
Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. eNOS and Its Regulation
eNOS Uncoupling
3. Hypoxia and Cardiovascular Diseases
4. Influence of Hypoxia on eNOS Expression
5. eNOS Activity in Hypoxia
6. eNOS Uncoupling Elicited by Hypoxia
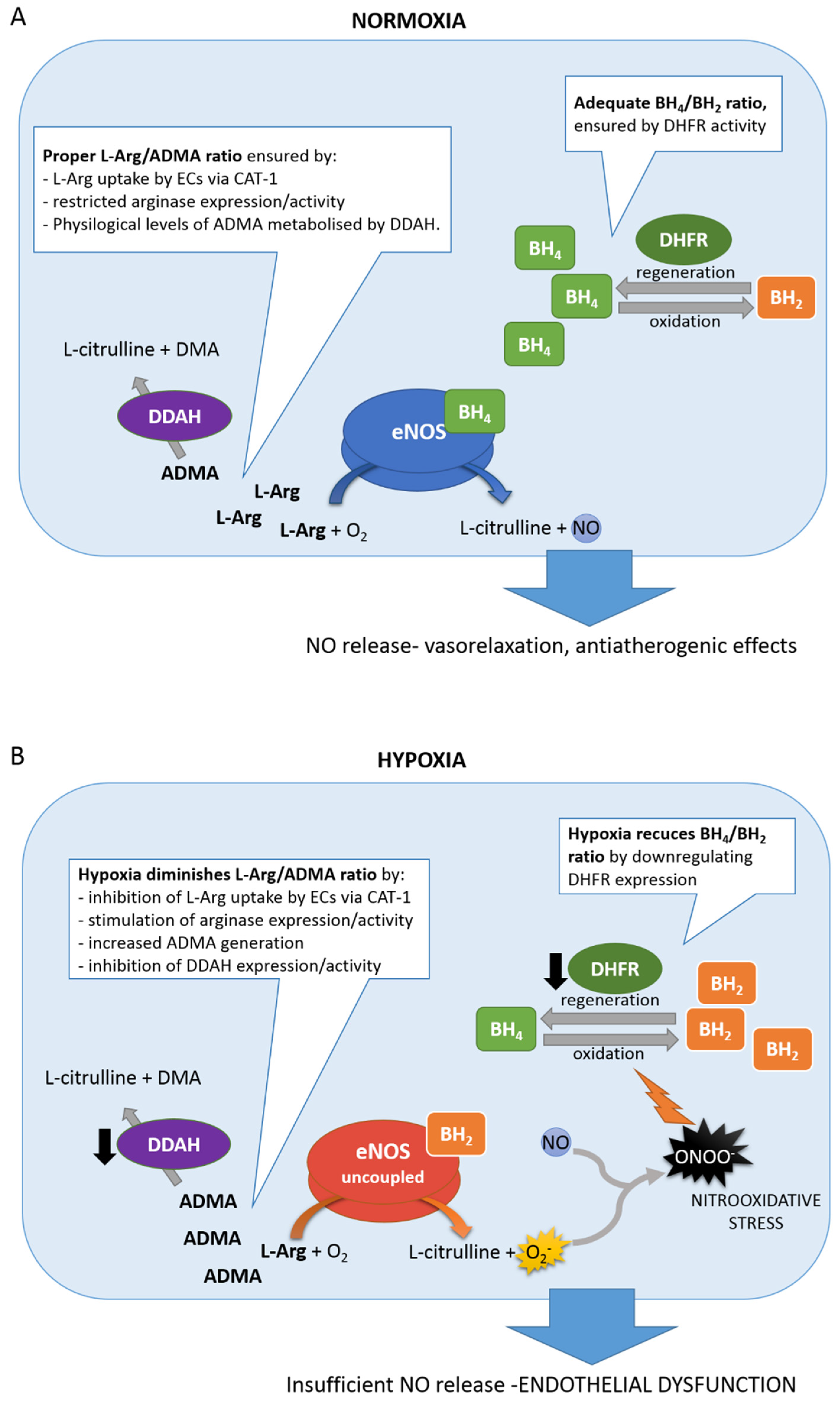
6.1. BH4/BH2 Ratio
6.2. L-Arg/ADMA Ratio
7. Hypoxia, Oxidative Stress and Endothelial Inflammation
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. BJA Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Chan, N. Endothelial function and nitric oxide: Clinical relevance. Heart 2001, 85, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Luks, A.M.; Swenson, E.R.; Bärtsch, P. Acute high-altitude sickness. Eur. Respir. Rev. 2017, 26, 160096. [Google Scholar] [CrossRef]
- Marchetti, M. COVID-19-driven endothelial damage: Complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann. Hematol. 2020, 99, 1701–1707. [Google Scholar] [CrossRef]
- Tuder, R.M.; Yun, J.H.; Bhunia, A.; Fijalkowska, I. Hypoxia and chronic lung disease. J. Mol. Med. 2007, 85, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Garvey, J.F.; Taylor, C.T.; McNicholas, W.T. Cardiovascular disease in obstructive sleep apnoea syndrome: The role of intermittent hypoxia and inflammation. Eur. Respir. J. 2009, 33, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- López-Barneo, J.; González-Rodríguez, P.; Gao, L.; Fernández-Agüera, M.C.; Pardal, R.; Ortega-Sáenz, P. Oxygen sensing by the carotid body: Mechanisms and role in adaptation to hypoxia. Am. J. Physiol. Physiol. 2016, 310, C629–C642. [Google Scholar] [CrossRef]
- Semenza, G.L.; Agani, F.; Feldser, D.; Iyer, N.; Kotch, L.; Laughner, E.; Yu, A. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv. Exp. Med. Biol. 2000, 475, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sforza, E.; Roche, F. Chronic intermittent hypoxia and obstructive sleep apnea: An experimental and clinical approach. Hypoxia (Auckl. N. Z.) 2016, 4, 99–108. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Physiol. 2002, 282, C227–C241. [Google Scholar] [CrossRef]
- Dewan, N.A.; Nieto, F.J.; Somers, V.K. Intermittent hypoxemia and OSA: Implications for comorbidities. Chest 2015, 147, 266–274. [Google Scholar] [CrossRef]
- Hunyor, I.; Cook, K.M. Models of intermittent hypoxia and obstructive sleep apnea: Molecular pathways and their contribution to cancer. Am. J. Physiol. Integr. Comp. Physiol. 2018, 315, R669–R687. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Ravenna, L.; Salvatori, L.; Russo, M.A. HIF3α: The little we know. FEBS J. 2016, 283, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial cell–specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor–2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Clin. Investig. 2007, 117, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Matouk, C.C.; Rachlis, A.; Lin, S.; Tai, S.C.; D’Abreo, C.; Marsden, P.A. The Expression of Endothelial Nitric-oxide Synthase Is Controlled by a Cell-specific Histone Code*. J. Biol. Chem. 2005, 280, 24824–24838. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.; Fish, J.E.; D’Abreo, C.; Lin, S.; Robb, G.B.; Teichert, A.-M.; Karantzoulis-Fegaras, F.; Keightley, A.; Steer, B.M.; Marsden, P.A. The Cell-specific Expression of Endothelial Nitric-oxide Synthase: A ROLE FOR DNA METHYLATION*. J. Biol. Chem. 2004, 279, 35087–35100. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef]
- Hill, B.G.; Dranka, B.P.; Bailey, S.M.; Lancaster, J.R.J.; Darley-Usmar, V.M. What part of NO don’t you understand? Some answers to the cardinal questions in nitric oxide biology. J. Biol. Chem. 2010, 285, 19699–19704. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W.S. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Blau, N.; Thöny, B. Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J. 2011, 438, 397–414. [Google Scholar] [CrossRef]
- Searles, C.D. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol. Cell Physiol. 2006, 291, C803–C816. [Google Scholar] [CrossRef]
- Dudzinski, D.M.; Michel, T. Life history of eNOS: Partners and pathways. Cardiovasc. Res. 2007, 75, 247–260. [Google Scholar] [CrossRef]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef]
- Mineo, C.; Shaul, P.W. Regulation of eNOS in caveolae. Adv. Exp. Med. Biol. 2012, 729, 51–62. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. eNOS phosphorylation in health and disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Investig. 2005, 85, 9–23. [Google Scholar] [CrossRef]
