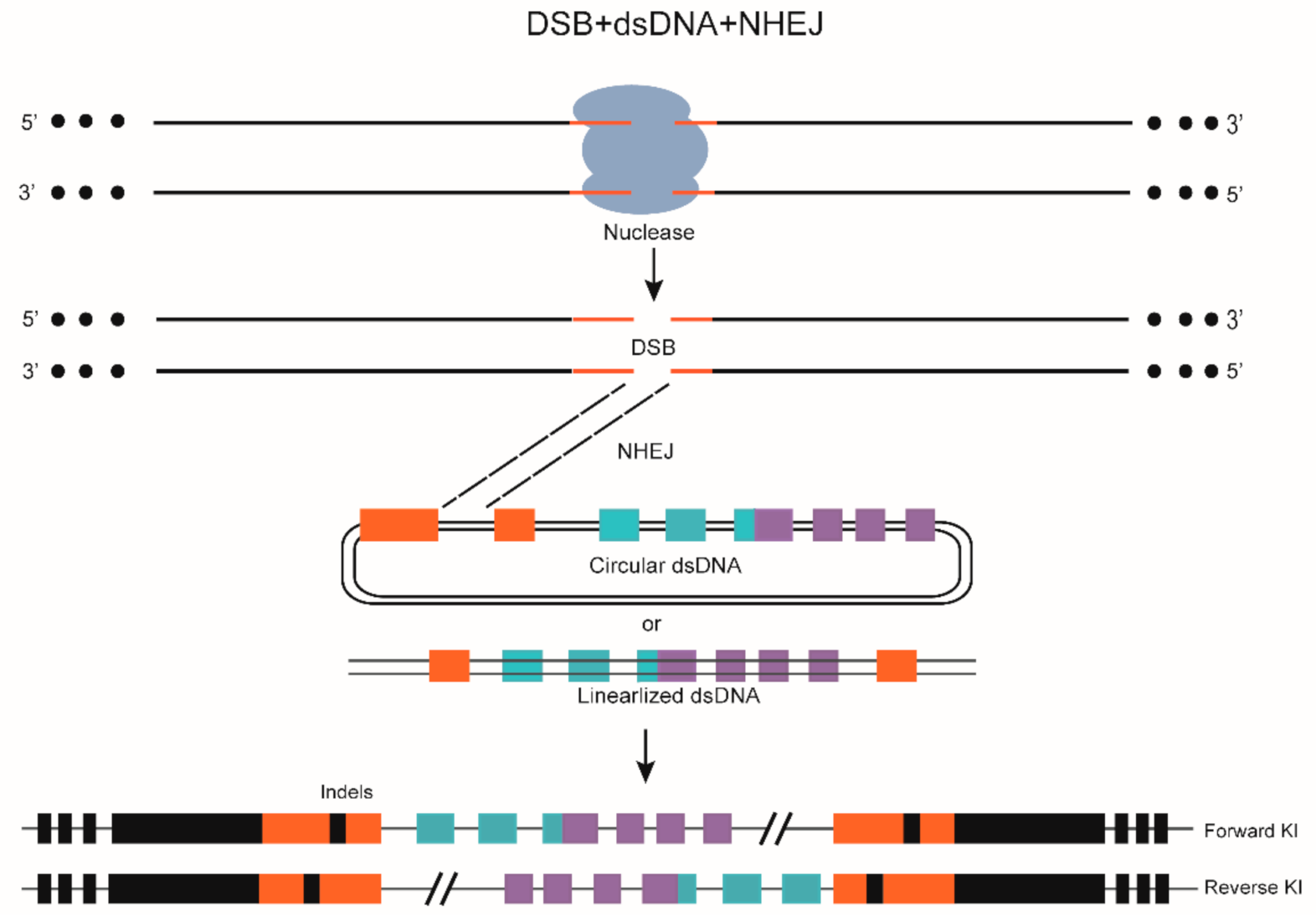{kind=link}
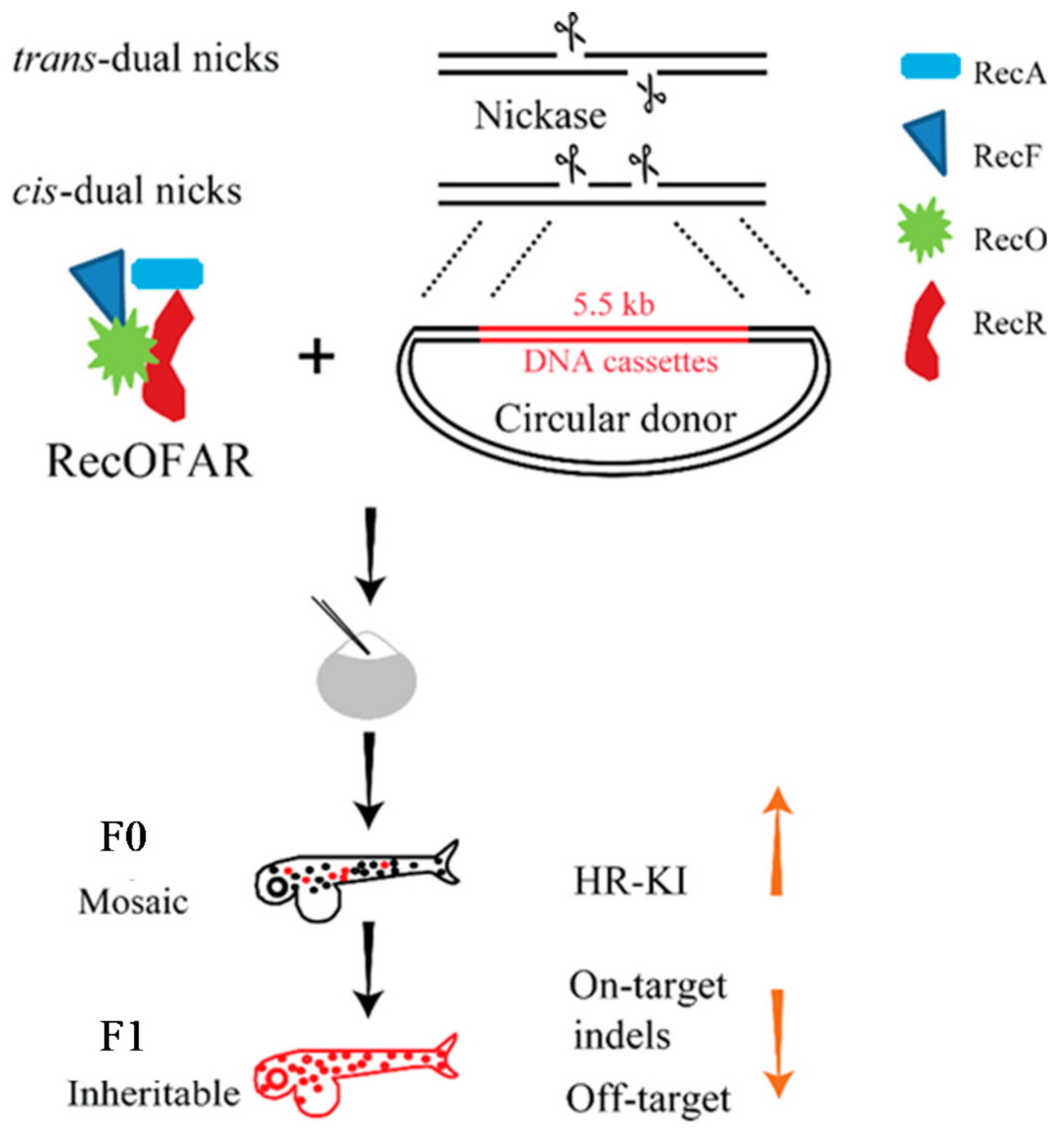{kind=link}
{kind=link}
{kind=link}
| Targeting System | Somatic KO Efficiency | Germline Transmission Rate | Reported in References |
|---|---|---|---|
| ZFN | 10–20% | ~30% (6/20) | [23] |
| ZFN | 0.5–2% | 1.3–25% | [24] |
| ZFN | 2–32% | ~20% | [25] |
| ZFN | 0.4–15.7% | ND | [27] |
| TALEN | 11–33% | ND | [31] |
| CRISPR/Cas9 | 2.7–72% | ND | [32] |
| ZFN | 3–20% | 6–50% | [51] |
| TALEN | 20–77% | ND | [52] |
| CRISPR/Cas9 | 1–27% | 22–33% | [57] |
| CRISPR/ErCas12a | 24–90% | ND | [60] |
| CRISPR/Cas9 | 2–100% | ND | [61] |
| CRISPR/Cas9 | 27–84% | ND | [62] |
6. Fixed Point-Oriented Reconstruction
7. DSB-Mediated Gene KI through NHEJ
8. DSB-HR-Mediated Gene KI
9. Inevitable Side Effects of DSBs
10. Nickase System
11. DNA Template-Free Techniques (Single Base Substitution)
12. Prime Editing Technology
13. Epilogue
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| ABE | adenine base editor |
| alt-NHEJ | alternative NHEJ |
| BE | base editor |
| Cas9n | modified Cas9 nickases |
| CBE | cytosine base editor |
| c-NHEJ | classical NHEJ |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CRISPR/Cas | clustered regularly interspaced short palindromic repeats and CRISPR-associated |
| crRNA | CRISPR RNA |
| dCas9 | dead Cas9 |
| DSB | double-strand break |
| dsDNA | double-stranded DNA |
| EGFP | enhanced green fluorescent protein |
| GCR | genetic compensation response |
| GFP | green fluorescent protein |
| gRNA | guide RNA |
| HMEJ | homology-mediated end joining |
| HR | homologous recombination |
| KI | knock-in |
| mloxP | modified loxP |
| MMEJ | microhomology-mediated end joining |
| MRN | MRE11/RAD/NBS1 |
| NEO | nickase-based homologous recombination enhanced by RecOFAR factors |
| NGS | next-generation sequencing |
| NHEJ | non-homologous end joining |
| NLS | nuclear localization signal |
| OPEN | oligomerized pool engineering |
| PCR | polymerase chain reaction |
| PE | prime editor |
| pegRNA | prime editing guide RNA |
| RACE | rapid amplification of cDNA ends |
| RFP | red fluorescent protein |
| RGNs | RNA-guided nucleases |
| RNP | ribonucleoprotein |
| sgRNA | single guide RNA |
| ssDNA | single-stranded DNA |
| ssODN | single-stranded oligonucleotide |
| TALE | transcription activator-like effector |
| TALEN | transcription activator-like effector nuclease |
| TF IIIA | transcription factor IIIA |
| TILLING | targeting induced local lesions in genomes |
| ZFN | zinc finger nucleases |
| ZFP | zinc finger protein |
| zLOST | zebrafish long single-stranded DNA template |
References
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; McLaren, S.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Houdebine, L.M.; Chourrout, D. Transgenesis in fish. Experientia 1991, 47, 891–897. [Google Scholar] [CrossRef]
- Jessen, J.R.; Meng, A.; McFarlane, R.J.; Paw, B.H.; Zon, L.I.; Smith, G.R.; Lin, S. Modification of bacterial artificial chromosomes through chi-stimulated homologous recombination and its application in zebrafish transgenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 5121–5126. [Google Scholar] [CrossRef] [Green Version]
