
Nitric Oxide-Dependent Pathways as Critical Factors in the Consequences and Recovery after Brain Ischemic Hypoxia



Abstract
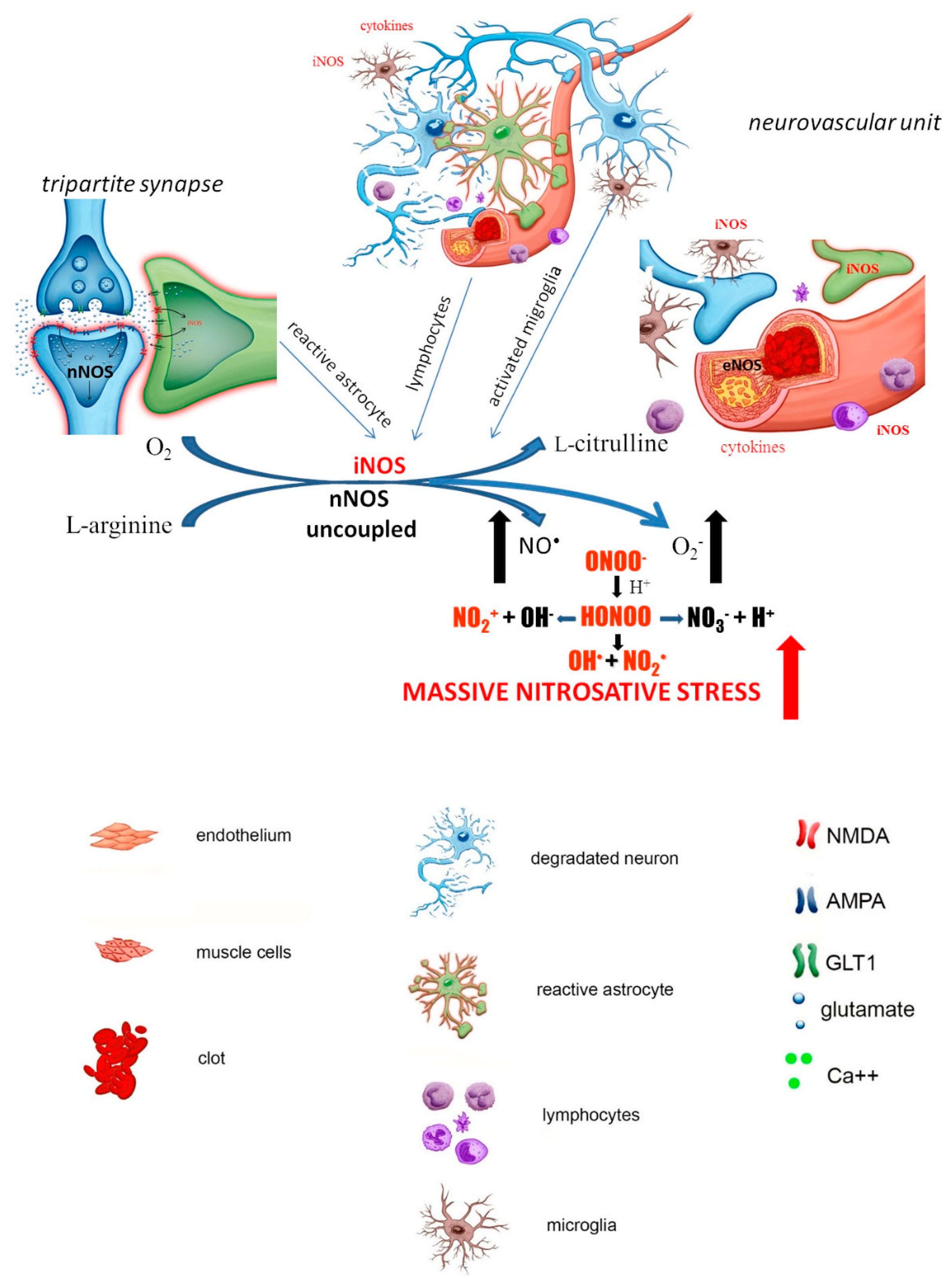
1. Brain Ischemic Stroke and the Role of NO Its Pathology
1.1. Early Stage of the Ischemia (Early Neuronal Damage)
1.2. Late Stage of the Ischemia (Delayed Neuronal Damage)
2. NO-Dependent Factors Aggravating Ischemic Cascade
2.1. S-Nitrosylation
2.2. Hypoxia-Inducible Factor 1α (HIF-1α)
3. Treatment Strategies
3.1. Free Radical Scavengers
3.2. Enhancement of NO Production
3.3. NOS Inhibitors

{kind=link}
{kind=link}
{kind=link}
| Compound | Effect | ||
|---|---|---|---|
| NO Donors | |||
| Pretreatment | LA-419 | in vivo:
| [156,157] |
| GSNO | in vivo:
| [169,170,171,172] | |
| ZJM-289 | in vitro:
| [155,173] | |
| SIN-1 | in vivo:
| [174,175] | |
| DETA NONOate | in vivo:
| [174] | |
| NBP | in vitro:
| [173,176] | |
| Spermine NONOate | in vivo:
| [152] | |
| sodium nitroprusside | in vivo:
| [152] | |
| Posttreatment | GSNO | in vivo:
| [171] |
| DETA NONOate | in vivo:
| [150] | |
| SIN-1 | in vivo:
| [153,154] | |
| sodium nitroprusside | in vivo:
| [153] | |
| NOS or nNOS inhibitors | |||
| Pretreatment | 7-NI | in vivo:
| [170,172,177] |
| L-NAME | in vivo:
| [174,178] | |
| Posttreatment | 7-NI | in vivo:
| [179] |
| L-NAME | in vivo:
| [178,180] | |
| iNOS inhibitors | |||
| Pretreatment | aminoguanidine | in vivo:
| [181] |
| Posttreatment | aminoguanidine | in vivo:
| [167,182,183] |
| 1400 W | in vivo:
| [184] | |
| S-methylisothiorea | in vivo:
| [185] |
3.4. HIF-1α
3.5. Combination Therapies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- GBD 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef]
- Müller, G.J.; Stadelmann, C.; Bastholm, L.; Elling, F.; Lassmann, H.; Johansen, F.F. Ischemia leads to apoptosis--and necrosis-like neuron death in the ischemic rat hippocampus. Brain Pathol. 2004, 14, 415–424. [Google Scholar] [CrossRef]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.-T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Zhang, S.J.; Steijaert, M.N.; Lau, D.; Schutz, G.; Delucinge-Vivier, C.; Descombes, P.; Bading, H. Decoding NMDA receptor signaling: Identification of genomic programs specifying neuronal survival and death. Neuron 2007, 53, 549–562. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef]
- Palmer, R.M.; Rees, D.D.; Ashton, D.S.; Moncada, S. L-arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 1988, 153, 1251–1256. [Google Scholar] [CrossRef]
- Katsuki, S.; Arnold, W.; Mittal, C.; Murad, F. Stimulation of guanylate cyclase by sodium nitroprusside, nitroglycerin and nitric oxide in various tissue preparations and comparison to the effects of sodium azide and hydroxylamine. J. Cyclic Nucleotide Res. 1977, 3, 23–35. [Google Scholar] [PubMed]
- Miki, N.; Kawabe, Y.; Kuriyama, K. Activation of cerebral guanylate cyclase by nitric oxide. Biochem. Biophys. Res. Commun. 1977, 75, 851–856. [Google Scholar] [CrossRef]
- Bon, C.L.M.; Garthwaite, J. On the role of nitric oxide in hippocampal long-term potentiation. J. Neurosci. 2003, 23, 1941–1948. [Google Scholar] [CrossRef]
- Kalinowski, L.; Malinski, T. Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: Relationship to endothelial dysfunction. Acta Biochim. Pol. 2004, 51, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.M.; Palmer, R.M.; Knowles, R.G.; Francis, P.L.; Ashton, D.S.; Moncada, S. Constitutive and inducible nitric oxide synthases incorporate molecular oxygen into both nitric oxide and citrulline. J. Biol. Chem. 1991, 266, 23790–23795. [Google Scholar] [CrossRef]
- Lelchuk, R.; Radomski, M.W.; Martin, J.F.; Moncada, S. Constitutive and inducible nitric oxide synthases in human megakaryoblastic cells. J. Pharmacol. Exp. Ther. 1992, 262, 1220–1224. [Google Scholar]
- Eliasson, M.J.; Huang, Z.; Ferrante, R.J.; Sasamata, M.; Molliver, M.E.; Snyder, S.H.; Moskowitz, M.A. Neuronal nitric oxide synthase activation and peroxynitriteformation in ischemic stroke linked to neural damage. J. Neurosci. 1999, 19, 5910–5918. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zweier, J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Giampietro, D.E.; Cherici, G.; Alesiani, M.; Carlà, V.; Moroni, F. Excitatory amino acid release from rat hippocampal slices as a consequence of free-radical formation. J. Neurochem. 1988, 51, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Dikshit, M. Vascular regulation by the L-arginine metabolites, nitric oxide and agmatine. Pharmacol. Res. 2004, 49, 397–414. [Google Scholar] [CrossRef]
- Dereski, M.O.; Chopp, M.; Knight, R.A.; Rodolosi, L.C.; Garcia, J.H. The heterogeneous temporal evolution of focal ischemic neuronal damage in the rat. Acta Neuropathol. 1993, 85, 327–333. [Google Scholar] [CrossRef]
- Ginsberg, M.D.; Busto, R. Rodent models of cerebral ischemia. Stroke 1989, 20, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Samdani, A.F.; Dawson, T.M.; Dawson, V.L. Nitric oxide synthase in models of focal ischemia. Stroke 1997, 28, 1283–1288. [Google Scholar] [CrossRef]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T.S. The role of the nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Almeida, A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim. Biophys. Acta 1999, 1411, 415–436. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Chen, Z.-Q.; Mou, R.-T.; Feng, D.-X.; Wang, Z.; Chen, G. The role of nitric oxide in stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar] [CrossRef]
- Heiss, W.D. Experimental evidence of ischemic thresholds and functional recovery. Stroke 1992, 23, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Furlan, M.; Marchal, G.; Viader, F.; Derlon, J.M.; Baron, J.C. Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann. Neurol. 1996, 40, 216–226. [Google Scholar] [CrossRef]
- Baird, A.E.; Benfield, A.; Schlaug, G.; Siewert, B.; Lövblad, K.O.; Edelman, R.R.; Warach, S. Enlargement of human cerebral ischemic lesion volumes measured by diffusion-weighted magnetic resonance imaging. Ann. Neurol. 1997, 41, 581–589. [Google Scholar] [CrossRef]
- Castillo, J.; Rama, R.; Dávalos, A. Nitric oxide-related brain damage in acute ischemic stroke. Stroke 2000, 31, 852–857. [Google Scholar] [CrossRef]
- Forster, C.; Clark, H.B.; Ross, M.E.; Iadecola, C. Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol. 1999, 97, 215–220. [Google Scholar] [CrossRef]
- Ketheeswaranathan, P.; Turner, N.A.; Spary, E.J.; Batten, T.F.C.; McColl, B.W.; Saha, S. Changes in glutamate transporter expression in mouse forebrain areas following focal ischemia. Brain Res. 2011, 1418, 93–103. [Google Scholar] [CrossRef]