- Balligand, J.-L.; Feron, O.; Dessy, C. eNOS Activation by Physical Forces: From Short-Term Regulation of Contraction to Chronic Remodeling of Cardiovascular Tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef]
- Duckles, S.P.; Miller, V.M. Hormonal modulation of endothelial NO production. Pflugers Arch. 2010, 459, 841–851. [Google Scholar] [CrossRef]
- Montagnani, M.; Chen, H.; Barr, V.A.; Quon, M.J. Insulin-stimulated Activation of eNOS Is Independent of Ca2+ but Requires Phosphorylation by Akt at Ser1179*. J. Biol. Chem. 2001, 276, 30392–30398. [Google Scholar] [CrossRef]
- Fisslthaler, B.; Benzing, T.; Busse, R.; Fleming, I. Insulin enhances the expression of the endothelial nitric oxide synthase in native endothelial cells: A dual role for Akt and AP-1. Nitric Oxide 2003, 8, 253–261. [Google Scholar] [CrossRef]
- Hiroi, Y.; Kim, H.-H.; Ying, H.; Furuya, F.; Huang, Z.; Simoncini, T.; Noma, K.; Ueki, K.; Nguyen, N.-H.; Scanlan, T.S.; et al. Rapid nongenomic actions of thyroid hormone. Proc. Natl. Acad. Sci. USA 2006, 103, 14104–14109. [Google Scholar] [CrossRef]
- Gaynullina, D.K.; Schubert, R.; Tarasova, O.S. Changes in Endothelial Nitric Oxide Production in Systemic Vessels during Early Ontogenesis-A Key Mechanism for the Perinatal Adaptation of the Circulatory System. Int. J. Mol. Sci. 2019, 20, 1421. [Google Scholar] [CrossRef]
- Hisamoto, K.; Ohmichi, M.; Kurachi, H.; Hayakawa, J.; Kanda, Y.; Nishio, Y.; Adachi, K.; Tasaka, K.; Miyoshi, E.; Fujiwara, N.; et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 2001, 276, 3459–3467. [Google Scholar] [CrossRef]
- Sumi, D.; Ignarro, L.J. Estrogen-related receptor α1 up-regulates endothelial nitric oxide synthase expression. Proc. Natl. Acad. Sci. USA 2003, 100, 14451–14456. [Google Scholar] [CrossRef]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 20, 295–302. [Google Scholar] [CrossRef]
- Quesada, A.; Sainz, J.; Wangensteen, R.; Rodriguez-Gomez, I.; Vargas, F.; Osuna, A. Nitric oxide synthase activity in hyperthyroid and hypothyroid rats. Eur. J. Endocrinol. 2002, 147, 117–122. [Google Scholar] [CrossRef]
- Yang, S.; Bae, L.; Zhang, L. Estrogen increases eNOS and NOx release in human coronary artery endothelium. J. Cardiovasc. Pharmacol. 2000, 36, 242–247. [Google Scholar] [CrossRef]
- Dore-Duffy, P.; Balabanov, R.; Beaumont, T.; Hritz, M.A.; Harik, S.I.; LaManna, J.C. Endothelial Activation Following Prolonged Hypobaric Hypoxia. Microvasc. Res. 1999, 57, 75–85. [Google Scholar] [CrossRef]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Shaul, P.W. Estrogen modulation of endothelial nitric oxide synthase. Endocr. Rev. 2002, 23, 665–686. [Google Scholar] [CrossRef]
- Averna, M.; Stifanese, R.; De Tullio, R.; Passalacqua, M.; Salamino, F.; Pontremoli, S.; Melloni, E. Functional role of HSP90 complexes with endothelial nitric-oxide synthase (eNOS) and calpain on nitric oxide generation in endothelial cells. J. Biol. Chem. 2008, 283, 29069–29076. [Google Scholar] [CrossRef]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef]
- Luo, S.; Lei, H.; Qin, H.; Xia, Y. Molecular mechanisms of endothelial NO synthase uncoupling. Curr. Pharm. Des. 2014, 20, 3548–3553. [Google Scholar] [CrossRef]
- Kalinowski, L.; Malinski, T. Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: Relationship to endothelial dysfunction. Acta Biochim. Pol. 2004, 51, 459–469. [Google Scholar] [CrossRef]
- Kalinowski, L.; Dobrucki, I.T.; Malinski, T. Race-specific differences in endothelial function: Predisposition of African Americans to vascular diseases. Circulation 2004, 109, 2511–2517. [Google Scholar] [CrossRef]
- Dobrucki, L.W.; Marsh, B.J.; Kalinowski, L. Elucidating structure-function relationships from molecule-to-cell-to-tissue: From research modalities to clinical realities. J. Physiol. Pharmacol. Off. J. Polish Physiol. Soc. 2009, 60 (Suppl. 4), 83–93. [Google Scholar]
- Chen, P.-S.; Chiu, W.-T.; Hsu, P.-L.; Lin, S.-C.; Peng, I.-C.; Wang, C.-Y.; Tsai, S.-J. Pathophysiological implications of hypoxia in human diseases. J. Biomed. Sci. 2020, 27, 63. [Google Scholar] [CrossRef]
- Barberà, J.A.; Peinado, V.I.; Santos, S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur. Respir. J. 2003, 21, 892–905. [Google Scholar] [CrossRef]