- Stuart, G.W.; Mcmurray, J.V.; Westerfield, M. Replication, integration and stable germ-line transmission of foreign sequences injected into early zebrafish embryos. Development 1988, 103, 403–412. [Google Scholar] [CrossRef]
- Grabher, C.; Joly, J.S.; Wittbrodt, J. Highly efficient zebrafish transgenesis mediated by the meganuclease I-SceI. Methods Cell Biol. 2004, 77, 381–401. [Google Scholar] [CrossRef]
- Soroldoni, D.; Hogan, B.M.; Oates, A.C. Simple and efficient transgenesis with meganuclease constructs in zebrafish. Methods Mol. Biol. 2009, 546, 117–130. [Google Scholar] [CrossRef]
- Rembold, M.; Lahiri, K.; Foulkes, N.S.; Wittbrodt, J. Transgenesis in fish: Efficient selection of transgenic fish by co-injection with a fluorescent reporter construct. Nat. Protoc. 2006, 1, 1133–1139. [Google Scholar] [CrossRef]
- Kawakami, K.; Largaespada, D.A.; Ivics, Z. Transposons as tools for functional genomics in vertebrate models. Trends Genet. 2017, 33, 784–801. [Google Scholar] [CrossRef]
- Mukherjee, K.; Liao, E.C. Generation and characterization of a zebrafish muscle specific inducible Cre line. Transgenic Res. 2018, 27, 559–569. [Google Scholar] [CrossRef]
- Suster, M.L.; Kikuta, H.; Urasaki, A.; Asakawa, K.; Kawakami, K. Transgenesis in zebrafish with the Tol2 transposon system. Methods Mol. Biol. 2009, 561, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Transposon tools and methods in zebrafish. Dev. Dyn. 2005, 234, 244–254. [Google Scholar] [CrossRef]
- Mosimann, C.; Puller, A.C.; Lawson, K.L.; Tschopp, P.; Amsterdam, A.; Zon, L.I. Site-directed zebrafish transgenesis into single landing sites with the phiC31 integrase system. Dev. Dyn. 2013, 242, 949–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gansner, J.M.; Dang, M.; Ammerman, M.; Zon, L.I. Transplantation in zebrafish. Methods Cell Biol. 2017, 138, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Mione, M.; Baldessari, D.; Deflorian, G.; Nappo, G.; Santoriello, C. How neuronal migration contributes to the morphogenesis of the CNS: Insights from the zebrafish. Dev. Neurosci. 2008, 30, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.N.; Moens, C.B. Approaching perfection: New developments in zebrafish genome engineering. Dev. Cell. 2016, 36, 595–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.S.; Lam, I.I.; Hilary, C.; Duong, D.N.; Deo, R.C.; Coughlin, S.R. A rapid method for directed gene knockout for screening in G0 zebrafish. Dev. Cell. 2018, 46, 112–125.e4. [Google Scholar] [CrossRef] [Green Version]
- Moens, C.B.; Donn, T.M.; Wolf-Saxon, E.R.; Ma, T.P. Reverse genetics in zebrafish by TILLING. Brief. Funct. Genom. Proteomic 2008, 7, 454–459. [Google Scholar] [CrossRef] [Green Version]
- McCallum, C.M.; Comai, L.; Greene, E.A.; Henikoff, S. Targeting induced local lesions IN genomes (TILLING) for plant functional genomics. Plant Physiol. 2000, 123, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Wienholds, E.; Schulte-Merker, S.; Walderich, B.; Plasterk, R.H. Target-selected inactivation of the zebrafish rag1 gene. Science 2002, 297, 99–102. [Google Scholar] [CrossRef]
