
Structural Changes in the Cap of Rv0183/mtbMGL Modulate the Shape of the Binding Pocket

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Expression and Purification
- K74A forward primer 5′-TAGCGGCGGTGCACGGGTCTTG-3′,
- K74Areverse primer 5′-CGCCCATGACCACGGTGA-3′,
- Q164A forward primer 5′-TTTACCGGTAGCGGAACTCGATTT-3′,
- Q164A reverse primer 5′-AAATCGAGTTCCGCTACCGGTAAA-3′,
- E165A forward primer 5′-CCGGTACAGGCACTCGATTTC-3′,
- E165A reverse primer 5′-GAAATCGAGTGCCTGTACCGG-3′,
- K249A forward primer 5′-GTACAGCTGGCGGAGTATCCG-3′,
- K249A reverse primer 5′-CGGATACTCCGCCAGCTGTAC-3′.
2.2. Crystallization, Data Processing and Refinement
2.3. MGH Activity Assay
2.4. Synthesis of Maglipan
3. Results
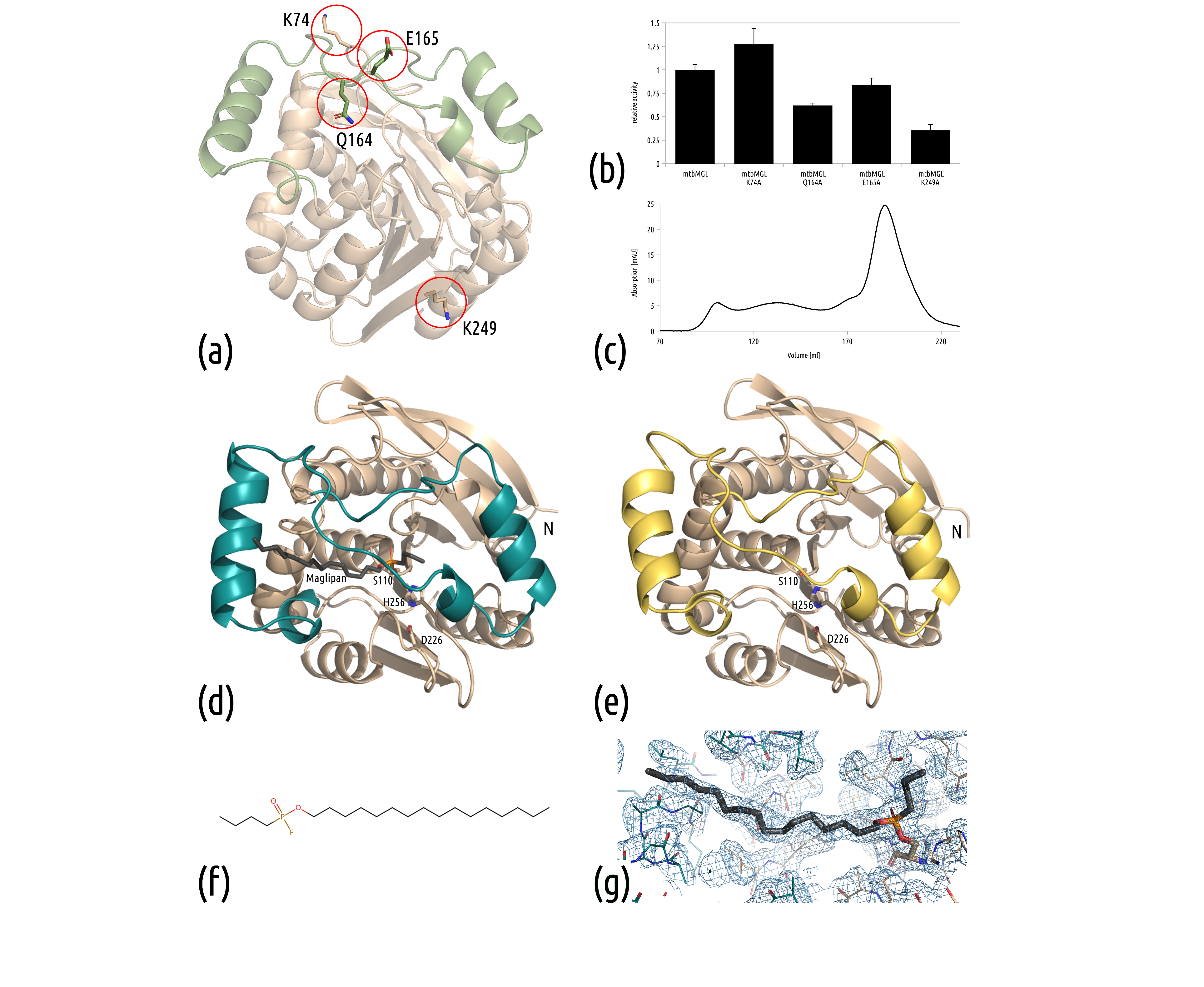
3.1. Surface Entropy Reduction (SER) Variants Remain Catalytically Active
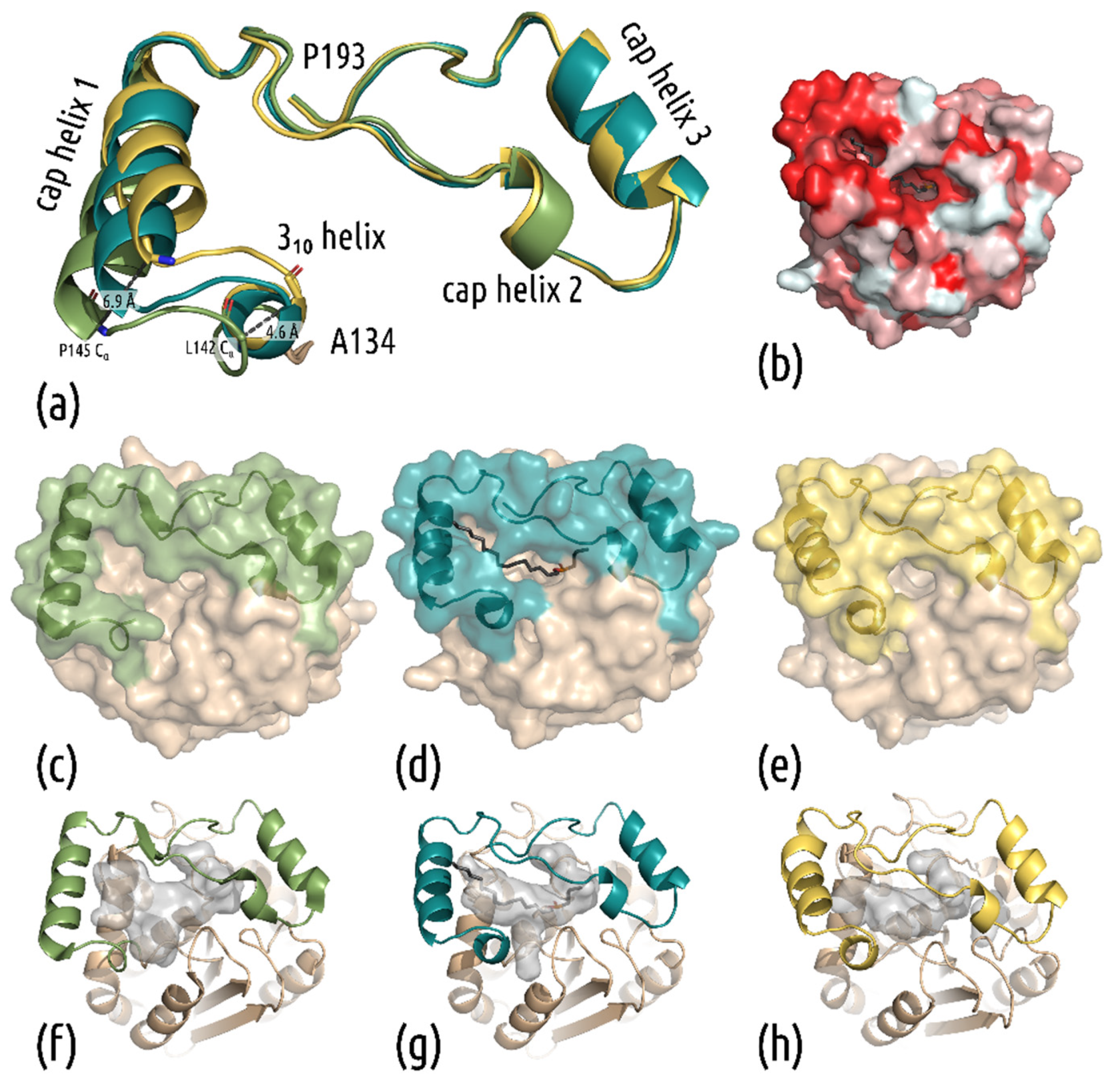
3.2. New Structures Reveal a Closed-Cap Conformation and Show the Bound Substrate Analog Maglipan
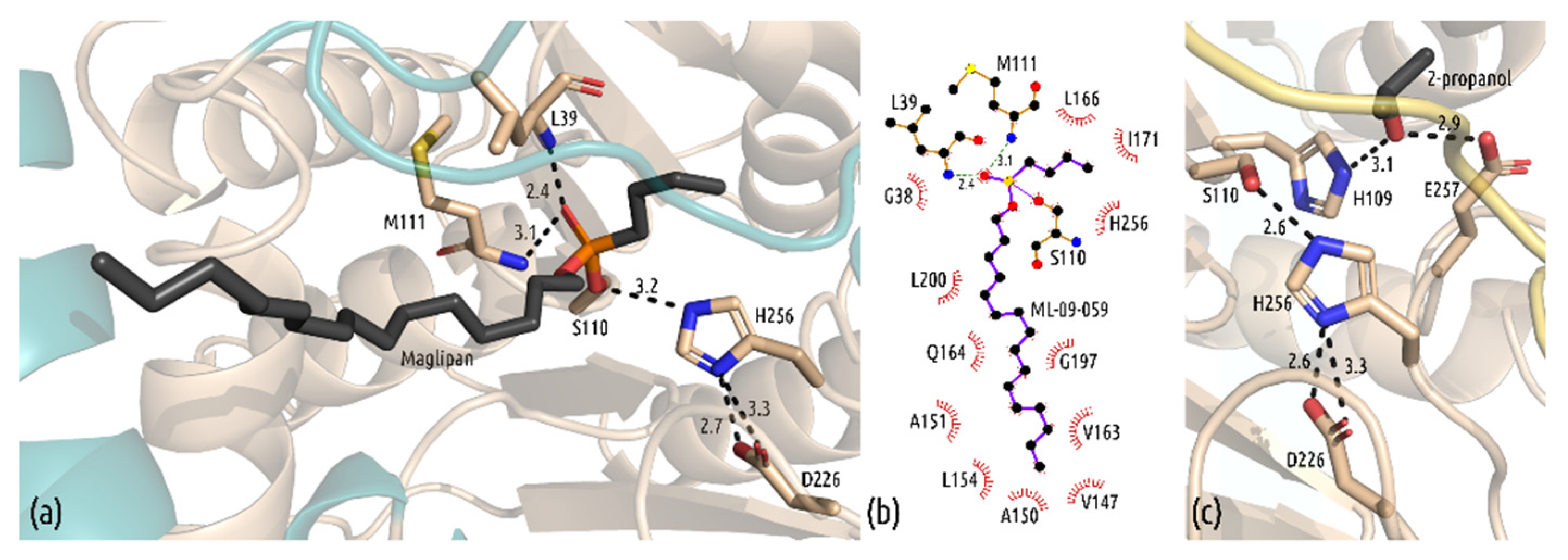
3.3. Interactions in the Active Site
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001313-1. [Google Scholar]
- Boom, W.H.; Schaible, U.E.; Achkar, J.M. The knowns and unknowns of latent Mycobacterium tuberculosis infection. J. Clin. Investig. 2021, 131, e136222. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.R.; Levine, S.R.; Beatty, K.E. Small-molecule probes reveal esterases with persistent activity in dormant and reactivating Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, O.A.; Arora, P.; Sridharan, V.; Tickoo, R.; Mohanty, D.; Gokhale, R.S. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature 2004, 428, 441–445. [Google Scholar] [CrossRef]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, P.E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.; Deb, C.; Dubey, V.S.; Sirakova, T.D.; Abomoelak, B.; Morbidoni, H.R.; Kolattukudy, P.E. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J. Bacteriol. 2004, 186, 5017–5030. [Google Scholar] [CrossRef] [Green Version]