- Sajkov, D.; McEvoy, R.D. Obstructive Sleep Apnea and Pulmonary Hypertension. Prog. Cardiovasc. Dis. 2009, 51, 363–370. [Google Scholar] [CrossRef]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.K.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J. Clin. Investig. 1999, 103, 691–696. [Google Scholar] [CrossRef]
- Brusselmans, K.; Compernolle, V.; Tjwa, M.; Wiesener, M.S.; Maxwell, P.H.; Collen, D.; Carmeliet, P. Heterozygous deficiency of hypoxia-inducible factor–2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Investig. 2003, 111, 1519–1527. [Google Scholar] [CrossRef]
- Tonelli, A.R.; Haserodt, S.; Aytekin, M.; Dweik, R.A. Nitric oxide deficiency in pulmonary hypertension: Pathobiology and implications for therapy. Pulm. Circ. 2013, 3, 20–30. [Google Scholar] [CrossRef]
- Kaneko, F.T.; Arroliga, A.C.; Dweik, R.A.; Comhair, S.A.; Laskowski, D.; Oppedisano, R.; Thomassen, M.J.; Erzurum, S.C. Biochemical Reaction Products of Nitric Oxide as Quantitative Markers of Primary Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 1998, 158, 917–923. [Google Scholar] [CrossRef]
- Budhiraja, R.; Parthasarathy, S.; Quan, S.F. Endothelial dysfunction in obstructive sleep apnea. J. Clin. Sleep Med. 2007, 3, 409–415. [Google Scholar] [CrossRef]
- Ip, M.S.; Lam, B.; Chan, L.Y.; Zheng, L.; Tsang, K.W.; Fung, P.C.; Lam, W.K. Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am. J. Respir. Crit. Care Med. 2000, 162, 2166–2171. [Google Scholar] [CrossRef]
- Schulz, R.; Schmidt, D.; Blum, A.; Lopes-Ribeiro, X.; Lücke, C.; Mayer, K.; Olschewski, H.; Seeger, W.; Grimminger, F. Decreased plasma levels of nitric oxide derivatives in obstructive sleep apnoea: Response to CPAP therapy. Thorax 2000, 55, 1046–1051. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kato, M.; Roberts-Thomson, P.; Phillips, B.G.; Haynes, W.G.; Winnicki, M.; Accurso, V.; Somers, V.K. Impairment of Endothelium-Dependent Vasodilation of Resistance Vessels in Patients with Obstructive Sleep Apnea. Circulation 2000, 102, 2607–2610. [Google Scholar] [CrossRef]
- Hultén, L.M.; Levin, M. The role of hypoxia in atherosclerosis. Curr. Opin. Lipidol. 2009, 20, 409–414. [Google Scholar] [CrossRef]
- Björnheden, T.; Levin, M.; Evaldsson, M.; Wiklund, O. Evidence of Hypoxic Areas Within the Arterial Wall In Vivo. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 870–876. [Google Scholar] [CrossRef]
- Sluimer, J.C.; Gasc, J.-M.; van Wanroij, J.L.; Kisters, N.; Groeneweg, M.; Sollewijn Gelpke, M.D.; Cleutjens, J.P.; van den Akker, L.H.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, Hypoxia-Inducible Transcription Factor, and Macrophages in Human Atherosclerotic Plaques Are Correlated With Intraplaque Angiogenesis. J. Am. Coll. Cardiol. 2008, 51, 1258–1265. [Google Scholar] [CrossRef]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Fish, J.E.; Yan, M.S.; Matouk, C.C.; St Bernard, R.; Ho, J.J.D.; Gavryushova, A.; Srivastava, D.; Marsden, P.A. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J. Biol. Chem. 2010, 285, 810–826. [Google Scholar] [CrossRef]
- Fish, J.E.; Matouk, C.C.; Yeboah, E.; Bevan, S.C.; Khan, M.; Patil, K.; Ohh, M.; Marsden, P.A. Hypoxia-inducible expression of a natural cis-antisense transcript inhibits endothelial nitric-oxide synthase. J. Biol. Chem. 2007, 282, 15652–15666. [Google Scholar] [CrossRef]
- McQuillan, L.P.; Leung, G.K.; Marsden, P.A.; Kostyk, S.K.; Kourembanas, S. Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am. J. Physiol. Circ. Physiol. 1994, 267, H1921–H1927. [Google Scholar] [CrossRef] [PubMed]
- Janaszak-Jasiecka, A.; Siekierzycka, A.; Bartoszewska, S.; Serocki, M.; Dobrucki, L.W.; Collawn, J.F.; Kalinowski, L.; Bartoszewski, R. eNOS expression and NO release during hypoxia is inhibited by miR-200b in human endothelial cells. Angiogenesis 2018, 21, 711–724. [Google Scholar] [CrossRef]
- Ho, J.J.D.; Robb, G.B.; Tai, S.C.; Turgeon, P.J.; Mawji, I.A.; Man, H.S.J.; Marsden, P.A. Active stabilization of human endothelial nitric oxide synthase mRNA by hnRNP E1 protects against antisense RNA and microRNAs. Mol. Cell. Biol. 2013, 33, 2029–2046. [Google Scholar] [CrossRef] [PubMed]
- Olszewska-Pazdrak, B.; Hein, T.W.; Olszewska, P.; Carney, D.H. Chronic hypoxia attenuates VEGF signaling and angiogenic responses by downregulation of KDR in human endothelial cells. Am. J. Physiol. Physiol. 2009, 296, C1162–C1170. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Zulueta, J.J.; Yu, F.S.; Peng, H.B.; Cote, C.G.; Hassoun, P.M. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J. Clin. Investig. 1995, 96, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, M.; Sun, J.; Hiroki, J.; Shimokawa, H.; Liao, J.K. Rho-Kinase Mediates Hypoxia-Induced Downregulation of Endothelial Nitric Oxide Synthase. Circulation 2002, 106, 57–62. [Google Scholar] [CrossRef]