- Raby, L.; Völkel, P.; Le Bourhis, X.; Angrand, P.O. Genetic engineering of zebrafish in cancer research. Cancers 2020, 12, 2168. [Google Scholar] [CrossRef]
- Miller, J.; Mclachlan, A.D.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar] [CrossRef]
- Meng, X.; Noyes, M.B.; Zhu, L.J.; Lawson, N.D.; Wolfe, S.A. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Smith, T.; McNulty, J.; Rayla, A.L.; Lakshmanan, A.; Siekmann, A.F.; Buffardi, M.; Meng, X.; Shin, J.; Padmanabhan, A.; et al. Evaluation and application of modularly assembled zinc-finger nucleases in zebrafish. Development 2011, 138, 4555–4564. [Google Scholar] [CrossRef] [Green Version]
- Doyon, Y.; McCammon, J.M.; Miller, J.C.; Faraji, F.; Ngo, C.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 702–708. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.E.; Maeder, M.L.; Pearlberg, J.; Joung, J.K.; Peterson, R.T.; Yeh, J.R. Targeted mutagenesis in zebrafish using customized zinc-finger nucleases. Nat. Protoc. 2009, 4, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Christensen, R.G.; Rayla, A.L.; Lakshmanan, A.; Stormo, G.D.; Wolfe, S.A. An optimized two-finger archive for ZFN-mediated gene targeting. Nat. Methods 2012, 9, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, E.J.; Kim, J.S. Construction of combinatorial libraries that encode zinc finger-based transcription factors. Methods Mol. Biol. 2010, 649, 133–147. [Google Scholar] [CrossRef]
- Ramirez, C.L.; Foley, J.E.; Wright, D.A.; Müller-Lerch, F.; Rahman, S.H.; Cornu, T.I.; Winfrey, R.J.; Sander, J.D.; Fu, F.; Townsend, J.A.; et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 2008, 5, 374–375. [Google Scholar] [CrossRef]
- Sander, J.D.; Dahlborg, E.J.; Goodwin, M.J.; Cade, L.; Zhang, F.; Cifuentes, D.; Curtin, S.; Blackburn, J.S.; Thibodeau-Beganny, S.; Qi, Y.; et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat. Methods 2011, 8, 67–69. [Google Scholar] [CrossRef]
- Sander, J.D.; Cade, L.; Khayter, C.; Reyon, D.; Peterson, R.T.; Joung, J.K.; Yeh, J. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 2011, 29, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Chen, S.; Oikonomou, G.; Chiu, C.N.; Niles, B.J.; Liu, J.; Lee, D.A.; Igor, A.; Prober, D.A. A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res. 2013, 41, 2769–2778. [Google Scholar] [CrossRef] [Green Version]
- Kay, S.; Hahn, S.; Marois, E.; Hause, G.; Bonas, U. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 2007, 318, 648–651. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Zu, Y.; Tong, X.; Wang, Z.; Liu, D.; Pan, R.; Li, Z.; Hu, Y.; Luo, Z.; Huang, P.; Wu, Q.; et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat. Methods 2013, 10, 329–331. [Google Scholar] [CrossRef]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.X.; Li, C.Y.; Hu, C.C.; Wang, Y.; Lin, J.; Jiang, Y.H.; Li, Q.; Xu, X. CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism. 2018, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.L.; Bian, W.P.; Wang, C.; Junaid, M.; Zou, J.X.; Pei, D.S. A novel technique based on in vitro oocyte injection to improve CRISPR/Cas9 gene editing in zebrafish. Sci. Rep. 2016, 6, 34555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojica, F.J.; Díez-Villaseñor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef]