- Delorme, V.; Diomandé, S.V.; Dedieu, L.; Cavalier, J.-F.; Carrière, F.; Kremer, L.; Leclaire, J.; Fotiadu, F.; Canaan, S. MmPPOX inhibits Mycobacterium tuberculosis lipolytic enzymes belonging to the hormone-sensitive lipase family and alters Mycobacterial growth. PLoS ONE 2012, 7, e46493. [Google Scholar] [CrossRef]
- Dhouib, R.; Ducret, A.; Hubert, P.; Carrière, F.; Dukan, S.; Canaan, S. Watching intracellular lipolysis in mycobacteria using time lapse fluorescence microscopy. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2011, 1811, 234–241. [Google Scholar] [CrossRef]
- Kremer, L.; Chastellier, C.D.; Dobson, G.; Gibson, K.J.C.; Bifani, P.; Balor, S.; Gorvel, J.-P.; Locht, C.; Minnikin, D.E.; Besra, G.S. Identification and structural characterization of an unusual mycobacterial monomeromycolyl-diacylglycerol. Mol. Microbiol. 2005, 57, 1113–1126. [Google Scholar] [CrossRef]
- Singh, V.K.; Srivastava, M.; Dasgupta, A.; Singh, M.P.; Srivastava, R.; Srivastava, B.S. Increased virulence of Mycobacterium tuberculosis H37Rv overexpressing LipY in a murine model. Tuberculosis 2014, 94, 252–261. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J.; Li, Z.; Xie, J.; Yang, Y.; Zhong, Y.; Wang, H. Expression and characterization of the carboxyl esterase Rv3487c from Mycobacterium tuberculosis. Protein Expr. Purif. 2005, 42, 59–66. [Google Scholar] [CrossRef]
- Arriaga-Guerrero, A.L.; Hernández-Luna, C.E.; Rigal-Leal, J.; Robles-González, R.J.; González-Escalante, L.A.; Silva-Ramírez, B.; Mercado-Hernández, R.; Vargas-Villarreal, J.; Bermúdez de León, M.; Peñuelas-Urquides, K. LipF increases rifampicin and streptomycin sensitivity in a Mycobacterium tuberculosis surrogate. BMC Microbiol. 2020, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Rao, R.N.; Reddy, J.R.C.; Prasad, R.; Kotturu, S.K.; Ghosh, S.; Mukhopadhyay, S. PE11, a PE/PPE family protein of Mycobacterium tuberculosis is involved in cell wall remodeling and virulence. Sci. Rep. 2016, 6, 21624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santucci, P.; Point, V.; Poncin, I.; Guy, A.; Crauste, C.; Serveau-Avesque, C.; Galano, J.M.; Spilling, C.D.; Cavalier, J.