- Mayhan, W.G.; Didion, S.P. Glutamate-induced disruption of the blood-brain barrier in rats. Role of nitric oxide. Stroke 1996, 27, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide Biol. Chem. 2009, 20, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Fabian, R.H.; Perez-Polo, J.R.; Kent, T.A. Perivascular nitric oxide and superoxide in neonatal cerebral hypoxia-ischemia. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1809–H1814. [Google Scholar] [CrossRef] [PubMed]
- Onodera, H.; Kogure, K.; Ono, Y.; Igarashi, K.; Kiyota, Y.; Nagaoka, A. Proto-oncogene c-fos is transiently induced in the rat cerebral cortex after forebrain ischemia. Neurosci. Lett. 1989, 98, 101–104. [Google Scholar] [CrossRef]
- Wessel, T.C.; Joh, T.H.; Volpe, B.T. In situ hybridization analysis of c-fos and c-jun expression in the rat brain following transient forebrain ischemia. Brain Res. 1991, 567, 231–240. [Google Scholar] [CrossRef]
- Jiang, X.; Mu, D.; Manabat, C.; Koshy, A.A.; Christen, S.; Täuber, M.G.; Vexler, Z.S.; Ferriero, D.M. Differential vulnerability of immature murinae neurons to oxygen-glucose deprivation. Exp. Neurol. 2004, 190, 224–232. [Google Scholar] [CrossRef]
- Grima, G.; Benz, B.; Do, K.Q. Glial-derived arginine, the nitric oxide precursor, protects neurons from NMDA-induced excitotoxicity. Eur. J. Neurosci. 2001, 14, 1762–1770. [Google Scholar] [CrossRef]
- Kondo, T.; Reaume, A.G.; Huang, T.T.; Carlson, E.; Murakami, K.; Chen, S.F.; Hoffman, E.K.; Scott, R.W.; Epstein, C.J.; Chan, P.H. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci. 1997, 17, 4180–4189. [Google Scholar] [CrossRef]
- Kondo, T.; Reaume, A.G.; Huang, T.T.; Murakami, K.; Carlson, E.; Chen, S.; Scott, R.W.; Epstein, C.J.; Chan, P.H. Edema formation exacerbates neurological and histological outcomes after focal cerebral ischemia in CuZn-superoxide dismutase gene knockout mutant mice. Acta Neurochir. Suppl. 1997, 70, 62–64. [Google Scholar]
- Chan, P.H.; Epstein, C.J.; Li, Y.; Huang, T.T.; Carlson, E.; Kinouchi, H.; Yang, G.; Kamii, H.; Mikawa, S.; Kondo, T.; et al. Transgenic mice and knockout mutants in the study of oxidative stress in brain injury. J. Neurotrauma 1995, 12, 815–824. [Google Scholar] [CrossRef]
- Léveillé, F.; Gaamouch, F.E.; Gouix, E.; Lecocq, M.; Lobner, D.; Nicole, O.; Buisson, A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008, 22, 4258–4271. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Doshi, S.; Spaethling, J.M.; Hockenberry, A.J.; Patel, T.P.; Geddes-Klein, D.M.; Lync, D.R.; Meaney, D.F. N-methyl-D-aspartate receptor mechanosensitivity is governed by C terminus of NR2B subunit. J. Biol. Chem. 2012, 287, 4348–4359. [Google Scholar] [CrossRef] [PubMed]
- Maneshi, M.M.; Maki, B.; Gnanasambandam, R.; Belin, S.; Popescu, G.K.; Sachs, F.; Hua, S.Z. Mechanical stress activates NMDA receptors in the absence of agonists. Sci. Rep. 2017, 7, 39610. [Google Scholar] [CrossRef]
- Amorini, A.M.; Lazzarino, G.; Pietro, V.D.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Severity of experimental traumatic brain injury modulates changes in concentrations of cerebral free amino acids. J. Cell. Mol. Med. 2017, 21, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Bowen, D.M.; DeKosky, S.T. Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport 1994, 6, 153–156. [Google Scholar] [CrossRef]
- Ito, Y.; Ohkubo, T.; Asano, Y.; Hattori, K.; Shimazu, T.; Yamazato, M.; Nagoya, H.; Kato, Y.; Araki, N. Nitric oxide production during cerebral ischemia and reperfusion in eNOS- and nNOS-knockout mice. Curr. Neurovasc. Res. 2010, 7, 23–31. [Google Scholar] [CrossRef]
- Wei, G.; Dawson, V.L.; Zweier, J.L. Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia. Biochim. Biophys. Acta 1999, 1455, 23–34. [Google Scholar] [CrossRef]
- Cui, X.; Chopp, M.; Zacharek, A.; Zhang, C.; Roberts, C.; Chen, J. Role of endothelial nitric oxide synthetase in arteriogenesis after stroke in mice. Neuroscience 2009, 159, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, C.; Arrick, D.M.; Yang, S.; Baluna, A.E.; Sun, H. Role of nitric oxide synthases in early blood-brain barrier disruption following transient focal cerebral ischemia. PLoS ONE 2014, 9, e93134. [Google Scholar] [CrossRef] [PubMed]
- Zarruk, J.G.; Greenhalgh, A.D.; David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp. Neurol. 2018, 301, 120–132. [Google Scholar] [CrossRef]
- Pei, X.; Li, Y.; Zhu, L.; Zhou, Z. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp. Cell Res. 2019, 382, 111474. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, D.B.; Wong, J.; Shapiro, A.; Durkin, J.P. Increase in surface expression of ICAM-1, VCAM-1 and E-selectin in human cerebromicrovascular endothelial cells subjected to ischemia-like insults. Acta Neurochir. Suppl. 1997, 70, 12–16. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Bushong, E.A.; Price, D.L.; Smarr, B.L.; Phung, V.; Terada, M.; Ellisman, M.H.; Pekny, M. Redefining the concept of reactive astrocytes as cells that remain within their unique do-mains upon reaction to injury. Proc. Natl. Acad. Sci. USA 2006, 103, 17513–17518. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Buskila, Y.; Farkash, S.; Hershfinkel, M.; Amitai, Y. Rapid and reactive nitric oxide production by astrocytes in mouse neocortical slices. Glia 2005, 52, 169–176. [Google Scholar] [CrossRef]
- Zhao, S.-C.; Ma, L.-S.; Chu, Z.-H.; Xu, H.; Wu, W.-Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef]
- Hewett, S.I.; Csernansky, C.A.; Choi, D.W. Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron 1994, 13, 487–494. [Google Scholar] [CrossRef]
- Garcia-Bonilla, L.; Moore, J.M.; Racchumi, G.; Zhou, P.; Butler, J.M.; Iadecola, C.; Anrather, J. Inducible Nitric Oxide Synthase in Neutrophils and Endothelium Contributes to Ischemic Brain Injury in Mice. J. Immunol. 2014, 193, 2531–2537. [Google Scholar] [CrossRef]
- Iadecola, C.; Zhang, F.; Xu, S.; Casey, R.; Ross, M.E. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Zhang, F.; Casey, R.; Nagayama, M.; Ross, M.E. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 1997, 17, 9157–9164. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.H.; Schmidley, J.W.; Fishman, R.A.; Longar, S.M. Brain injury, edema, and vascular permeability changes induced by oxygen-derived free radicals. Neurology 1984, 34, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Gasche, Y.; Fujimura, M.; Morita-Fujimura, Y.; Copin, J.C.; Kawase, M.; Massengale, J.; Chan, P.H. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: A possible role in blood-brain barrier dysfunction. Int. Soc. Cereb. Blood Flow Metab. 1999, 19, 1020–1028. [Google Scholar] [CrossRef]
