
Efficacy of Novel Aminooxyacetic Acid Prodrugs in Colon Cancer Models: Towards Clinical Translation of the Cystathionine β-Synthase Inhibition Concept

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Measurement of the Catalytic Activity of Recombinant Human CBS
2.3. Effect of Test Compounds on Cell Proliferation and Viability
2.4. Ex Vivo Assessment of Tumor and Liver CBS Activity Following In Vivo Treatment with Test Compounds
2.5. Assessment of the Efficacy of YD0251 and AOAA in Mice Bearing HCT116 Subcutaneous Xenografts
2.6. Assessment of the Efficacy of YD0251 in Mice Bearing Subcutaneous Patient-Derived Tumor Xenografts (PDTXs)
2.7. Assessment of the Safety and Tolerability of YD0251 in Mice
2.8. Sample Preparation and Detection of Transsulfuration Pathway Metabolites by Mass Spectrometry
2.9. Statistics
3. Results and Discussion
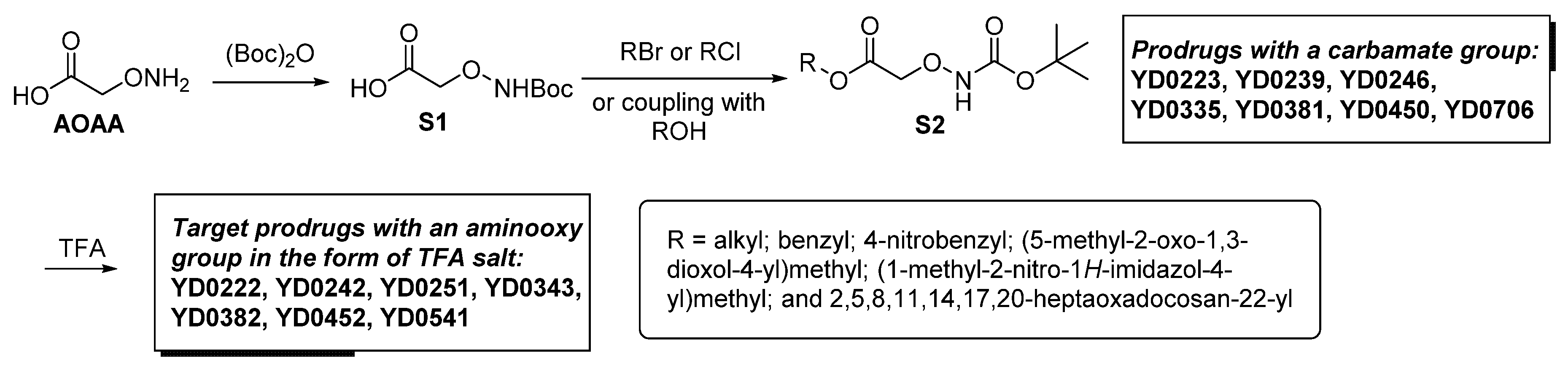
3.1. AOAA-Based Prodrugs Are Weak CBS Inhibitors in the Absence of Bioactivation
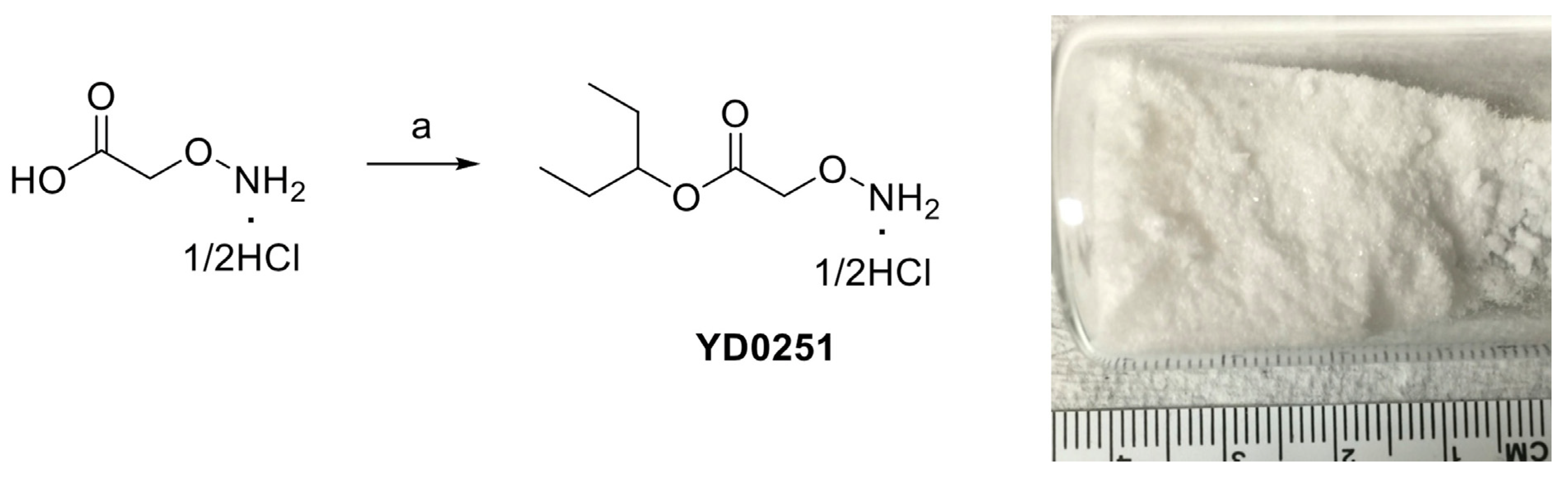