-F.; Canaan, S. LipG a bifunctional phospholipase/thioesterase involved in mycobacterial envelope remodeling. Biosci. Rep. 2018, 38, BSR20181953. [Google Scholar] [CrossRef] [Green Version]
- Dhouib, R.; Laval, F.; Carrière, F.; Daffé, M.; Canaan, S. A monoacylglycerol lipase from Mycobacterium smegmatis involved in bacterial cell interaction. J. Bacteriol. 2010, 192, 4776–4785. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, K.A.; Hu, W.; Li, G.; Lu, Y.; Richardson, E.J.; Loman, N.J.; Huang, H.; Besra, G.S. Anti-tubercular derivatives of rhein require activation by the monoglyceride lipase Rv0183. Cell Surf. 2020, 6, 100040. [Google Scholar] [CrossRef] [PubMed]
- Côtes, K.; Dhouib, R.; Douchet, I.; Chahinian, H.; de Caro, A.; Carrière, F.; Canaan, S. Characterization of an exported monoglyceride lipase from Mycobacterium tuberculosis possibly involved in the metabolism of host cell membrane lipids. Biochem. J. 2007, 408, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Aschauer, P.; Zimmermann, R.; Breinbauer, R.; Pavkov-Keller, T.; Oberer, M. The crystal structure of monoacylglycerol lipase from M. tuberculosis reveals the basis for specific inhibition. Sci. Rep. 2018, 8, 8948. [Google Scholar] [CrossRef]
- Labar, G.; Bauvois, C.; Borel, F.; Ferrer, J.-L.; Wouters, J.; Lambert, D.M. Crystal structure of the human Monoacylglycerol lipase, a key actor in Endocannabinoid signaling. ChemBioChem 2010, 11, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, P.; Rengachari, S.; Lichtenegger, J.; Schittmayer, M.; Das, K.M.P.; Mayer, N.; Breinbauer, R.; Birner-Gruenberger, R.; Gruber, C.C.; Zimmermann, R.; et al. Crystal structure of the Saccharomyces cerevisiae monoglyceride lipase Yju3p. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Rengachari, S.; Aschauer, P.; Schittmayer, M.; Mayer, N.; Gruber, K.; Breinbauer, R.; Birner-Gruenberger, R.; Dreveny, I.; Oberer, M. Conformational plasticity and ligand binding of bacterial monoacylglycerol lipase. J. Biol. Chem. 2013, 288, 31093–31104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labar, G.; Brandt, N.; Flaba, A.; Wouters, J.; Leherte, L. Structure and dynamics of an archeal monoglyceride lipase from Palaeococcus ferrophilus as revealed by crystallography and In Silico analysis. Biomolecules 2021, 11, 533. [Google Scholar] [CrossRef]
- Riegler-Berket, L.; Leitmeier, A.; Aschauer, P.; Dreveny, I.; Oberer, M. Identification of lipases with activity towards monoacylglycerol by criterion of conserved cap architectures. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 679–687. [Google Scholar] [CrossRef] [Green Version]
- Nasr, M.L.; Shi, X.; Bowman, A.L.; Johnson, M.; Zvonok, N.; Janero, D.R.; Vemuri, V.K.; Wales, T.E.; Engen, J.R.; Makriyannis, A. Membrane phospholipid bilayer as a determinant of monoacylglycerol lipase kinetic profile and conformational repertoire. Protein Sci. 2013, 22, 774–787. [Google Scholar] [CrossRef]
- Tyukhtenko, S.; Ma, X.; Rajarshi, G.; Karageorgos, I.; Anderson, K.W.; Hudgens, J.W.; Guo, J.J.; Nasr, M.L.; Zvonok, N.; Vemuri, K.; et al. Conformational gating, dynamics and allostery in human monoacylglycerol lipase. Sci. Rep. 2020, 10, 18531. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, L.; Cooper, D.R.; Derewenda, Z.S.; Eisenberg, D. Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 2007, 16, 1569–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golovanov, A.P.; Hautbergue, G.M.; Wilson, S.A.; Lian, L.-Y. A simple method for improving protein solubility and long-term stability. J. Am. Chem. Soc. 2004, 126, 8933–8939. [Google Scholar] [CrossRef] [PubMed]