- Guo, M.; Cox, B.; Mahale, S.; Davis, W.; Carranza, A.; Hayes, K.; Sprague, S.; Jimenez, D.; Ding, Y. Pre-ischemic exercise reduces matrix metalloproteinase-9 expression and ameliorates blood-brain barrier dysfunction in stroke. Neuroscience 2008, 151, 340–351. [Google Scholar] [CrossRef]
- Broughton, B.R.S.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef]
- Iadecola, C.; Alexander, M. Cerebral ischemia and inflammation. Curr. Opin. Neurol. 2001, 14, 89–94. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Lipton, S.A.; Choi, Y.B.; Pan, Z.H.; Lei, S.Z.; Chen, H.S.; Sucher, N.J.; Loscalzo, J.; Singel, D.J.; Stamler, J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 1993, 364, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Marozkina, N.V.; Gaston, B. S-Nitrosylation signaling regulates cellular protein interactions. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Neuronal protection and destruction by NO. Cell Death Differ. 1999, 6, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Marshall, H.E. S-nitrosylation in the regulation of gene transcription. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Murray, C.I.; Venkatraman, V.; Crowgey, E.L.; Rainer, P.P.; Cole, R.N.; Bomgarden, R.D.; Rogers, J.C.; Balkan, W.; Hare, J.M.; et al. Dual Labeling Biotin Switch Assay to Reduce Bias Derived From Different Cysteine Subpopulations: A Method to Maximize S-Nitrosylation Detection. Circ. Res. 2015, 117, 846–857. [Google Scholar] [CrossRef]
- Doulias, P.-T.; Greene, J.L.; Greco, T.M.; Tenopoulou, M.; Seeholzer, S.H.; Dunbrack, R.L.; Ischiropoulos, H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. USA 2010, 107, 16958–16963. [Google Scholar] [CrossRef]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.-Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef]
- Chen, T.; Cao, L.; Dong, W.; Luo, P.; Liu, W.; Qu, Y.; Fei, Z. Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. Neurochem. Res. 2012, 37, 983–990. [Google Scholar] [CrossRef]
- Qu, Z.W.; Miao, W.Y.; Hu, S.Q.; Li, C.; Zhuo, X.L.; Zong, Y.Y.; Wu, Y.P.; Zhang, G.Y. N-Methyl-D-Aspartate Receptor-Dependent Denitrosylation of Neuronal Nitric Oxide Synthase Increase the Enzyme Activity. PLoS ONE 2012, 7, e52788. [Google Scholar] [CrossRef][Green Version]
- Ravi, K.; Brennan, L.A.; Levic, S.; Ross, P.A.; Black, S.M. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc. Natl. Acad. Sci. USA 2004, 101, 2619–2624. [Google Scholar] [CrossRef] [PubMed]
- Erwin, P.A.; Lin, A.J.; Golan, D.E.; Michel, T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 2005, 280, 19888–19894. [Google Scholar] [CrossRef]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Feng, J.J.; Wu, Y.P.; Zhang, G.Y. Cerebral ischemia-reperfusion induces GAPDH S-nitrosylation and nuclear translocation. Biochemistry (Moscow) 2012, 77, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002, 297, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell. Signal. 2011, 23, 317–323. [Google Scholar] [CrossRef]
- Tanaka, R.; Mochizuki, H.; Suzuki, A.; Katsube, N.; Ishitani, R.; Mizuno, Y.; Urabe, T. Induction of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression in rat brain after focal ischemia/reperfusion. J. Cereb. Blood Flow Metab. 2002, 22, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Helton, R.; Cui, J.; Scheel, J.R.; Ellison, J.A.; Ames, C.; Gibson, C.; Blouw, B.; Ouyang, L.; Dragatsis, I.; Zeitlin, S.; et al. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxic-ischemic damage. J. Neurosci. 2005, 25, 4099–4107. [Google Scholar] [CrossRef] [PubMed]
- Barteczek, P.; Li, L.; Ernst, A.-S.; Böhler, L.-I.; Marti, H.H.; Kunze, R. Neuronal HIF-1α and HIF-2α deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Chen, R.L.; Ogunshola, O.O.; Yeoh, K.K.; Jani, A.; Papadakis, M.; Nagel, S.; Schofield, C.J.I.; Buchan, A.M. HIF prolyl hydroxylase inhibition prior to transient focal cerebral ischaemia is neuroprotective in mice. J. Neurochem. 2014, 131, 177–189. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef]
- Khan, M.; Dhammu, T.S.; Dhaindsa, T.S. An NO/GSNO-based Neuroregeneration Strategy for Stroke Therapy. J. Neurol. Neurosci. 2015, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Khan, H.; Singh, I.; Singh, A.K. Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury. Neural Regen. Res. 2017, 12, 696–701. [Google Scholar] [CrossRef]
- Khan, M.; Dhammu, T.S.; Baarine, M.; Kim, J.; Paintlia, M.K.; Singh, I.; Singh, A.K. GSNO promotes functional recovery in experimental TBI by stabilizing HIF-1α. Behav. Brain Res. 2016, 340, 63–70. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Ogle, M.E.; Gu, X.; Espinera, A.R.; Wei, L. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1α. Neurobiol. Dis. 2012, 45, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.A.; Alba, J.D.; Leza, J.C.; Lorenzo, P.; Fernández, A.P.; Bentura, M.L.; Boscá, L.; Rodrigo, J.; Lizasoain, I. Neuronal expression of inducible nitric oxide synthase after oxygen and glucose deprivation in rat forebrain slices. Eur. J. Neurosci. 1998, 10, 445–456. [Google Scholar] [CrossRef]
- Matrone, C.; Pignataro, G.; Molinaro, P.; Irace, C.; Scorziello, A.; Di Renzo, G.F.; Annunziato, L.; Renzo, G.F.D.; Annunziato, L. HIF-1alpha reveals a binding activity to the promoter of iNOS gene after permanent middle cerebral artery occlusion. J. Neurochem. 2004, 90, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yan, J.; Shi, H. Role of Hypoxia Inducible Factor 1 in Hyperglycemia-Exacerbated Blood-Brain Barrier Disruption in Ischemic Stroke. Neurobiol. Dis. 2016, 95, 82–92. [Google Scholar] [CrossRef]
- Li, Q.F.; Xu, H.; Sun, Y.; Hu, R.; Jiang, H. Induction of inducible nitric oxide synthase by isoflurane post-conditioning via hypoxia inducible factor-1α during tolerance against ischemic neuronal injury. Brain Res. 2012, 1451, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Baranova, O.; Miranda, L.F.; Pichiule, P.; Dragatsis, I.; Johnson, R.S.; Chavez, J.C. Neuron-specific inactivation of the hypoxia inducible factor 1α increases brain injury in a mouse model of transient focal cerebral ischemia. J. Neurosci. 2007, 27, 6320–6332. [Google Scholar] [CrossRef]
- Kunze, R.; Zhou, W.; Veltkamp, R.; Wielockx, B.; Breier, G.; Marti, H.H. Neuron-specific prolyl-4-hydroxylase domain 2 knockout reduces brain injury after transient cerebral ischemia. Stroke 2012, 43, 2748–2756. [Google Scholar] [CrossRef]
- Bok, S.; Kim, Y.E.; Woo, Y.; Kim, S.; Kang, S.J.; Lee, Y.; Park, S.K.; Weissman, I.L.; Ahn, G.O. Hypoxia-inducible factor-1a regulates microglial functions affecting neuronal survival in the acute phase of ischemic stroke in mice. Oncotarget 2017, 8, 111508–111521. [Google Scholar] [CrossRef]