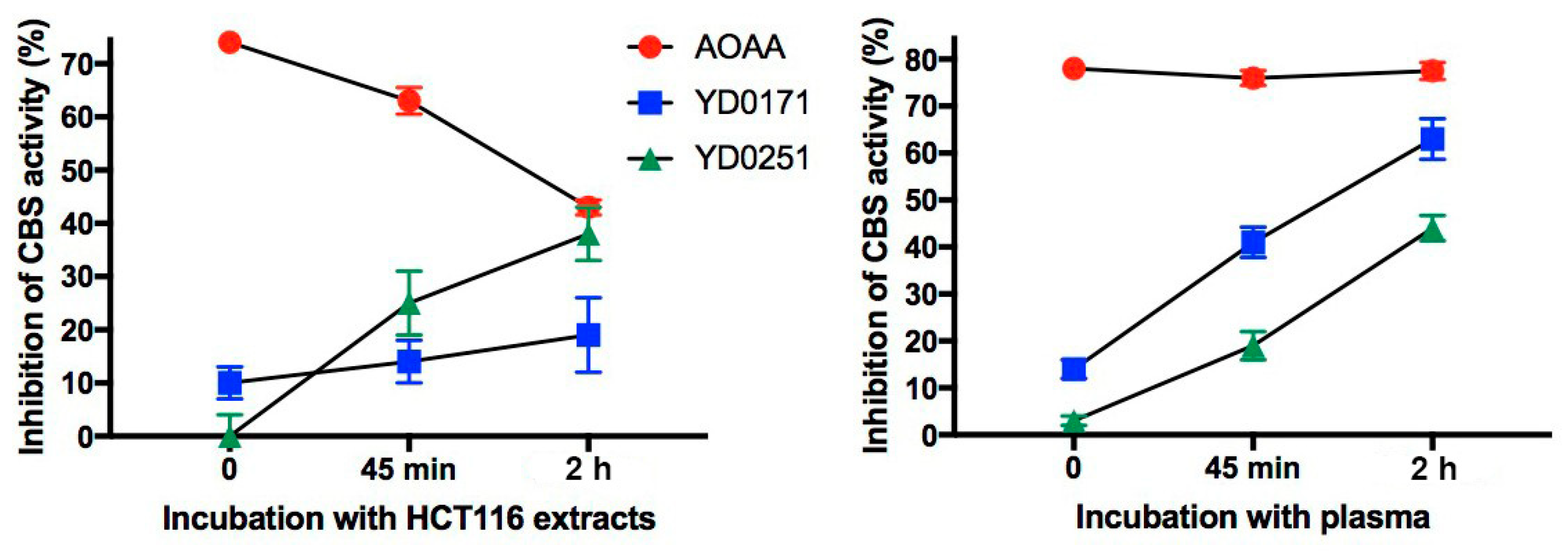
3.2. Bioactivation Enhances the Inhibitory Activity of the AOAA-Based Prodrugs YD0171 and YD0251
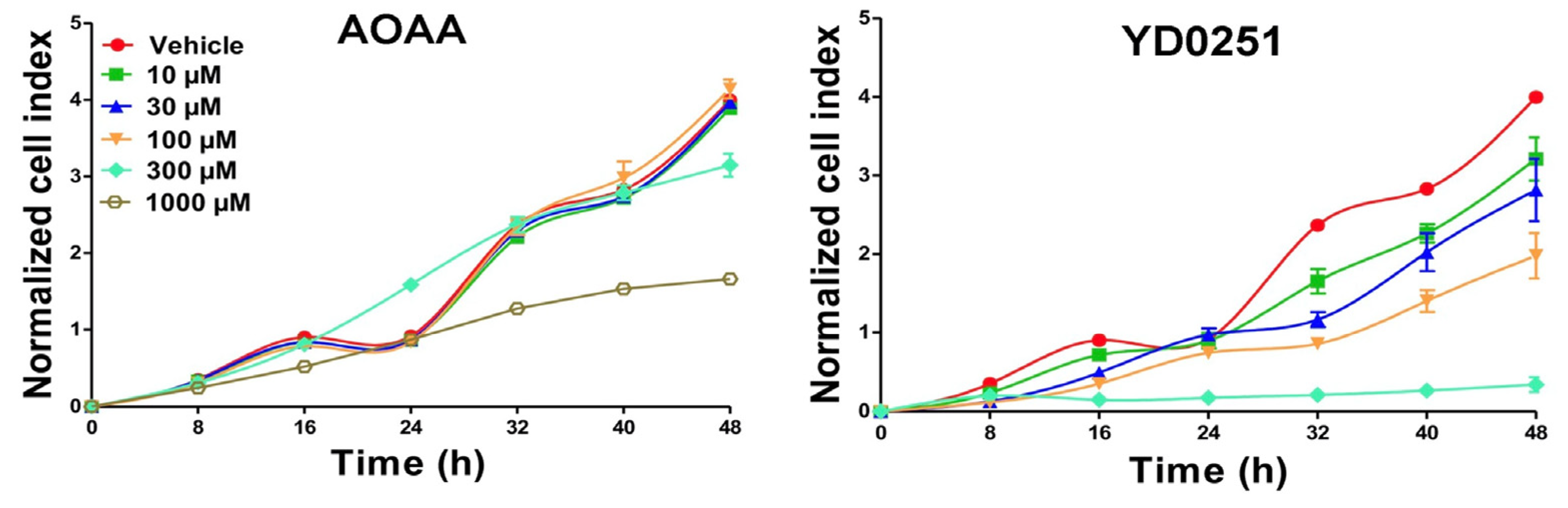
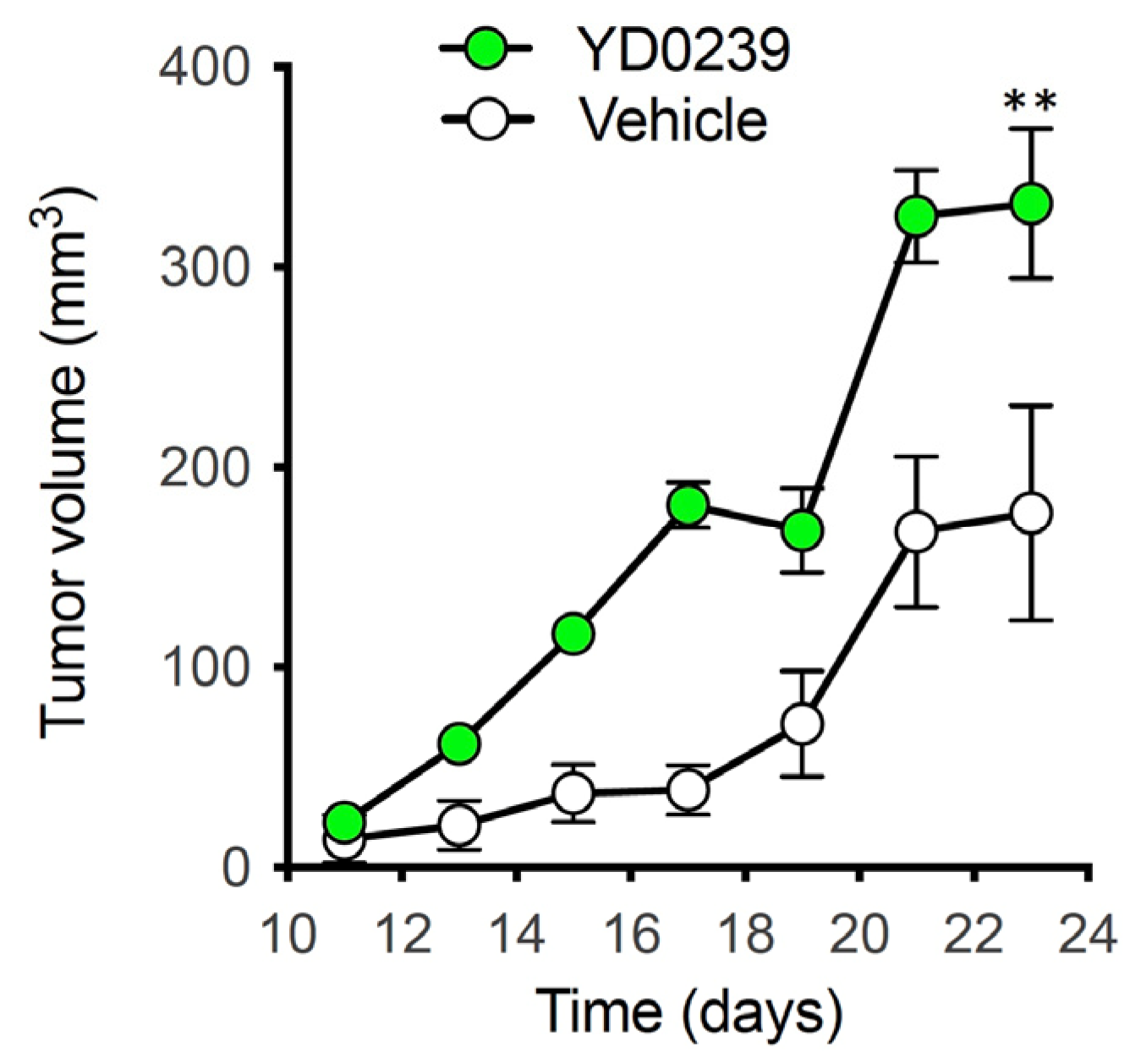
3.3. AOAA Prodrugs Show Enhanced Antiproliferative Efficacy Compared to AOAA In Vitro