- Zhang, J.H.; Adikaram, P.; Pandey, M.; Genis, A.; Simonds, W.F. Optimization of genome editing through CRISPR-Cas9 engineering. Bioengineered 2016, 7, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Zhao, X.; Zhang, Q.; Li, W.; Zu, Y. Comparison of various nuclear localization signal-fused Cas9 proteinS and Cas9 mRNA for genome editing in zebrafish. G3 (Bethesda) 2018, 8, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Chen, C.; Han, Y.; Chen, Z.; Lu, X.; Liang, F.; Li, S.; Qin, W.; Lin, S. Expanding CRISPR/Cas9 genome editing capacity in zebrafish using SaCas9. G3 (Bethesda) 2016, 6, 2517–2521. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Brown, W.; Bardhan, A.; Delaney, M.; Deiters, A. Spatiotemporal control of CRISPR/Cas9 function in cells and zebrafish using light-activated guide RNA. Angew. Chem. Int. Ed. 2019, 59, 8998–9003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z.; Ge, W. An efficient platform for generating somatic point mutations with germline transmission in the zebrafish by CRISPR/Cas9-mediatedGene editing. J. Biol. Chem. 2018, 293, 6611–6622. [Google Scholar] [CrossRef] [Green Version]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef]
- Foley, J.E.; Yeh, J.R.; Maeder, M.L.; Reyon, D.; Sander, J.D.; Peterson, R.T.; Joung, J.K. Rapid mutation of endogenous zebrafish genes using zinc finger nucleases made by Oligomerized Pool ENgineering (OPEN). PLoS ONE 2009, 4, e4348. [Google Scholar] [CrossRef] [Green Version]
- Moore, F.E.; Reyon, D.; Sander, J.D.; Martinez, S.A.; Blackburn, J.S.; Khayter, C.; Ramirez, C.L.; Joung, J.K.; Langenau, D.M. Improved somatic mutagenesis in zebrafish using transcription activator-like effector nucleases (TALENs). PLoS ONE 2012, 7, e37877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, G.K.; Pei, W.; Lafave, M.C.; Idol, J.; Xu, L.; Gallardo, V.; Carrington, B.; Bishop, K.; Jones, M.P.; Li, M. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015, 25, 1030–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhou, Y.; Qi, X.; Chen, J.; Chen, W.; Qiu, G.; Wu, Z.; Wu, N. CRISPR/Cas9 in zebrafish: An efficient combination for human genetic diseases modeling. Hum. Genet. 2017, 136, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshijima, K.; Jurynec, M.J.; Grunwald, D.J. Precise editing of the zebrafish genome made simple and efficient. Dev. Cell. 2016, 36, 654–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jao, L.E.; Wente, S.R.; Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl. Acad. Sci. USA 2013, 110, 13904–13909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.H.; Zhang, G.J. Generating stable knockout zebrafish lines by deleting large chromosomal fragments using multiple gRNAs. G3 (Bethesda) 2020, 10, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Ablain, J.; Durand, E.M.; Yang, S.; Zhou, Y.; Zon, L.I. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev. Cell. 2015, 32, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Jaenisch, R.; Zhang, F.; Gage, F. Genome editing in neurosciences. Res. Perspect. Neurosci. 2017. [CrossRef]