- Huang, T.; Huang, W.; Zhang, Z.; Yu, L.; Xie, C.; Zhu, D.; Peng, Z.; Chen, J. Hypoxia-inducible factor-1α upregulation in microglia following hypoxia protects against ischemia-induced cerebral infarction. Neuroreport 2014, 25, 1122–1128. [Google Scholar] [CrossRef]
- Hsiao, G.; Lee, J.-J.J.; Chen, Y.-C.C.; Lin, J.-H.H.; Shen, M.-Y.Y.; Lin, K.-H.H.; Chou, D.-S.S.; Sheu, J.-R.R. Neuroprotective effects of PMC, a potent α-tocopherol derivative, in brain ischemia-reperfusion: Reduced neutrophil activation and anti-oxidant actions. Biochem. Pharmacol. 2007, 73, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Luciano, J.A.; Tan, T.; Zhang, Q.; Huang, E.; Scholz, P.; Weiss, H.R. Hypoxia inducible factor-1 improves the actions of nitric oxide and natriuretic peptides after simulated ischemia-reperfusion. Cell. Physiol. Biochem. 2008, 21, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Shi, H. Hypoxia Inducible Factor 1 as a Therapeutic Target in Ischemic Stroke. Curr. Med. Chem. 2009, 16, 4593. [Google Scholar] [CrossRef]
- Aminova, L.R.; Chavez, J.C.; Lee, J.; Ryu, H.; Kung, A.; Lamanna, J.C.; Ratan, R.R. Prosurvival and prodeath effects of hypoxia-inducible factor-1alpha stabilization in a murine hippocampal cell line. J. Biol. Chem. 2005, 280, 3996–4003. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Turley, H.; Allen, W.E.; Willam, C.; Eckardt, K.U.; Talks, K.L.; Wood, S.M.; Gatter, K.C.; Harris, A.L.; Pugh, C.W.; et al. Induction of endothelial PAS domain protein-1 by hypoxia: Characterization and comparison with hypoxia-inducible factor-1alpha. Blood 1998, 92, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef]
- Brusselmans, K.; Compernolle, V.; Tjwa, M.; Wiesener, M.S.; Maxwell, P.H.; Collen, D.; Carmeliet, P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Investig. 2003, 111, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Halterman, M.W.; Miller, C.C.; Federoff, H.J. Hypoxia-Inducible Factor-1α Mediates Hypoxia-Induced Delayed Neuronal Death That Involves p53. J. Neurosci. 1999, 19, 6818–6824. [Google Scholar] [CrossRef]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol. Cell. Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef]
- Renton, A.; Llanos, S.; Lu, X. Hypoxia induces p53 through a pathway distinct from most DNA-damaging and stress-inducing agents. Carcinogenesis 2003, 24, 1177–1182. [Google Scholar] [CrossRef]
- Zivin, J.A. Factors determining the therapeutic window for stroke. Neurology 1998, 50, 599–603. [Google Scholar] [CrossRef]
- Jaffer, H.; Morris, V.B.; Stewart, D.; Labhasetwar, V. Advances in stroke therapy. Drug Deliv. Transl. Res. 2011. [Google Scholar] [CrossRef]
- Wung, B.S.; Cheng, J.J.; Shyue, S.K.; Wang, D.L. NO modulates monocyte chemotactic prootein-1 expression in endothelial cells under cyclic strain. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1941–1947. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Begum, N.; Sandu, O.A.; Duddy, N. Negative regulation of Rho signaling by insulin and its impact on actin cytoskeleton organization in vascular smooth muscle cells: Role of nitric oxide and cyclic guanosine monophosphate signaling pathways. Diabetes 2002, 51, 2256–2263. [Google Scholar] [CrossRef]
- Hirabayashi, H.; Takizawa, S.; Fukuyama, N.; Nakazawa, H.; Shinohara, Y. Nitrotyrosine generation via inducible nitric oxide synthase in vascular wall in focal ischemia-reperfusion. Brain Res. 2000, 852, 319–325. [Google Scholar] [CrossRef]
- Gow, A.; Duran, D.; Thom, S.R.; Ischiropoulos, H. Carbon Dioxide Enhancement of Peroxynitrite-Mediated Protein Tyrosine Nitration. Arch. Biochem. Biophys. 1996, 333, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Sanjuán-Pla, A.; Cervera, A.M.; Apostolova, N.; Garcia-Bou, R.; Víctor, V.M.; Murphy, M.P.; McCreath, K.J. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1alpha. FEBS Lett. 2005, 579, 2669–26674. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Tancharoen, S.; Takeshige, N.; Yoshitomi, M.; Morioka, M.; Murai, Y.; Tanaka, E. The efficacy of edaravone (radicut), a free radical scavenger, for cardiovascular disease. Int. J. Mol. Sci. 2013, 14, 13909–13930. [Google Scholar] [CrossRef]
- Satoh, K.; Ikeda, Y.; Shioda, S.; Tobe, T.; Yoshikawa, T. Edarabone scavenges nitric oxide. Redox Rep. 2002, 7, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Banno, M.; Mizuno, T.; Kato, H.; Zhang, G.; Kawanokuchi, J.; Wang, J.; Kuno, R.; Jin, S.; Takeuchi, H.; Suzumura, A. The radical scavenger edaravone prevents oxidative neurotoxicity induced by peroxynitrite and activated microglia. Neuropharmacology 2005, 48, 283–290. [Google Scholar] [CrossRef]
- Watanabe, T.; Yuki, S.; Egawa, M.; Nishi, H. Protective effects of MCI-186 on cerebral ischemia: Possible involvement of free radical scavenging and antioxidant actions. J. Pharmacol. Exp. Ther. 1994, 268, 1597–1604. [Google Scholar]
- Feng, S.; Yang, Q.; Liu, M.; Li, W.; Yuan, W.; Zhang, S.; Wu, B.; Li, J. Edaravone for acute ischaemic stroke. Cochrane Database Syst. Rev. 2011, 12, CD007230. [Google Scholar] [CrossRef]
- Otomo, E.; Tohgi, H.; Kogure, K.; Hirai, S.; Takakura, K.; Terashi, A.; Gotoh, F.; Maruyama, S.; Tazaki, Y.; Shinohara, Y.; et al. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction: Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [Google Scholar]
- The RANTTAS Investigators. A randomized trial of tirilazad mesylate in patients with acute stroke (RANTTAS). Stroke 1996, 27, 1453–1458. [Google Scholar] [CrossRef]
- The Tirilazad International Steering Committee. Tirilazad for acute ischaemic stroke. Cochrane Database Syst. Rev. 2001, CD002087. [Google Scholar] [CrossRef]
- Saito, I.; Asano, T.; Sano, K.; Takakura, K.; Abe, H.; Yoshimoto, T.; Kikuchi, H.; Ohta, T.; Ishibashi, S. Neuroprotective effect of an antioxidant, ebselen, in patients with delayed neurological deficits after aneurysmal subarachnoid hemorrhage. Neurosurgery 1998, 42, 269–277. [Google Scholar] [CrossRef]
- Ogawa, A.; Yoshimoto, T.; Kikuchi, H.; Sano, K.; Saito, I.; Yamaguchi, T.; Yasuhara, H. Ebselen in acute middle cerebral artery occlusion: A placebo-controlled, double-blind clinical trial. Cerebrovasc. Dis. 1999, 9, 112–118. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, E.; Rosenblatt, S.; Moskowitz, M.A. l-Arginine dilates rat pial arterioles by nitric oxide-dependent mechanisms and increases blood flow during focal cerebral ischaemia. Br. J. Pharmacol. 1992, 107, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, E.; Huang, Z.; Moskowitz, M.A. L-Arginine decreases infarct size caused by middle cerebral arterial occlusion in SHR. Am. J. Physiol. Hear. Circ. Physiol. 1992, 263, H1632–H1635. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, E.; Moskowitz, M.A.; Huang, Z.; Yoshida, T.; Irikura, K.; Dalkara, T. L-arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke 1994, 25, 429–435. [Google Scholar] [CrossRef]