- Giaid, A.; Saleh, D. Reduced Expression of Endothelial Nitric Oxide Synthase in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Wang, B.; Yan, B.; Song, D.; Ye, X.; Liu, S.F. Chronic intermittent hypoxia down-regulates endothelial nitric oxide synthase expression by an NF-κB-dependent mechanism. Sleep Med. 2013, 14, 165–171. [Google Scholar] [CrossRef]
- Vega-Tapia, F.; Peñaloza, E.; Krause, B.J. Specific arterio-venous transcriptomic and ncRNA-RNA interactions in human umbilical endothelial cells: A meta-analysis. iScience 2021, 24, 102675. [Google Scholar] [CrossRef]
- Krause, B.J.; Costello, P.M.; Muñoz-Urrutia, E.; Lillycrop, K.A.; Hanson, M.A.; Casanello, P. Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics 2013, 8, 944–952. [Google Scholar] [CrossRef]
- Peñaloza, E.; Soto-Carrasco, G.; Krause, B.J. MiR-21-5p directly contributes to regulating eNOS expression in human artery endothelial cells under normoxia and hypoxia. Biochem. Pharmacol. 2020, 182, 114288. [Google Scholar] [CrossRef]
- Sugimoto, K.; Yokokawa, T.; Misaka, T.; Nakazato, K.; Ishida, T.; Takeishi, Y. Senescence Marker Protein 30 Deficiency Exacerbates Pulmonary Hypertension in Hypoxia-Exposed Mice. Int. Heart J. 2019, 60, 1430–1434. [Google Scholar] [CrossRef]
- Xu, X.-F.; Ma, X.-L.; Shen, Z.; Wu, X.-L.; Cheng, F.; Du, L.-Z. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 2010, 28, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Gloe, T.; Pohl, U. Hypoxia-induced upregulation of eNOS gene expression is redox-sensitive: A comparison between hypoxia and inhibitors of cell metabolism. J. Cell. Physiol. 2001, 188, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Bird, I.M.; Magness, R.R.; Longo, L.D.; Zhang, L. Upregulation of eNOS in pregnant ovine uterine arteries by chronic hypoxia. Am. J. Physiol. Circ. Physiol. 2001, 280, H812–H820. [Google Scholar] [CrossRef]
- Su, Y.; Block, E.R. Role of calpain in hypoxic inhibition of nitric oxide synthase activity in pulmonary endothelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2000, 278, L1204–L1212. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Aschner, J.L.; Kaplowitz, M.R.; Summar, M.; Fike, C.D. Tetrahydrobiopterin oral therapy recouples eNOS and ameliorates chronic hypoxia-induced pulmonary hypertension in newborn pigs. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L743–L753. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L1096–L1100. [Google Scholar] [CrossRef]
- Kheirandish-Gozal, L.; Khalyfa, A.; Gozal, D.; Bhattacharjee, R.; Wang, Y. Endothelial dysfunction in children with obstructive sleep apnea is associated with epigenetic changes in the eNOS gene. Chest 2013, 143, 971–977. [Google Scholar] [CrossRef]
- Kalinowski, L.; Janaszak-Jasiecka, A.; Siekierzycka, A.; Bartoszewska, S.; Woźniak, M.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. Posttranscriptional and transcriptional regulation of endothelial nitric-oxide synthase during hypoxia: The role of microRNAs. Cell. Mol. Biol. Lett. 2016, 21, 16. [Google Scholar] [CrossRef]
- Sun, H.-X.; Zeng, D.-Y.; Li, R.-T.; Pang, R.-P.; Yang, H.; Hu, Y.-L.; Zhang, Q.; Jiang, Y.; Huang, L.-Y.; Tang, Y.-B.; et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension 2012, 60, 1407–1414. [Google Scholar] [CrossRef]
- Rippe, C.; Blimline, M.; Magerko, K.A.; Lawson, B.R.; LaRocca, T.J.; Donato, A.J.; Seals, D.R. MicroRNA changes in human arterial endothelial cells with senescence: Relation to apoptosis, eNOS and inflammation. Exp. Gerontol. 2012, 47, 45–51. [Google Scholar] [CrossRef]
- Zhang, W.; Yan, L.; Li, Y.; Chen, W.; Hu, N.; Wang, H.; Ou, H. Roles of miRNA-24 in regulating endothelial nitric oxide synthase expression and vascular endothelial cell proliferation. Mol. Cell. Biochem. 2015, 405, 281–289. [Google Scholar] [CrossRef]
- Sugimoto, M.; Nakayama, M.; Goto, T.M.; Amano, M.; Komori, K.; Kaibuchi, K. Rho-kinase phosphorylates eNOS at threonine 495 in endothelial cells. Biochem. Biophys. Res. Commun. 2007, 361, 462–467. [Google Scholar] [CrossRef]