3.4. Ex Vivo Assessment of Tumor and Liver CBS Activity after In Vivo Treatment with AOAA Prodrugs
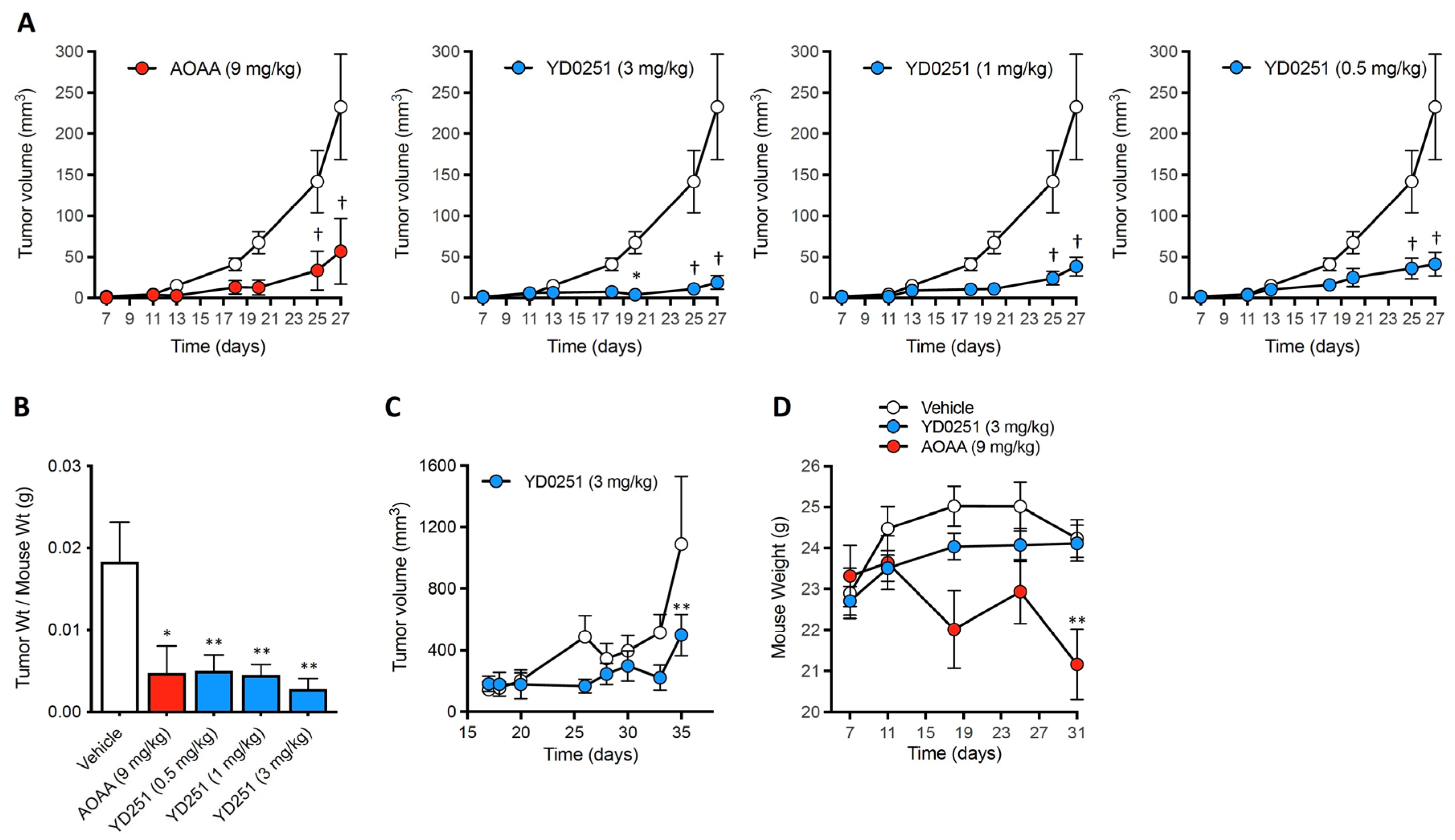
3.5. YD0251 Exhibits Improved Antitumor Potency In Vivo When Compared to AOAA