- Zhao, H.; Asai, S.; Ishikawa, K. Neither L-NAME nor L-arginine changes extracellular glutamate elevation and anoxic depolarization during global ischemia and reperfusion in rat. Neuroreport 1999, 10, 313–318. [Google Scholar] [CrossRef]
- Kurt, T.; Oǧuzhanoǧlu, A.; Ortaç, R.; Turman, B.; Adigüzel, E. Effects of L-arginine on the brain ischaemia-reperfusion damage in rats: An investigation by somatosensory evoked potentials and histopathology. Neurosci. Res. Commun. 2002, 31, 175–182. [Google Scholar] [CrossRef]
- Zhao, X.; Ross, M.E.; Iadecola, C. L-arginine increases ischemic injury in wild-type mice but not in iNOS-deficient mice. Brain Res. 2003, 966, 308–311. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Warner, T.D.; Ishii, K.; Sheng, H.; Murad, F. Insulin secretion from pancreatic B cells caused by L-arginine-derived nitrogen oxides. Science 1992, 255, 721–723. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamatani, K.; Hara, M.; Sasaki, H. Gliclazide directly suppresses arginine-induced glucagon secretion. Diabetes Res. Clin. Pract. 1994, 24, 143–151. [Google Scholar] [CrossRef]
- Alba-Roth, J.; Müller, O.A.; Schopohl, J.; Von Werder, K. Arginine stimulates growth hormone secretion by suppressing endogenous somatostatin secretion. J. Clin. Endocrinol. Metab. 1988, 67, 1186–1189. [Google Scholar] [CrossRef]
- Strasser, A.; McCarron, R.M.; Ishii, H.; Stanimirovic, D.; SpatZ, M. L-arginine induces dopamine release from the striatum in vivo. Neuroreport 1994, 5, 2298–2300. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Regunathan, S.; Barrow, C.J.; Eshraghi, J.; Cooper, R.; Reis, D.J. Agmatine: An endogenous clonidine-displacing substance in the brain. Science 1994, 263, 966–969. [Google Scholar] [CrossRef]
- Lortie, M.J.; Novotny, W.F.; Peterson, O.W.; Vallon, V.; Malvey, K.; Mendonca, M.; Satriano, J.; Insel, P.; Thomson, S.C.; Blantz, R.C. Agmatine, a bioactive metabolite of arginine. Production, degradation, and functional effects in the kidney of the rat. J. Clin. Investig. 1996, 97, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Galea, E.; Feinstein, D.; Reis, D.J.; Regunathan, S. Metabolism of agmatine in macrophages: Modulation by lipopolysaccharide and inhibitory cytokines. Biochem. J. 1998, 330, 1405–1409. [Google Scholar] [CrossRef]
- Galea, E.; Regunathan, S.; Eliopoulos, V.; Feinstein, D.L.; Reis, D.J. Inhibition of mammalian nitric oxide synthases by agmatine, an endogenous polyamine formed by decarboxylation of arginine. Biochem. J. 1996, 316, 247–249. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, L.; Zhang, Z.; Wang, Y.; Lu, M.; LaPointe, M.; Chopp, M. A nitric oxide donor induces neurogenesis and reduces functional deficits after stroke in rats. Ann. Neurol. 2001, 50, 602–611. [Google Scholar] [CrossRef]
- Zhang, F.; Ladecola, C. Nitroprusside improves blood flow and reduces brain damage after focal ischemia. Neuroreport 1993, 4, 559–562. [Google Scholar] [CrossRef]
- Salom, J.B.; Ortí, M.; Centeno, J.M.; Torregrosa, G.; Alborch, E. Reduction of infarct size by the NO donors sodium nitroprusside and spermine/NO after transient focal cerebral ischemia in rats. Brain Res. 2000, 865, 149–156. [Google Scholar] [CrossRef]
- Zhang, F.; White, J.G.; Iadecola, C. Nitric oxide donors increase blood flow and reduce brain damage in focal ischemia: Evidence that nitric oxide is beneficial in the early stages of cerebral ischemia. J. Cereb. Blood Flow Metab. 1994, 14, 217–226. [Google Scholar] [CrossRef]
- Zhang, F.; Iadecola, C. Reduction of focal cerebral ischemic damage by delayed treatment with nitric oxide donors. J. Cereb. Blood Flow Metab. 1994, 14, 574–580. [Google Scholar] [CrossRef]
- Zhuang, P.; Ji, H.; Zhang, Y.H.; Min, Z.L.; Ni, Q.G.; You, R. ZJM-289, a novel nitric oxide donor, alleviates the cerebral ischaemic-reperfusion injury in rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, e121–e127. [Google Scholar] [CrossRef]
- Martínez-Murillo, R.; Fernández, A.P.; Serrano, J.; Rodrigo, J.; Salas, E.; Mourelle, M.; Martínez, A. The nitric oxide donor LA 419 decreases brain damage in a focal ischemia model. Neurosci. Lett. 2007, 415, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Serrano, J.; Fernández, A.P.; Martínez-Murillo, R.; Alonso, D.; Rodrigo, J.; Salas, E.; Mourelle, M.; Martínez, A. The nitric oxide donor LA 419 decreases ischemic brain damage. Int. J. Mol. Med. 2007, 19, 229–236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Butterworth, R.J.; Cluckie, A.; Jackson, S.H.D.; Buxton-Thomas, M.; Bath, P.M.W. Pathophysiological assessment of nitric oxide (given as sodium nitroprusside) in acute ischaemic stroke. Cerebrovasc. Dis. 1998, 8, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Rosenwasser, R.H.; Armonda, R.A.; Harrop, J.; Mitchell, W.; Galaria, I. Safety of intrathecal sodium nitroprusside for the treatment and prevention of refractory cerebral vasospasm and ischemia in humans. Stroke 1999, 30, 1409–1416. [Google Scholar] [CrossRef]
- Khan, M.; Jatana, M.; Elango, C.; Singh Paintlia, A.; Singh, A.K.; Singh, I. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide 2006, 15, 114–124. [Google Scholar] [CrossRef]
- Khan, M.; Sekhon, B.; Giri, S.; Jatana, M.; Gilg, A.G.; Ayasolla, K.; Elango, C.; Singh, A.K.; Singh, I. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J. Cereb. Blood Flow Metab. 2005, 25, 177–192. [Google Scholar] [CrossRef]
- Barakat, W.; Fahmy, A.; Askar, M.; El-Kannishy, S. Effectiveness of arginase inhibitors against experimentally induced stroke. Naunyn. Schmiedebergs. Arch. Pharmacol. 2018, 391, 603–612. [Google Scholar] [CrossRef]
- Jung, C.S.; Iuliano, B.A.; Harvey-White, J.; Espey, M.G.; Oldfield, E.H.; Pluta, R.M. Association between cerebrospinal fluid levels of asymmetric dimethyl-L-arginine, an endogenous inhibitor of endothelial nitric oxide synthase, and cerebral vasospasm in a primate model of subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Willmot, M.; Gibson, C.; Gray, L.; Murphya, S.; Batha, P.; Murphy, S.; Bath, P. Nitric oxide synthase inhibitors in experimental ischemic stroke and their effects on infarct size and cerebral blood flow: A systematic review. Free Radic. Biol. Med. 2005, 39, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Margaill, I.; Allix, M.; Boulu, R.G.; Plotkine, M. Dose- and time-dependence of L-NAME neuroprotection in transient focal cerebral ischaemia in rats. Br. J. Pharmacol. 1997, 120, 160–163. [Google Scholar] [CrossRef]