- Toporsian, M.; Govindaraju, K.; Nagi, M.; Eidelman, D.; Thibault, G.; Ward, M.E. Downregulation of Endothelial Nitric Oxide Synthase in Rat Aorta After Prolonged Hypoxia In Vivo. Circ. Res. 2000, 86, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Jelic, S.; Padeletti, M.; Kawut, S.M.; Higgins, C.; Canfield, S.M.; Onat, D.; Colombo, P.C.; Basner, R.C.; Factor, P.; LeJemtel, T.H. Inflammation, Oxidative Stress, and Repair Capacity of the Vascular Endothelium in Obstructive Sleep Apnea. Circulation 2008, 117, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Arnet, U.A.; McMillan, A.; Dinerman, J.L.; Ballermann, B.; Lowenstein, C.J. Regulation of Endothelial Nitric-oxide Synthase during Hypoxia. J. Biol. Chem. 1996, 271, 15069–15073. [Google Scholar] [CrossRef] [PubMed]
- Le Cras, T.D.; Xue, C.; Rengasamy, A.; Johns, R.A. Chronic hypoxia upregulates endothelial and inducible NO synthase gene and protein expression in rat lung. Am. J. Physiol. 1996, 270, L164–L170. [Google Scholar] [CrossRef]
- Le Cras, T.D.; Tyler, R.C.; Horan, M.P.; Morris, K.G.; Tuder, R.M.; McMurtry, I.F.; Johns, R.A.; Abman, S.H. Effects of chronic hypoxia and altered hemodynamics on endothelial nitric oxide synthase expression in the adult rat lung. J. Clin. Investig. 1998, 101, 795–801. [Google Scholar] [CrossRef][Green Version]
- Ostergaard, L.; Stankevicius, E.; Andersen, M.R.; Eskildsen-Helmond, Y.; Ledet, T.; Mulvany, M.J.; Simonsen, U. Diminished NO release in chronic hypoxic human endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2894–H2903. [Google Scholar] [CrossRef]
- Ghosh, S.; Gupta, M.; Xu, W.; Mavrakis, D.A.; Janocha, A.J.; Comhair, S.A.A.; Haque, M.M.; Stuehr, D.J.; Yu, J.; Polgar, P.; et al. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L1199–L1205. [Google Scholar] [CrossRef]
- Murata, T.; Sato, K.; Hori, M.; Ozaki, H.; Karaki, H. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J. Biol. Chem. 2002, 277, 44085–44092. [Google Scholar] [CrossRef]
- Thöny, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347, 1–16. [Google Scholar] [CrossRef]
- Wang, S.; Xu, J.; Song, P.; Wu, Y.; Zhang, J.; Chul Choi, H.; Zou, M.-H. Acute inhibition of guanosine triphosphate cyclohydrolase 1 uncouples endothelial nitric oxide synthase and elevates blood pressure. Hypertension 2008, 52, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Moens, A.L.; Kass, D.A. Tetrahydrobiopterin and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2439–2444. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Levy, B.D.; Michel, T. Tetrahydrobiopterin recycling, a key determinant of endothelial nitric-oxide synthase-dependent signaling pathways in cultured vascular endothelial cells. J. Biol. Chem. 2009, 284, 12691–12700. [Google Scholar] [CrossRef]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef]
- Chalupsky, K.; Kračun, D.; Kanchev, I.; Bertram, K.; Görlach, A. Folic Acid Promotes Recycling of Tetrahydrobiopterin and Protects Against Hypoxia-Induced Pulmonary Hypertension by Recoupling Endothelial Nitric Oxide Synthase. Antioxid. Redox Signal. 2015, 23, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Francis, B.N.; Hale, A.; Channon, K.M.; Wilkins, M.R.; Zhao, L. Effects of tetrahydrobiopterin oral treatment in hypoxia-induced pulmonary hypertension in rat. Pulm. Circ. 2014, 4, 462–470. [Google Scholar] [CrossRef]
- Koubský, K.; Ďurišová, J.; Miková, D.; Herget, J. Chronic hypoxia inhibits tetrahydrobiopterin-induced NO production in rat lungs. Respir. Physiol. Neurobiol. 2013, 185, 547–552. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef]
- Rajapakse, N.W.; Mattson, D.L. Role of L-arginine in nitric oxide production in health and hypertension. Clin. Exp. Pharmacol. Physiol. 2009, 36, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.A.; May, J.M. Coordinate regulation of L-arginine uptake and nitric oxide synthase activity in cultured endothelial cells. Free Radic. Biol. Med. 2002, 32, 122–131. [Google Scholar] [CrossRef]
- Bode-Böger, S.M.; Scalera, F.; Ignarro, L.J. The l-arginine paradox: Importance of the l-arginine/asymmetrical dimethylarginine ratio. Pharmacol. Ther. 2007, 114, 295–306. [Google Scholar] [CrossRef]
- Closs, E.I.; Simon, A.; Vékony, N.; Rotmann, A. Plasma Membrane Transporters for Arginine. J. Nutr. 2004, 134, 2752S–2759S. [Google Scholar] [CrossRef]
- Zharikov, S.I.; Block, E.R. Characterization of L-arginine uptake by plasma membrane vesicles isolated from cultured pulmonary artery endothelial cells. Biochim. Biophys. Acta 1998, 1369, 173–183. [Google Scholar] [CrossRef][Green Version]