- Wierson, W.A.; Simone, B.W.; WareJoncas, Z.; Mann, C.; Welker, J.M.; Kar, B.; Emch, M.J.; Friedberg, I.; Gendron, W.A.C.; Barry, M.A.; et al. Expanding the CRISPR toolbox with ErCas12a in zebrafish and human cells. CRISPR J. 2019, 2, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, B.; Luo, L.; Shi, D.L.; Wang, H.; Cui, Z.; Huang, H.; Cao, Y.; Shu, X.; Zhang, W.; et al. Systematic genome editing of the genes on zebrafish Chromosome 1 by CRISPR/Cas9. Genome Res. 2019, 30, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Isiaku, A.I.; Zhang, Z.; Pazhakh, V.; Manley, H.R.; Thompson, E.R.; Fox, L.C.; Yerneni, S.; Blombery, P.; Lieschke, G.J. Transient, flexible gene editing in zebrafish neutrophils and macrophages for determination of cell-autonomous functions. Dis. Model Mech. 2021, 14, dmm047431. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Chen, J. Premature termination codon-bearing mRNA mediates genetic compensation response. Zebrafish 2020, 17, 157–162. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Günther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef]
- Ma, Z.; Zhu, P.; Shi, H.; Guo, L.; Zhang, Q.; Chen, Y.; Chen, S.; Zhang, Z.; Peng, J.; Chen, J. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 2019, 568, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Krüger, I.; Thompson, L.H.; Löbrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [Green Version]
- Pei, D.S.; Strauss, P.R. Zebrafish as a model system to study DNA damage and repair. Mutat. Res. 2013, 743–744, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, H.; Zhang, B.; Lin, S. TALEN- and CRISPR-enhanced DNA homologous recombination for gene editing in zebrafish. Methods Cell Biol. 2016, 135, 107–120. [Google Scholar] [CrossRef]
- Weterings, E.; Chen, D.J. The endless tale of non-homologous end-joining. Cell Res. 2008, 18, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcvey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devkota, S. The road less traveled: Strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Rep. 2018, 51, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, A.; Hisano, Y.; Ota, S.; Taimatsu, K. Site-specific integration of exogenous genes using genome editing technologies in zebrafish. Int. J. Mol. Sci. 2016, 17, 727. [Google Scholar] [CrossRef] [Green Version]
- Auer, T.O.; Duroure, K.; De Cian, A.; Concordet, J.P.; Bene, F.D. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014, 24, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Hisano, Y.; Kawahara, A.; Higashijima, S.I. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci. Rep. 2014, 4, 6545. [Google Scholar] [CrossRef] [Green Version]
- Hisano, Y.; Sakuma, T.; Nakade, S.; Ohga, R.; Ota, S.; Okamoto, H.; Yamamoto, T.; Kawahara, A. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 2015, 5, 8841. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.D.; Ray, G.J.; Dewitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Li, K.; Wang, G.; Andersen, T.; Zhou, P.; Pu, W.T. Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS ONE 2014, 9, e105779. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Stieger, K. Optimizing the DNA donor template for homology-directed repair of double-strand breaks. Mol. Ther. Nucleic. Acids 2017, 7, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irion, U.; Krauss, J.; Nüsslein-Volhard, C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 2014, 141, 4827–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinapoli, S.E.; Martinez-McFaline, R.; Gribbin, C.K.; Wrighton, P.J.; Balgobin, C.A.; Nelson, I.; Leonard, A.; Maskin, C.R.; Shwartz, A.; Quenzer, E.D.; et al. Synthetic CRISPR/Cas9 reagents facilitate genome editing and homology directed repair. Nucleic Acids Res. 2020, 48, e38. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Cui, X.; Zhu, Z.; Hu, W. Non-homologous end joining plays a key role in transgene concatemer formation in transgenic zebrafish embryos. Int. J. Biol. Sci. 2010, 6, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013, 5, a012757. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Jette, N.; Lees-Miller, S.P. Non-homologous end joining: Emerging themes and unanswered questions. DNA Repair. 2014, 17, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, B.B.; Ren, Y.G.; Gu, S.Y.; Xiang, Y.H.; Huang, C.; Du, J.L. Intron targeting-mediated and endogenous gene integrity-maintaining knockin in zebrafish using the CRISPR/Cas9 system. Cell Res. 2015, 25, 634–637. [Google Scholar] [CrossRef]
- Kesavan, G.; Chekuru, A.; Machate, A.; Brand, M. CRISPR/Cas9-mediated zebrafish knock-in as a novel strategy to study midbrain-hindbrain boundary development. Front Neuroanat. 2017, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.J.; Bian, W.P.; Liu, Y.; Huang, H.-Y.; Yin, Q.; Yang, X.-J.; Pei, D.-S. CRISPR/Cas9-based genome engineering of zebrafish using a seamless integration strategy. FASEB J. 2018, 32, 5132–5142. [Google Scholar] [CrossRef] [Green Version]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G.; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, G.A.; Liao, M.; You, Z.; Lissouba, A.; Chen, B.E.; Drapeau, P. Homology directed knockin of point mutations in the zebrafish tardbp and fus genes in ALS using the CRISPR/Cas9 system. PLoS ONE 2016, 11, e0150188. [Google Scholar] [CrossRef]
- Bai, H.; Liu, L.; An, K.; Lu, X.; Harrison, M.; Zhao, Y.; Yan, R.; Lu, Z.; Li, S.; Lin, S.; et al. CRISPR/Cas9-mediated precise genome modification by a long ssDNA template in zebrafish. BMC Genom. 2020, 21, 67. [Google Scholar] [CrossRef]
- Eschstruth, A.; Schneider-Maunoury, S.; Giudicelli, F. Creation of zebrafish knock-in reporter lines in the nefma gene by Cas9-mediated homologous recombination. Genesis 2019, 58, e23340. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Chen, W.; Liu, Z.; Yu, G.; Chen, Y.; Cai, Y.; Sun, L.; Xu, W.; Zhong, L.; Gao, C.; et al. Efficient and risk-reduced genome editing using double nicks enhanced by bacterial recombination factors in multiple species. Nucleic Acids Res. 2020, 48, e57. [Google Scholar] [CrossRef]
- Zhang, Y.; Qin, W.; Lu, X.; Xu, J.; Huang, H.; Bai, H.; Li, S.; Lin, S. Programmable base editing of zebrafish genome using a modified CRISPR-Cas9 system. Nat. Commun. 2017, 8, 118. [Google Scholar] [CrossRef]