- Fukuda, S.; Hashimoto, N.; Naritomi, H.; Nagata, I.; Nozaki, K.; Kondo, S.; Kurino, M.; Kikuchi, H. Prevention of rat cerebral aneurysm formation by inhibition of nitric oxide synthase. Circulation 2000, 101, 2532–2538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Casey, R.M.; Ross, M.E.; Iadecola, C. Aminoguanidine ameliorates and L-arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke 1996, 27, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Kidd, G.A.; Hong, H.; Majid, A.; Kaufman, D.I.; Chen, A.F. Inhibition of brain GTP cyclohydrolase I and tetrahydrobiopterin attenuates cerebral infarction via reducing inducible NO synthase and peroxynitrite in ischemic stroke. Stroke 2005, 36, 2705–2711. [Google Scholar] [CrossRef]
- Yin, X.; Yan, J.; Hou, X.; Wu, S.; Zgang, G. Neuroprotection of S-nitrosoglutathione against ischemic injury by down-regulating Fas S-nitrosylation and downstream signaling. Neuroscience 2013, 248, 209–298. [Google Scholar] [CrossRef]
- Yu, L.-M.; Zhang, T.-Y.; Yin, X.-H.; Yang, Q.; Lu, F.; Yan, J.-Z.; Li, C. Denitrosylation of nNOS induced by cerebral ischemia-reperfusion contributes to nitrosylation of CaMKII and its inhibition of autophosphorylation in hippocampal CA1. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7674–7683. [Google Scholar]
- Khan, M.; Dhammu, T.S.; Qiao, F.; Kumar, P.; Singh, A.K.; Singh, I. S-Nitrosoglutathione Mimics the Beneficial Activity of Endothelial Nitric Oxide Synthase-Derived Nitric Oxide in a Mouse Model of Stroke. J. Stroke Cerebrovasc. Dis. 2019, 28, 104470. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.W.; Hao, L.Y.; Qia, S.H. Inhibition on the S-nitrosylation of MKK4 can protect hippocampal CA1 neurons in rat cerebral ischemia/reperfusion. Brain Res. Bull. 2016, 124, 123–128. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, C.; Wang, X.; Chen, L.; Ji, H.; Zhang, Y. (S)-ZJM-289, a nitric oxide-releasing derivative of 3-n-butylphthalide, protects against ischemic neuronal injury by attenuating mitochondrial dysfunction and associated cell death. Neurochem. Int. 2012, 60, 134–144. [Google Scholar] [CrossRef]
- Coert, B.A.; Anderson, R.E.; Meyer, F.B. A comparative study of the effects of two nitric oxide synthase inhibitors and two nitric oxide donors on temporary focal cerebral ischemia in the Wistar rat. J. Neurosurg. 1999, 90, 332–338. [Google Scholar] [CrossRef]
- Coert, B.A.; Anderson, R.E.; Meyer, F.B. Effects of the nitric oxide donor 3-morpholinosydnonimine (SIN-1) in focal cerebral ischemia dependent on intracellular brain pH. J. Neurosurg. 2002, 97, 914–921. [Google Scholar] [CrossRef]
- Zhang, P.; Guo, Z.; Xu, Y.; Li, Y.; Song, J. N-Butylphthalide (NBP) ameliorated cerebral ischemia reperfusion-induced brain injury via HGF-regulated TLR4/NF-κB signaling pathway. Biomed. Pharmacother. 2016, 83, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.H.; Kaku, T.; Hada, J.; Hayashi, Y. 7-Nitroindazole reduces nitric oxide concentration in rat hippocampus after transient forebrain ischemia. Eur. J. Pharmacol. 1999, 380, 117–121. [Google Scholar] [CrossRef]
- Pramila, B.; Kalaivani, P.; Anita, A.; Saravana, C. l-NAME combats excitotoxicity and recuperates neurological deficits in MCAO/R rats. Pharmacol. Biochem. Behav. 2015, 134, 246–253. [Google Scholar] [CrossRef]
- Nanri, K.; Montécot, C.; Springhetti, V.; Seylaz, J.; Pinard, E. The Selective Inhibitor of Neuronal Nitric Oxide Synthase, 7-Nitroindazole, Reduces the Delayed Neuronal Damage Due to Forebrain Ischemia in Rats. Stroke 1998, 29, 1248–1254. [Google Scholar] [CrossRef]
- Ding-Zhou, L.; Marchand-Verrecchia, C.; Croci, N.; Plotkine, M.; Margaill, I. L-NAME reduces infarction, neurological deficit and blood-brain barrier disruption following cerebral ischemia in mice. Eur. J. Pharmacol. 2002, 457, 137–146. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, Y.; Gu, Y.; Xu, C. Neuroprotective actions of aminoguanidine involve reduced the activation of calpain and caspase-3 in a rat model of stroke. Neurochem. Int. 2010, 56, 634–641. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Liu, S.H.; Sun, H.S.; Lu, Y.M. Expression of inducible nitric oxide synthase after focal cerebral ischemia stimulates neurogenesis in the adult rodent dentate gyrus. J. Neurosci. 2003, 23, 233–239. [Google Scholar] [CrossRef]
- Sugimoto, K.; Iadecola, C. Effects of aminoguanidine on cerebral ischemia in mice: Comparison between mice with and without inducible nitric oxide synthase gene. Neurosci. Lett. 2002, 331, 25–28. [Google Scholar] [CrossRef]
- Pérez-Asensio, F.J.; Hurtado, O.; Burguete, M.C.; Moro, M.A.; Salom, J.B.; Lizasoain, I.; Torregrosa, G.; Leza, J.C.; Alborch, E.; Castillo, J.; et al. Inhibition of iNOS activity by 1400W decreases glutamate release and ameliorates stroke outcome after experimental ischemia. Neurobiol. Dis. 2005, 18, 375–384. [Google Scholar] [CrossRef]
- Zheng, L.; Ding, J.; Wang, J.; Zhou, C.; Zhang, W. Effects and Mechanism of Action of Inducible Nitric Oxide Synthase on Apoptosis in a Rat Model of Cerebral Ischemia-Reperfusion Injury. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2016, 299, 246–255. [Google Scholar] [CrossRef]
- Nagel, S.; Papadakis, M.; Chen, R.; Hoyte, L.C.; Brooks, K.J.; Gallichan, D.; Sibson, N.R.; Pugh, C.; Buchan, A.M. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2011, 31, 132–143. [Google Scholar] [CrossRef]
- Prass, K.; Ruscher, K.; Karsch, M.; Isaev, N.; Megow, D.; Priller, J.; Scharff, A.; Dirnagl, U.; Meisel, A. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J. Cereb. Blood Flow Metab. 2002, 22, 520–525. [Google Scholar] [CrossRef]
- Reischl, S.; Li, L.; Walkinshaw, G.; Flippin, L.A.; Marti, H.H.; Kunze, R. Inhibition of HIF prolyl-4-hydroxylases by FG-4497 reduces brain tissue injury and edema formation during ischemic stroke. PLoS ONE 2014, 9, e84767. [Google Scholar] [CrossRef]
- Siddiq, A.; Ayoub, I.A.; Chavez, J.C.; Aminova, L.; Shah, S.; LaManna, J.C.; Patton, S.M.; Connor, J.R.; Cherny, R.A.; Volitakis, I.; et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition: A target for neuroprotection in the central nervous system. J. Biol. Chem. 2005, 280, 41732–41743. [Google Scholar] [CrossRef] [PubMed]
- Harten, S.K.; Ashcroft, M.; Maxwell, P.H. Prolyl hydroxylase domain inhibitors: A route to HIF activation and neuroprotection. Antioxid. Redox Signal. 2010, 12, 459–480. [Google Scholar] [CrossRef]
- Zaman, K.; Ryu, H.; Hall, D.; O’Donovan, K.; Lin, K.I.; Miller, M.P.; Marquis, J.C.; Baraban, J.M.; Semenza, G.L.; Ratan, R.R. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and ery. J. Neurosci. 1999, 19, 9821–9830. [Google Scholar] [CrossRef][Green Version]
- Yin, W.; Clare, K.; Zhang, Q.; Volkow, N.D.; Du, C. Chronic cocaine induces HIF-VEGF pathway activation along with angiogenesis in the brain. PLoS ONE 2017, 12, e0175499. [Google Scholar] [CrossRef]