- McDonald, K.K.; Zharikov, S.; Block, E.R.; Kilberg, M.S. A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the “arginine paradox”. J. Biol. Chem. 1997, 272, 31213–31216. [Google Scholar] [CrossRef] [PubMed]
- Block, E.R.; Herrera, H.; Couch, M. Hypoxia inhibits L-arginine uptake by pulmonary artery endothelial cells. Am. J. Physiol. 1995, 269, L574–L580. [Google Scholar] [CrossRef]
- Zharikov, S.I.; Block, E.R. Association of l-arginine transporters with fodrin: Implications for hypoxic inhibition of arginine uptake. Am. J. Physiol. Cell. Mol. Physiol. 2000, 278, L111–L117. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Chen, B.; Chicoine, L.G.; Nelin, L.D. Overexpression of cationic amino acid transporter-1 increases nitric oxide production in hypoxic human pulmonary microvascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2011, 38, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, S.D.; Yu, H.; Grody, W.W.; Kern, R.M.; Yoo, P.; Iyer, R.K. Arginases I and II: Do their functions overlap? Mol. Genet. Metab. 2004, 81 (Suppl. 1), S38–S44. [Google Scholar] [CrossRef]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef]
- Prieto, C.P.; Krause, B.J.; Quezada, C.; San Martin, R.; Sobrevia, L.; Casanello, P. Hypoxia-reduced nitric oxide synthase activity is partially explained by higher arginase-2 activity and cellular redistribution in human umbilical vein endothelium. Placenta 2011, 32, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ming, X.-F. Arginase: The Emerging Therapeutic Target for Vascular Oxidative Stress and Inflammation. Front. Immunol. 2013, 4, 149. [Google Scholar] [CrossRef] [PubMed]
- Krotova, K.; Patel, J.M.; Block, E.R.; Zharikov, S. Hypoxic upregulation of arginase II in human lung endothelial cells. Am. J. Physiol. Cell Physiol. 2010, 299, C1541–C1548. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bhatta, A.; Toque, H.A.; Rojas, M.; Yao, L.; Xu, Z.; Patel, C.; Caldwell, R.B.; Caldwell, R.W. Arginase inhibition enhances angiogenesis in endothelial cells exposed to hypoxia. Microvasc. Res. 2015, 98, 1–8. [Google Scholar] [CrossRef]
- Liang, X.; Arullampalam, P.; Yang, Z.; Ming, X.-F. Hypoxia Enhances Endothelial Intercellular Adhesion Molecule 1 Protein Level Through Upregulation of Arginase Type II and Mitochondrial Oxidative Stress. Front. Physiol. 2019, 10, 1003. [Google Scholar] [CrossRef]
- Pandey, D.; Nomura, Y.; Rossberg, M.C.; Hori, D.; Bhatta, A.; Keceli, G.; Leucker, T.; Santhanam, L.; Shimoda, L.A.; Berkowitz, D.; et al. Hypoxia Triggers SENP1 (Sentrin-Specific Protease 1) Modulation of KLF15 (Kruppel-Like Factor 15) and Transcriptional Regulation of Arg2 (Arginase 2) in Pulmonary Endothelium. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 913–926. [Google Scholar] [CrossRef]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaço, R.D.D.R.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.J.M.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.; Vallance, P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc. Res. 1999, 43, 542–548. [Google Scholar] [CrossRef]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; Van-Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, R.; Ratnatunga, C.; et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: Implications for endothelial function in human atherosclerosis. Eur. Heart J. 2009, 30, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Strobel, J.; Mieth, M.; Endress, B.; Auge, D.; Koenig, J.; Fromm, M.; Maas, R. Interaction of the cardiovascular risk marker asymmetric dimethylarginine (ADMA) with the human cationic amino acid transporter 1 (CAT1). J. Mol. Cell. Cardiol. 2012, 53, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Closs, E.I.; Basha, F.Z.; Habermeier, A.; Förstermann, U. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide Biol. Chem. 1997, 1, 65–73. [Google Scholar] [CrossRef]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The Role of Asymmetric Dimethylarginine (ADMA) in Endothelial Dysfunction and Cardiovascular Disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef]
- Lu, C.-W.; Xiong, Y.; He, P. Dimethylarginine dimethylaminohydrolase-2 overexpression improves impaired nitric oxide synthesis of endothelial cells induced by glycated protein. Nitric Oxide 2007, 16, 94–103. [Google Scholar] [CrossRef]