- Qin, W.; Lu, X.; Liu, Y.; Bai, H.; Li, S.; Lin, S. Precise A•T to G•C base editing in the zebrafish genome. BMC Biol. 2018, 16, 139. [Google Scholar] [CrossRef]
- Carrington, B.; Weinstein, R.N.; Sood, R. BE4max and AncBE4max are efficient in germline conversion of C:G to T:A base pairs in zebrafish. Cells 2020, 9, 1690. [Google Scholar] [CrossRef] [PubMed]
- Petri, K.; Zhang, W.; Ma, J.; Schmidts, A.; Lee, H.; Horng, J.E.; Kim, D.Y.; Kurt, I.C.; Clement, K.; Hsu, J.Y.; et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat. Biotechnol. 2021. [Google Scholar] [CrossRef]
- Wierson, W.A.; Welker, J.M.; Almeida, M.P.; Mann, C.M.; Webster, D.A.; Torrie, M.E.; Weiss, T.J.; Kambakam, S.; Vollbrecht, M.K.; Lan, M.; et al. Efficient targeted integration directed by short homology in zebrafish and mammalian cells. eLife. 2020, 9, e53968. [Google Scholar] [CrossRef]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell. 2012, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Celli, G.B.; Denchi, E.L.; de Lange, T.D. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 2006, 8, 885–890. [Google Scholar] [CrossRef]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell. 2016, 64, 940–950. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Kaini, P.; Sander, J.D.; Joung, J.K.; Peterson, R.T.; Yeh, J.R. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS ONE 2013, 8, e68708. [Google Scholar] [CrossRef]
- Boel, A.; De Saffel, H.; Steyaert, W.; Callewaert, B.; De Paepe, A.; Coucke, P.J.; Willaert, A. CRISPR/Cas9-mediated homology-directed repair by ssODNs in zebrafish induces complex mutational patterns resulting from genomic integration of repair-template fragments. Dis. Model Mech. 2018, 11, dmm035352. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J. Genome editing with RNA-guided Cas9 nuclease in Zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, Y.; Han, B.; Li, L.; Li, M.; Lu, X.; Chen, C.; Lu, M.; Zhang, Y.; Jia, X.; et al. One-step efficient generation of dual-function conditional knockout and geno-tagging alleles in zebrafish. eLife 2019, 8, e48081. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksoy, Y.A.; Nguyen, D.T.; Chow, S.; Chung, R.S.; Guillemin, G.J.; Cole, N.J.; Hesselson, D. Chemical reprogramming enhances homology-directed genome editing in zebrafish embryos. Commun. Biol. 2019, 2, 198. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Shang, D.; Ying, R.; Cheng, H.; Zhou, R. An optimized base editor with efficient C-to-T base editing in zebrafish. BMC Biol. 2020, 18, 190. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Yoshioka, S.; Nishida, K.; Hosokawa, H.; Kakizuka, A.; Maegawa, S. In vivo targeted single-nucleotide editing in zebrafish. Sci. Rep. 2018, 8, 11423. [Google Scholar] [CrossRef] [Green Version]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef]
- Park, S.H.; Beal, P.A. Off-target editing by CRISPR-guided DNA base editors. Biochemistry 2019, 58, 3727–3734. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Amacher, S.L. Emerging gene knockout technology in zebrafish: Zinc-finger nucleases. Brief. Funct. Genom. Proteomic 2008, 7, 460–464. [Google Scholar] [CrossRef] [Green Version]