- Zhang, B.; Tanaka, J.; Yang, L.; Yang, L.; Sakanaka, M.; Hata, R.; Maeda, N.; Mitsuda, N. Protective effect of vitamin E against focal brain ischemia and neuronal death through induction of target genes of hypoxia-inducible factor-1. Neuroscience 2004, 126, 433–440. [Google Scholar] [CrossRef]
- Chern, C.M.; Liou, K.T.; Wang, Y.H.; Liao, J.F.; Yen, J.C.; Shen, Y.C. Andrographolide inhibits PI3K/AKT-dependent NOX2 and iNOS expression protecting mice against hypoxia/ischemia-induced oxidative brain injury. Planta Med. 2011, 77, 1669–1679. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulationand destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Piret, J.-P.; Mottet, D.; Raes, M.; Michiels, C. Is HIF-1alpha apro- or an anti-apoptotic protein? Biochem. Pharmacol. 2002, 64, 889–892. [Google Scholar] [CrossRef]
- Chang, Y.; Hsieh, C.Y.; Peng, Z.A.; Yen, T.L.; Hsiao, G.; Chou, D.S.; Chen, C.M.; Sheu, J.R. Neuroprotective mechanisms of puerarin in middle cerebral artery occlusion-induced brain infarction in rats. J. Biomed. Sci. 2009, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hu, Q.; Yan, J.; Lei, J.; Qin, L.; Shi, X.; Luan, L.; Yang, L.; Wang, K.; Han, J.; et al. Multiple effects of 2ME2 and D609 on the cortical expression of HIF-1α and apoptotic genes in a middle cerebral artery occlusion-induced focal ischemia rat model. J. Neurochem. 2007, 102, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, M.; Gidday, J.M.; Aimee, Y.Y.; Semenza, G.L.; Ferriero, D.M.; Sharp, F.R. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann. Neurol. 2000, 48, 285–296. [Google Scholar] [CrossRef]
- Freret, T.; Valable, S.; Chazalviel, L.; Saulnier, R.; Mackenzie, E.T.; Petit, E.; Bernaudin, M.; Boulouard, M.; Schumann-Bard, P. Delayed administration of deferoxamine reduces brain damage and promotes functional recovery after transient focal cerebral ischemia in the rat. Eur. J. Neurosci. 2006, 23, 1757–1765. [Google Scholar] [CrossRef]
- Zhao, Y.; Rempe, D.A. Prophylactic neuroprotection against stroke: Low-dose, prolonged treatment with deferoxamine or deferasirox establishes prolonged neuroprotection independent of HIF-1 function. J. Cereb. Blood Flow Metab. 2011, 31, 1412–1423. [Google Scholar] [CrossRef]
- Sharp, F.R.; Bergeron, M.; Bernaudin, M. Hypoxia-inducible factor in brain. In Hypoxia. Advances in Experimental Medicine and Biology; Roach, R.C., Wagner, P.D., Hackett, P., Eds.; Springer: Boston, MA, USA, 2001; Volume 502, pp. 273–291. [Google Scholar]
- Jones, N.M.; Bergeron, M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J. Cereb. Blood Flow Metab. 2001, 21, 1105–1114. [Google Scholar] [CrossRef]
- Kovalenko, T.N.; Ushakova, G.A.; Osadchenko, I.; Skibo, G.G.; Pierzynowski, S.G. The neuroprotective effect of 2-oxoglutarate in the experimental ischemia of hippocampus. J. Physiol. Pharmacol. 2011, 62, 239–246. [Google Scholar] [PubMed]
- Zhou, J.; Li, J.; Rosenbaum, D.M.; Zhuang, J.; Poon, C.; Qin, P.; Rivera, K.; Lepore, J.; Willette, R.N.; Hu, E.; et al. The prolyl 4-hydroxylase inhibitor GSK360A decreases post-stroke brain injury and sensory, motor, and cognitive behavioral deficits. PLoS ONE 2017, 12, e0184049. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Wilson, J.W.; Schofield, C.J.; Chen, R. Hypoxia-inducible factor (HIF) prolyl hydroxylase inhibitors induce autophagy and have a protective effect in an in-vitro ischaemia model. Sci. Rep. 2020, 10, 1597. [Google Scholar] [CrossRef]
- Sakanaka, M.; Wen, T.C.; Matsuda, S.; Masuda, S.; Morishita, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, M.; Marti, H.H.; Roussel, S.; Divoux, D.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1999, 19, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Sirén, A.L.; Fratelli, M.; Brines, M.; Goemans, C.; Casagrande, S.; Lewczuk, P.; Keenan, S.; Gleiter, C.; Pasquali, C.; Capobianco, A.; et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc. Natl. Acad. Sci. USA 2001, 98, 4044–4049. [Google Scholar] [CrossRef] [PubMed]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; De Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef]
- Sadamoto, Y.; Igase, K.; Sakanaka, M.; Sato, K.; Otsuka, H.; Sakaki, S.; Masuda, S.; Sasaki, R. Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochem. Biophys. Res. Commun. 1998, 253, 26–32. [Google Scholar] [CrossRef]
- Gan, Y.; Xing, J.; Jing, Z.; Anne Stetler, R.; Zhang, F.; Luo, Y.; Ji, X.; Gao, Y.; Cao, G. Mutant erythropoietin without erythropoietic activity is neuroprotective against ischemic brain injury. Stroke 2012, 43, 3071–3077. [Google Scholar] [CrossRef]
- Zhu, L.; Bai, X.; Wang, S.; Hu, Y.; Wang, T.; Qian, L.; Jiang, L. Recombinant human erythropoietin augments angiogenic responses in a neonatal rat model of cerebral unilateral hypoxia-ischemia. Neonatology 2014, 106, 143–148. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Sinn, D.I.; Jung, K.H.; Kim, E.H.; Kim, S.J.; Kim, J.M.; Ko, S.Y.; Kim, M.; Roh, J.K. Erythropoietin reduces perihematomal inflammation and cell death with eNOS and STAT3 activations in experimental intracerebral hemorrhage. J. Neurochem. 2006, 96, 1728–1739. [Google Scholar] [CrossRef]
- Sun, Y.; Jin, K.; Xie, L.; Childs, J.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Investig. 2003, 111, 1843–1851. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Van Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef]
- Amin, N.; Chen, S.; Ren, Q.; Tan, X.; Botchway, B.O.A.; Hu, Z.; Chen, F.; Ye, S.; Du, X.; Chen, Z.; et al. Hypoxia Inducible Factor-1α Attenuates Ischemic Brain Damage by Modulating Inflammatory Response and Glial Activity. Cells 2021, 10, 1359. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhou, B.; Taheri, S.; Shi, H. Differential Effects of HIF-1 Inhibition by YC-1 on the Overall Outcome and Blood-Brain Barrier Damage in a Rat Model of Ischemic Stroke. PLoS ONE 2011, 6, e27798. [Google Scholar] [CrossRef] [PubMed]
- El Khashab, I.H.; Abdelsalam, R.M.; Elbrairy, A.I.; Attia, A.S. Chrysin attenuates global cerebral ischemic reperfusion injury via suppression of oxidative stress, inflammation and apoptosis. Biomed. Pharmacother. 2019, 112, 108619. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, L.; Xiao, J.; Wang, C.; Jiang, W.; Zhang, R.; Hao, J. Chrysin Protects against Focal Cerebral Ischemia/Reperfusion Injury in Mice through Attenuation of Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2014, 15, 20913–201926. [Google Scholar] [CrossRef]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF-1α inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol. Dis. 2008, 31, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhou, J.; Li, X.; Xiao, Y.; Zhang, J.; Yang, Y.; Feng, L.; Kang, Y.J. The Association of Suppressed Hypoxia-Inducible Factor-1 Transactivation of Angiogenesis With Defective Recovery From Cerebral Ischemic Injury in Aged Rats. Front. Aging Neurosci. 2021, 13, 648115. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Hasselblatt, M.; Dembowski, C.; Cepek, L.; Lewczuk, P.; Stiefel, M.; Rustenbeck, H.H.; Breiter, N.; Jacob, S.; Knerlich, F.; et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol. Med. 2002, 8, 495–505. [Google Scholar] [CrossRef]