- Palm, F.; Onozato, M.L.; Luo, Z.; Wilcox, C.S. Dimethylarginine dimethylaminohydrolase (DDAH): Expression, regulation, and function in the cardiovascular and renal systems. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3227–H3245. [Google Scholar] [CrossRef]
- Lüneburg, N.; Harbaum, L.; Hennigs, J.K. The Endothelial ADMA/NO Pathway in Hypoxia-Related Chronic Respiratory Diseases. BioMed Res. Int. 2014, 2014, 501612. [Google Scholar] [CrossRef]
- Yildirim, A.O.; Bulau, P.; Zakrzewicz, D.; Kitowska, K.E.; Weissmann, N.; Grimminger, F.; Morty, R.E.; Eickelberg, O. Increased protein arginine methylation in chronic hypoxia: Role of protein arginine methyltransferases. Am. J. Respir. Cell Mol. Biol. 2006, 35, 436–443. [Google Scholar] [CrossRef]
- Hannemann, J.; Zummack, J.; Hillig, J.; Böger, R. Metabolism of asymmetric dimethylarginine in hypoxia: From bench to bedside. Pulm. Circ. 2020, 10, 31–41. [Google Scholar] [CrossRef]
- İn, E.; Özdemir, C.; Kaman, D.; Sökücü, S.N. Heat Shock Proteins, L-Arginine, and Asymmetric Dimethylarginine Levels in Patients with Obstructive Sleep Apnea Syndrome. Arch. Bronconeumol. 2015, 51, 544–550. [Google Scholar] [CrossRef]
- Fang, Z.; Huang, Y.; Tang, L.; Hu, X.; Shen, X.; Tang, J.; Zhou, S. Asymmetric Dimethyl-l-Arginine is a Biomarker for Disease Stage and Follow-Up of Pulmonary Hypertension Associated with Congenital Heart Disease. Pediatr. Cardiol. 2015, 36, 1062–1069. [Google Scholar] [CrossRef]
- Millatt, L.J.; Whitley, G.S.; Li, D.; Leiper, J.M.; Siragy, H.M.; Carey, R.M.; Johns, R.A. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia-induced pulmonary hypertension. Circulation 2003, 108, 1493–1498. [Google Scholar] [CrossRef]
- Pekarova, M.; Koudelka, A.; Kolarova, H.; Ambrozova, G.; Klinke, A.; Cerna, A.; Kadlec, J.; Trundova, M.; Sindlerova Svihalkova, L.; Kuchta, R.; et al. Asymmetric dimethyl arginine induces pulmonary vascular dysfunction via activation of signal transducer and activator of transcription 3 and stabilization of hypoxia-inducible factor 1-alpha. Vasc. Pharmacol. 2015, 73, 138–148. [Google Scholar] [CrossRef]
- Iannone, L.; Zhao, L.; Dubois, O.; Duluc, L.; Rhodes, C.; Wharton, J.; Wilkins, M.; Leiper, J.; Wojciak-Stothard, B. MiRNA-21/DDAH1 pathway regulates pulmonary vascular responses to hypoxia. Biochem. J. 2014, 462, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, F.I.; Vallance, P.; Haworth, S.G.; Leiper, J.M. Metabolism of Asymmetric Dimethylarginines Is Regulated in the Lung Developmentally and With Pulmonary Hypertension Induced by Hypobaric Hypoxia. Circulation 2003, 107, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.; Kiss, L.; Ghofrani, H.A.; Voswinckel, R.; Haredza, P.; Klepetko, W.; Aigner, C.; Fink, L.; Muyal, J.P.; Weissmann, N.; et al. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1175–1177. [Google Scholar] [CrossRef]
- Hernansanz-Agustín, P.; Izquierdo-Álvarez, A.; Sánchez-Gómez, F.J.; Ramos, E.; Villa-Piña, T.; Lamas, S.; Bogdanova, A.; Martínez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 2014, 71, 146–156. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Manish, M.; Markus, R.; Peter, K.; Simone, H.; Eva, D.; Parag, G.; Anne-Christin, S.; Theo, S.R.; Ardeschir, G.H.; Grazyna, K.; et al. Hypoxia-Dependent Regulation of Nonphagocytic NADPH Oxidase Subunit NOX4 in the Pulmonary Vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.-S.; Wang, Y.-X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCɛ signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef]
- Crabtree, M.J.; Tatham, A.L.; Al-Wakeel, Y.; Warrick, N.; Hale, A.B.; Cai, S.; Channon, K.M.; Alp, N.J. Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: Insights from cells with tet-regulated GTP cyclohydrolase I expression. J. Biol. Chem. 2009, 284, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Münzel, T. ADMA and oxidative stress. Atheroscler. Suppl. 2003, 4, 41–51. [Google Scholar] [CrossRef]
- Thengchaisri, N.; Hein, T.W.; Wang, W.; Xu, X.; Li, Z.; Fossum, T.W.; Kuo, L. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Romero, M.J.; Shatanawi, A.; Alkilany, A.M.; Caldwell, R.B.; Caldwell, R.W. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br. J. Pharmacol. 2012, 165, 506–519. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Sehnert, B.; Burkhardt, H.; Wessels, J.T.; Schröder, A.; May, M.J.; Vestweber, D.; Zwerina, J.; Warnatz, K.; Nimmerjahn, F.; Schett, G.; et al. NF-κB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-κB in immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 16556–16561. [Google Scholar] [CrossRef] [PubMed]