- Bolen, D.W.; Baskakov, I.V. The osmophobic effect: Natural selection of a thermodynamic force in protein folding11Edited by D. Draper. J. Mol. Biol. 2001, 310, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Cianci, M.; Bourenkov, G.; Pompidor, G.; Karpics, I.; Kallio, J.; Bento, I.; Roessle, M.; Cipriani, F.; Fiedler, S.; Schneider, T.R. P13, the EMBL macromolecular crystallography beamline at the low-emittance PETRA III ring for high- and low-energy phasing with variable beam focusing. J. Synchrotron Rad. 2017, 24, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Burkhardt, A.; Pakendorf, T.; Reime, B.; Meyer, J.; Fischer, P.; Stübe, N.; Panneerselvam, S.; Lorbeer, O.; Stachnik, K.; Warmer, M.; et al. Status of the crystallography beamlines at PETRA III. Eur. Phys. J. Plus 2016, 131, 56. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and Evaluation of a New Integration Package. Acta Crystallogr. Sect. D Biol. Crystallogr. 2018, 74, 85–97. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Aschauer, P.; Pavkov-Keller, T.; Oberer, M. Crystal Strukture of Rv0183, a Monoglyceride Lipase from Mycobacterium Tuberculosis; Worldwide Protein Data Bank: Piscataway, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, N.W.; Grosse-Kunstleve, R.W.; Adams, P.D. electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Rengachari, S.; Bezerra, G.A.; Riegler-Berket, L.; Gruber, C.C.; Sturm, C.; Taschler, U.; Boeszoermenyi, A.; Dreveny, I.; Zimmermann, R.; Gruber, K.; et al. The structure of monoacylglycerol lipase from Bacillus sp. H257 reveals unexpected conservation of the cap architecture between bacterial and human enzymes. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.R.; Boczek, T.; Grelewska, K.; Pinkowska, M.; Sikorska, M.; Zawadzki, M.; Derewenda, Z. Protein crystallization by surface entropy reduction: Optimization of the SER strategy. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Schalk-Hihi, C.; Schubert, C.; Alexander, R.; Bayoumy, S.; Clemente, J.C.; Deckman, I.; DesJarlais, R.L.; Dzordzorme, K.C.; Flores, C.M.; Grasberger, B.; et al. Crystal structure of a soluble form of human monoglyceride lipase in complex with an inhibitor at 1.35 Å resolution. Protein Sci. 2011, 20, 670–683. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, T.; Augé, F.; Houtmann, J.; Rak, A.; Vallée, F.; Mikol, V.; Berne, P.F.; Michot, N.; Cheuret, D.; Hoornaert, C.; et al. Structural basis for human monoglyceride lipase inhibition. J. Mol. Biol. 2010, 396, 663–673. [Google Scholar] [CrossRef]