- Auer, T.O.; Del Bene, F.D. CRISPR/Cas9 and TALEN-mediated knock-in approaches in zebrafish. Methods 2014, 69, 142–150. [Google Scholar] [CrossRef] [PubMed]
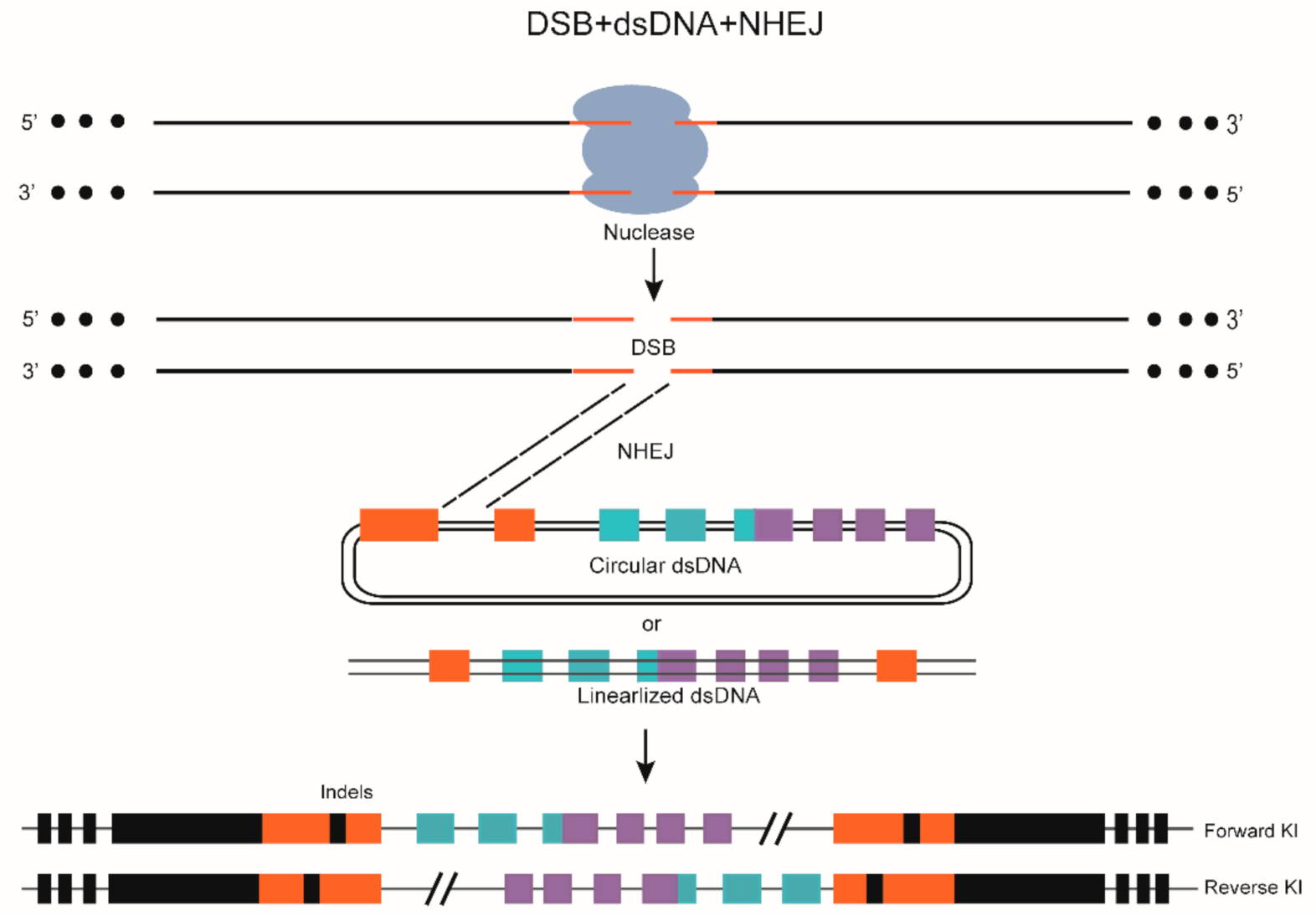


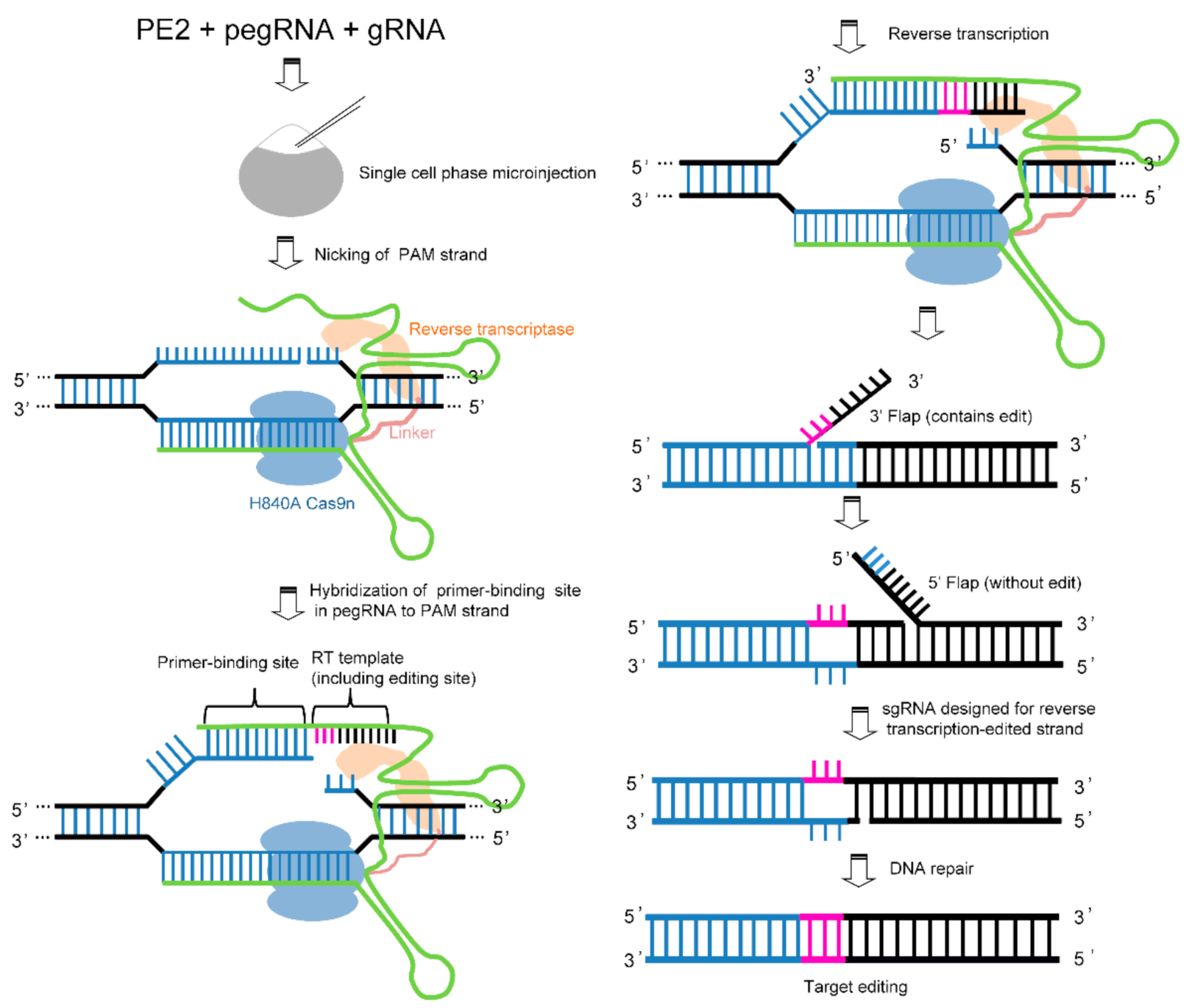
| Targeting System | Programmable Manner | Integration Mechanism | Donor Type | Insertion | Germline Transmission Rate | Disadvantage(s) | Advantage(s) | References |
|---|---|---|---|---|---|---|---|---|
| TALENs | DSB | HR | Linearizeds DNA | EGFP | ~1.5% | Disruption of endogenous gene/Low efficiency | Large fragment insertion | [38] |
| CRISPR/Cas9 | DSB | NHEJ | Plasmid | Gal4/RFP | ~12% | Plasmid backbone insertion/unwanted indels | Large fragment insertion/easy donor design | [74] |
| CRISPR/Cas9 | DSB | HR | Plasmid | Single base | ~11% | Short-fragment insertion/unwanted indels | Correction of mismatches/target mutation | [79] |
| CRISPR/Cas9 | DSB | NHEJ | Plasmid | EGFP | ~12% (after GFP pre-screen) | Plasmid backbone insertion/unwanted indels | Large fragment insertion | [86] |
| CRISPR/Cas9 | DSB | NHEJ | Plasmid | Venus | ~20% | Unwanted indels | Large fragment insertion | [87] |
| CRISPR/Cas9 | DSB | MMEJ | Plasmid | mCheery | ~20.7% | Unwanted indels | Large fragment Insertion | [88] |
| TALENs | DSB | NHEJ/HR | ssDNA | LoxP | ~10% | Short -fragment insertion/unwanted indels | Easy to synthesize and manipulate | [89] |
| CRISPR/Cas9 | DSB | HR | ssDNA | Single base | ~2.1% | Short-fragment insertion/unwanted indels | Correction of mismatches/point mutation | [90] |
| CRISPR/Cas9 | DSB | HDR | ssDNA | Single base | 31.8% | Unwanted indels | Correction of mismatches/point mutation | [91] |
| CRISPR/Cas9 | DSB | HR | Plasmid | KalTA4 | 8% | Short-fragment insertion/unwanted indels | Large fragment insertion | [92] |
| CRISPR/Cas9 | Nick | HR | Plasmid | GFAP | 11.1% | Difficult donor design | Precise and large fragment insertion/without DSB | [93] |
| CRISPR/Cas9 | Nick | BE system | / | C:G to T:A | 7–37% | Unwanted indels/off-target risk | Without DNA template and DSB | [94] |
| CRISPR/Cas9 | Nick | ABEmax | / | A-G | 25–58% | Unwanted indels/off-target risk | Without DNA template and DSB | [95] |
| CRISPR/Cas9 | Nick | AncBE4max | / | C:G to T:A | 7.9% | Unwanted indels /off-target risk | Without DNA template and DSB | [96] |
| CRISPR/Cas9 | Nick | PE system | PegRNA | Short-fragment insertions/deletions | 30% (somatic mutations) | Short-fragment editing/unwanted indels | Without DNA template and DSB | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Jia, Z.; Zhang, S.; He, X. Progress in Gene-Editing Technology of Zebrafish. Biomolecules 2021, 11, 1300. https://doi.org/10.3390/biom11091300
Li Y, Jia Z, Zhang S, He X. Progress in Gene-Editing Technology of Zebrafish. Biomolecules. 2021; 11(9):1300. https://doi.org/10.3390/biom11091300
Chicago/Turabian StyleLi, Yanling, Zhipeng Jia, Shuchao Zhang, and Xiaozhen He. 2021. "Progress in Gene-Editing Technology of Zebrafish" Biomolecules 11, no. 9: 1300. https://doi.org/10.3390/biom11091300
APA StyleLi, Y., Jia, Z., Zhang, S., & He, X. (2021). Progress in Gene-Editing Technology of Zebrafish. Biomolecules, 11(9), 1300. https://doi.org/10.3390/biom11091300