- Read, M.A.; Whitley, M.Z.; Williams, A.J.; Collins, T. NF-kappa B and I kappa B alpha: An inducible regulatory system in endothelial activation. J. Exp. Med. 1994, 179, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Barbaux, S.; Tiret, L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003, 170, 191–203. [Google Scholar] [CrossRef]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Rocha, S. Hypoxia Induced NF-κB. Cells 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Ohga, E.; Nagase, T.; Tomita, T.; Teramoto, S.; Matsuse, T.; Katayama, H.; Ouchi, Y. Increased levels of circulating ICAM-1, VCAM-1, and L-selectin in obstructive sleep apnea syndrome. J. Appl. Physiol. 1999, 87, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Tschöp, M.; Fischer, R.; Bidlingmaier, C.; Riepl, R.; Tschöp, K.; Hautmann, H.; Endres, S.; Toepfer, M. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine 2000, 12, 246–252. [Google Scholar] [CrossRef]
- Williams, A.; Scharf, S.M. Obstructive sleep apnea, cardiovascular disease, and inflammation—Is NF-kappaB the key? Sleep Breath. 2007, 11, 69–76. [Google Scholar] [CrossRef]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Luboshits, G.; Barshack, I.; Polak-Charcon, S.; Tzahor, E.; Keren, G.; George, J. Hypoxia-Inducible Factor-1α and -2α Additively Promote Endothelial Vasculogenic Properties. J. Vasc. Res. 2009, 46, 299–310. [Google Scholar] [CrossRef]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone Jr, M.A.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef]
- Yoshizumi, M.; Perrella, M.A.; Burnett, J.C.J.; Lee, M.E. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res. 1993, 73, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Gertzberg, N.; Johnson, A. TNF-alpha induces a decrease in eNOS promoter activity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L452–L459. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Rath, S.; Adhami, V.M.; Mukhtar, H. Hypoxia driven glycation: Mechanisms and therapeutic opportunities. Semin. Cancer Biol. 2018, 49, 75–82. [Google Scholar] [CrossRef]
- Chang, J.S.; Wendt, T.; Qu, W.; Kong, L.; Zou, Y.S.; Schmidt, A.M.; Yan, S.-F. Oxygen Deprivation Triggers Upregulation of Early Growth Response-1 by the Receptor for Advanced Glycation End Products. Circ. Res. 2008, 102, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, L.G.; Kaneko, M.; Ananthakrishnan, R.; Harja, E.; Lee, L.K.; Hwang, Y.C.; Lerner, S.; Bakr, S.; Li, Q.; Lu, Y.; et al. Receptor for advanced-glycation end products: Key modulator of myocardial ischemic injury. Circulation 2006, 113, 1226–1234. [Google Scholar] [CrossRef]
- Kamide, T.; Kitao, Y.; Takeichi, T.; Okada, A.; Mohri, H.; Schmidt, A.M.; Kawano, T.; Munesue, S.; Yamamoto, Y.; Yamamoto, H.; et al. RAGE mediates vascular injury and inflammation after global cerebral ischemia. Neurochem. Int. 2012, 60, 220–228. [Google Scholar] [CrossRef]
- Woźniak, M.; Konopka, C.J.; Płoska, A.; Hedhli, J.; Siekierzycka, A.; Banach, M.; Bartoszewski, R.; Dobrucki, L.W.; Kalinowski, L.; Dobrucki, I.T. Molecularly targeted nanoparticles: An emerging tool for evaluation of expression of the receptor for advanced glycation end products in a murine model of peripheral artery disease. Cell. Mol. Biol. Lett. 2021, 26, 10. [Google Scholar] [CrossRef]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive Oxygen Species Activate the HIF-1α Promoter Via a Functional NFκB Site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janaszak-Jasiecka, A.; Siekierzycka, A.; Płoska, A.; Dobrucki, I.T.; Kalinowski, L. Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules 2021, 11, 982. https://doi.org/10.3390/biom11070982
Janaszak-Jasiecka A, Siekierzycka A, Płoska A, Dobrucki IT, Kalinowski L. Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules. 2021; 11(7):982. https://doi.org/10.3390/biom11070982
Chicago/Turabian StyleJanaszak-Jasiecka, Anna, Anna Siekierzycka, Agata Płoska, Iwona T. Dobrucki, and Leszek Kalinowski. 2021. "Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability" Biomolecules 11, no. 7: 982. https://doi.org/10.3390/biom11070982
APA StyleJanaszak-Jasiecka, A., Siekierzycka, A., Płoska, A., Dobrucki, I. T., & Kalinowski, L. (2021). Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules, 11(7), 982. https://doi.org/10.3390/biom11070982