- Butler, C.R.; Beck, E.M.; Harris, A.; Huang, Z.; McAllister, L.A.; am Ende, C.W.; Fennell, K.; Foley, T.L.; Fonseca, K.; Hawrylik, S.J.; et al. Azetidine and piperidine carbamates as efficient, covalent inhibitors of monoacylglycerol lipase. J. Med. Chem. 2017, 60, 9860–9873. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Biagi, M.; Spinelli, V.; Di Stefano, M.; Macchia, M.; Minutolo, F.; Granchi, C.; Poli, G.; Tuccinardi, T. Discovery of Monoacylglycerol Lipase (MAGL) inhibitors based on a pharmacophore-guided virtual screening study. Molecules 2020, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Gütschow, M.; Vanden Eynde, J.J.; Jampilek, J.; Kang, C.; Mangoni, A.A.; Fossa, P.; Karaman, R.; Trabocchi, A.; Scott, P.J.H.; Reynisson, J.; et al. Breakthroughs in medicinal chemistry: New targets and mechanisms, new drugs, new hopes–7. Molecules 2020, 25, 2968. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| 7OZM | 7P0Y | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Data Collection | ||||||||||||
| Wavelength (Å) | 1.072 | 1.0332 | ||||||||||
| Resolution range (Å) | 39.77–2.15 (2.23–2.15) | 40.78–2.25 (2.33–2.25) | ||||||||||
| Space group | P 2 21 21 | P 21 2 21 | ||||||||||
| Unit cell | a | b | c | α | β | γ | a | b | c | α | β | γ |
| (Å, °) | 40.50 | 82.24 | 90.86 | 90 | 90 | 90 | 74.69 | 82.60 | 93.78 | 90 | 90 | 90 |
| Total reflections | 54,614 (5426) | 145,938 (14,206) | ||||||||||
| Unique reflections | 16,663 (1655) | 28,192 (2766) | ||||||||||
| Multiplicity | 3.3 (3.3) | 5.2 (5.1) | ||||||||||
| Completeness (%) | 97.22 (97.93) | 99.65 (99.50) | ||||||||||
| Mean I/sigma(I) | 7.27 (1.43) | 20.99 (1.19) | ||||||||||
| Wilson B-factor | 29.19 | 36.18 | ||||||||||
| R-merge | 0.1415 (0.7517) | 0.149 (0.7195) | ||||||||||