- Tsai, T.H.; Lu, C.H.; Wallace, C.G.; Chang, W.N.; Chen, S.F.; Huang, C.R.; Tsai, N.W.; Lan, M.Y.; Sung, P.H.; Liu, C.F.; et al. Erythropoietin improves long-term neurological outcome in acute ischemic stroke patients: A randomized, prospective, placebo-controlled clinical trial. Crit. Care 2015, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Bath, P.M.W.; Bayraktutan, U. Current therapeutic strategies to mitigate the eNOS dysfunction in ischaemic stroke. Cell. Mol. Neurobiol. 2012, 32, 319–336. [Google Scholar] [CrossRef]
- Zhao, X.; Strong, R.; Piriyawat, P.; Palusinski, R.; Grotta, J.C.; Aronowski, J. Caffeinol at the receptor level: Anti-ischemic effect of n-methyl-d-aspartate receptor blockade is potentiated by caffeine. Stroke 2010, 41, 363–367. [Google Scholar] [CrossRef]
- Dong, Z.S.W.; Cao, Z.P.; Shang, Y.J.; Liu, Q.Y.; Wu, B.Y.; Liu, W.X.; Li, C.H. Neuroprotection of cordycepin in NMDA-induced excitotoxicity by modulating adenosine A1 receptors. Eur. J. Pharmacol. 2019, 853, 325–335. [Google Scholar] [CrossRef]
- Imai, K.; Mori, T.; Izumoto, H.; Takabatake, N.; Kunieda, T.; Watanabe, M. Hyperbaric oxygen combined with intravenous edaravone for treatment of acute embolic stroke: A pilot clinical trial. Neurol. Med. Chir. 2006, 46, 373–378. [Google Scholar] [CrossRef][Green Version]
- Kimura, K.; Aoki, J.; Sakamoto, Y.; Kobayashi, K.; Sakai, K.; Inoue, T.; Iguchi, Y.; Shibazaki, K. Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients—A preliminary study. J. Neurol. Sci. 2012, 313, 132–136. [Google Scholar] [CrossRef]
- Aoki, J.; Kimura, K.; Morita, N.; Harada, M.; Metoki, N.; Tateishi, Y.; Todo, K.; Yamagami, H.; Hayashi, K.; Terasawa, Y.; et al. YAMATO Study (Tissue-Type Plasminogen Activator and Edaravone Combination Therapy). Stroke 2017, 48, 712–719. [Google Scholar] [CrossRef]
- Grotta, J.; Combination Therapy Stroke Trial Investigators. Combination therapy stroke trial: Recombinant tissue-type plasminogen activator with/without lubeluzole. Cerebrovasc. Dis. 2001, 12, 258–263. [Google Scholar] [CrossRef]
- Montaner, J.; Bustamante, A.; García-Matas, S.; Martínez-Zabaleta, M.; Jiménez, C.; De La Torre, J.; Rubio, F.R.; Segura, T.; Masjuán, J.; Cánovas, D.; et al. Combination of thrombolysis and statins in acute stroke is safe: Results of the STARS randomized trial (Stroke Treatment With Acute Reperfusion and Simvastatin). Stroke 2016, 47, 2870–2873. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Chandra, A.; Geng, X.; Cheng, Z.; Tong, Y.; Du, H.; Ding, Y. Low dose concomitant treatment with chlorpromazine and promethazine is safe in acute ischemic stroke. J. Neurosurg. Sci. 2019, 63, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Vellimana, A.K.; Milner, E.; Azad, T.D.; Harries, M.D.; Zhou, M.-L.L.; Gidday, J.M.; Han, B.H.; Zipfel, G.J. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage-induced cerebral vasospasm. Stroke 2011, 42, 776–782. [Google Scholar] [CrossRef]
- Chen, S.; Li, N.; Deb-Chatterji, M.; Dong, Q.; Kielstein, J.T.; Weissenborn, K.; Worthmann, H. Asymmetric Dimethyarginine as marker and mediator in Ischemic stroke. Int. J. Mol. Sci. 2012, 13, 15983–16004. [Google Scholar] [CrossRef] [PubMed]



| Compound | Effect | ||
|---|---|---|---|
| HIF-1α Stabilizing Agents | |||
| Pretreatment | Deferoxamine (DFO)-iron chelator | in vivo:
| [101,187,200,201,202,203] |
| Cobalt chloride (CoCl2) | in vivo:
| [200,203,204] | |
Prolyl hydroxylase (PHD) inhibitors
| in vivo:
| [87,186,188,189,205,206,207] | |
| Posttreatment | Prolyl hydroxylase inhibitors
| in vivo:
| [188,205] |
| HIF-1α dependent proteins | |||
| Pretreatment | Erythropoietin (EPO) | in vivo:
| [208,209,210,211] |
| Posttreatment | Erythropoietin (EPO), or its analogs (MEPO, S104I-EPO) | in vivo:
| [211,212,213,214,215] |
| Vascular endothelial growth factor (VEGF) | in vivo:
| [216,217] | |
| Direct and indirect HIF-1α inhibitors | |||
| Pretreatment | Acriflavine | in vivo:
| [218] |
| 2,2,5,7,8-Pentamethyl-6-hydroxychromane (PMC) | in vivo:
| [105] | |
| YC-1 | in vivo:
| [219] | |
| Chrysin | in vivo:
| [220,221] | |
| Posttreatment | 2ME2 | in vivo:
| [199,222] |
| D609 | in vivo:
| [199] | |
| Chetomin | in vivo:
| [223] | |
| Clinical trials | |||
| rhEPO (double-blind placebo controlled proof-of-concept trial; i.v.) |
| [224] | |
| EPO (prospective, randomized, placebo-controlled trial; s.c.) |
| [225] |
| Mode of Action | Combined Treatment | Effect | |
|---|---|---|---|
| Free radical scavenger | Edaravone + Hyperbaric oxygen + Heparin (pilot trial) |
| [229] |
| Edaravone + t-PA (pilot trial) |
| [230] | |
| Edaravone + t-PA (multicenter, prospective, randomized and open-label trial) |
| [231] | |
| NMDA antagonist, VGSC and VGCC blocker, inhibitor of NO synthesis | Lubeluzole + t-PA (feasibility, safety and efficacy trial—uncompleted) |
| [232] |
| Statin | Simvastatin + t-PA (phase IV, prospective, randomized, double-blind, placebo-controlled trial) |
| [233] |
| Typical antipsychotic + First-generation antihistamine | Chlorpromazine + Promethazine + SOC (pilot trial) |
| [234] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wierońska, J.M.; Cieślik, P.; Kalinowski, L. Nitric Oxide-Dependent Pathways as Critical Factors in the Consequences and Recovery after Brain Ischemic Hypoxia. Biomolecules 2021, 11, 1097. https://doi.org/10.3390/biom11081097
Wierońska JM, Cieślik P, Kalinowski L. Nitric Oxide-Dependent Pathways as Critical Factors in the Consequences and Recovery after Brain Ischemic Hypoxia. Biomolecules. 2021; 11(8):1097. https://doi.org/10.3390/biom11081097
Chicago/Turabian StyleWierońska, Joanna M, Paulina Cieślik, and Leszek Kalinowski. 2021. "Nitric Oxide-Dependent Pathways as Critical Factors in the Consequences and Recovery after Brain Ischemic Hypoxia" Biomolecules 11, no. 8: 1097. https://doi.org/10.3390/biom11081097
APA StyleWierońska, J. M., Cieślik, P., & Kalinowski, L. (2021). Nitric Oxide-Dependent Pathways as Critical Factors in the Consequences and Recovery after Brain Ischemic Hypoxia. Biomolecules, 11(8), 1097. https://doi.org/10.3390/biom11081097