| R-meas | 0.1669 (0.8893) | 0.1654 (0.8019) | ||||||||||
| R-pim | 0.08599 (0.4619) | 0.07091 (0.3494) | ||||||||||
| CC1/2 | 0.991 (0.578) | 0.99 (0.77) | ||||||||||
| CC* | 0.998 (0.856) | 0.998 (0.933) | ||||||||||
| Refinement | ||||||||||||
| No. of reflections | 16,658 (1665 for test set) | 28,118 (3571 for test set) | ||||||||||
| Rwork/Rfree | 0.1950/0.2500 | 0.2534/0.2951 | ||||||||||
| Non-solvent atoms | 2126 | 4240 | ||||||||||
| Solvent atoms | 133 | 141 | ||||||||||
| RMS (bonds, Å) | 0.007 | 0.003 | ||||||||||
| RMS (angles, °) | 0.86 | 0.61 | ||||||||||
| Ramachandran- favored (%) | 97.1 | 97.05 | ||||||||||
| Ramachandran- allowed (%) | 2.54 | 2.95 | ||||||||||
| Ramachandran outliers (%) | 0.36 | 0 | ||||||||||
| Rotamer outliers (%) | 0.45 | 0.92 | ||||||||||
| Clashscore | 3.53 | 8.18 | ||||||||||
| Average B-factor (Å2) | 30.0 | 45.6 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grininger, C.; Leypold, M.; Aschauer, P.; Pavkov-Keller, T.; Riegler-Berket, L.; Breinbauer, R.; Oberer, M. Structural Changes in the Cap of Rv0183/mtbMGL Modulate the Shape of the Binding Pocket. Biomolecules 2021, 11, 1299. https://doi.org/10.3390/biom11091299
Grininger C, Leypold M, Aschauer P, Pavkov-Keller T, Riegler-Berket L, Breinbauer R, Oberer M. Structural Changes in the Cap of Rv0183/mtbMGL Modulate the Shape of the Binding Pocket. Biomolecules. 2021; 11(9):1299. https://doi.org/10.3390/biom11091299
Chicago/Turabian StyleGrininger, Christoph, Mario Leypold, Philipp Aschauer, Tea Pavkov-Keller, Lina Riegler-Berket, Rolf Breinbauer, and Monika Oberer. 2021. "Structural Changes in the Cap of Rv0183/mtbMGL Modulate the Shape of the Binding Pocket" Biomolecules 11, no. 9: 1299. https://doi.org/10.3390/biom11091299
APA StyleGrininger, C., Leypold, M., Aschauer, P., Pavkov-Keller, T., Riegler-Berket, L., Breinbauer, R., & Oberer, M. (2021). Structural Changes in the Cap of Rv0183/mtbMGL Modulate the Shape of the Binding Pocket. Biomolecules, 11(9), 1299. https://doi.org/10.3390/biom11091299