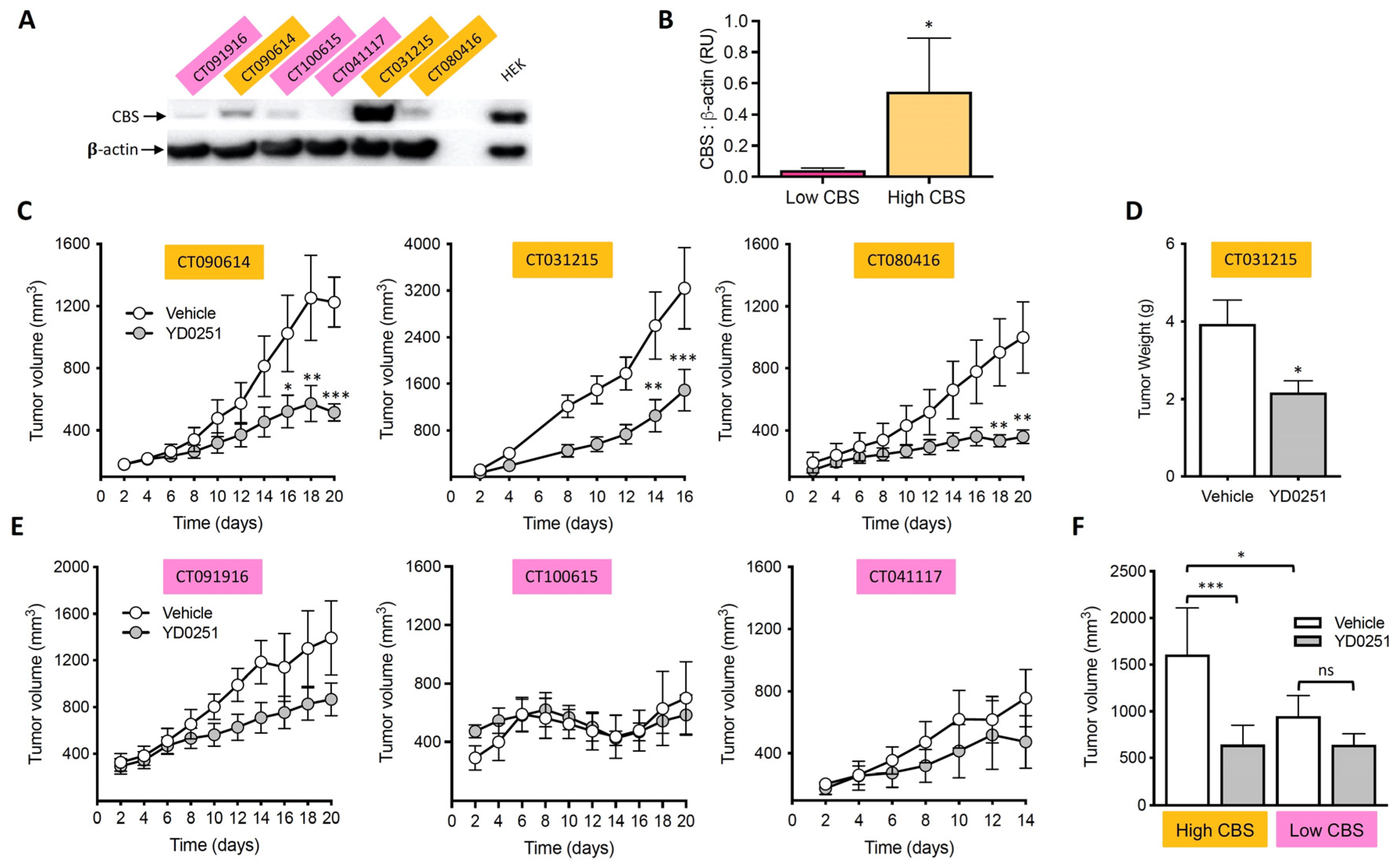
3.6. YD0251 Inhibits Patient-Derived Tumor Xenograft Growth
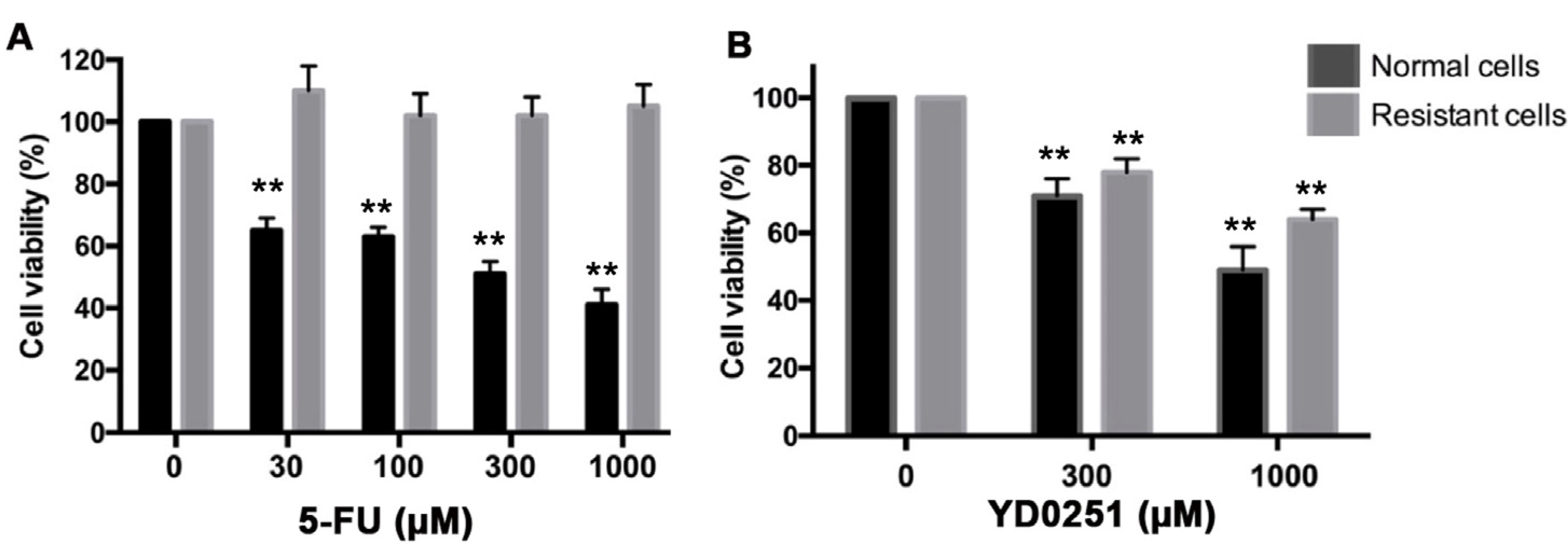
3.7. YD0251 Exerts Antiproliferative Effects in Multidrug-Resistant Colon Cancer Cells
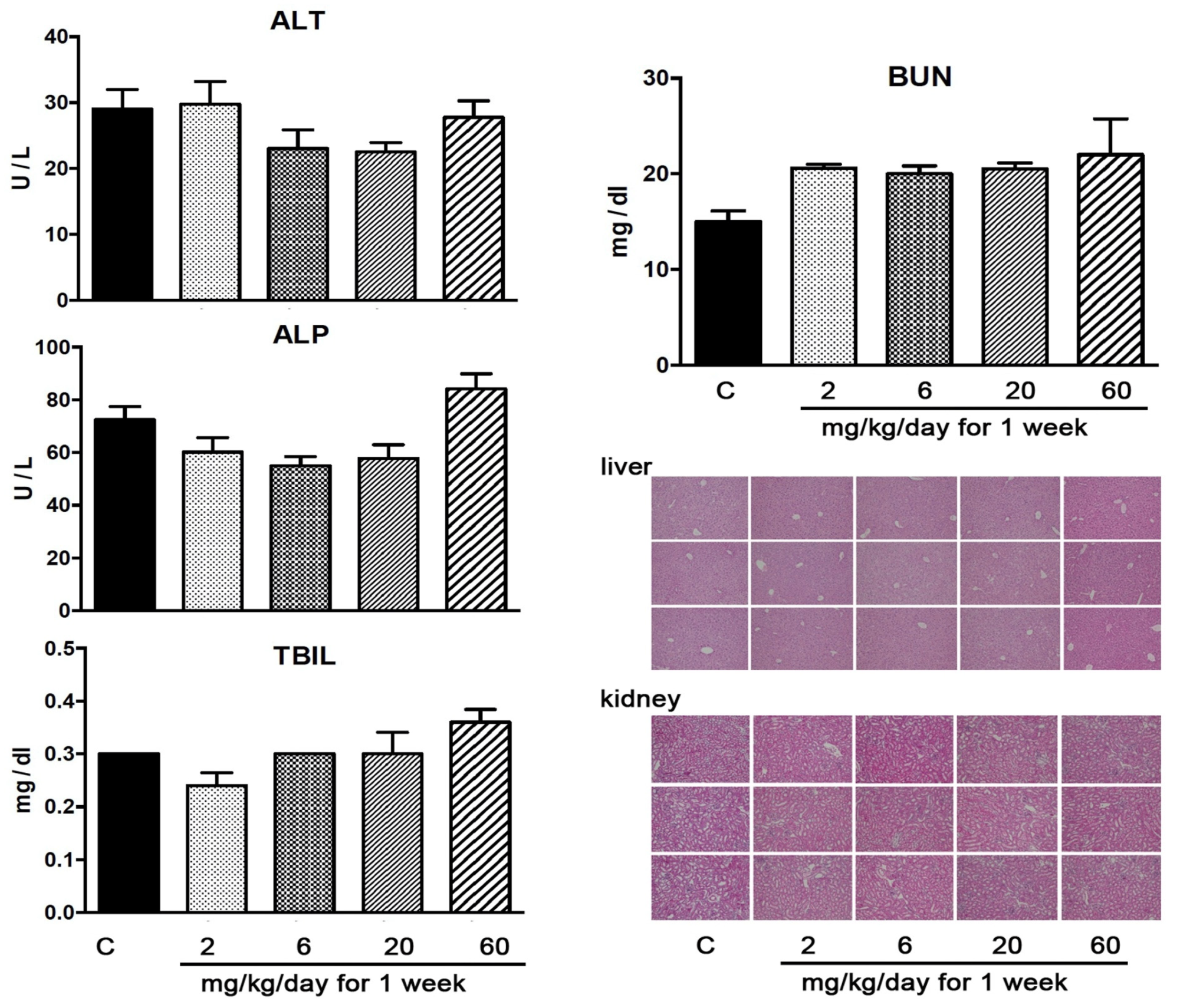
3.8. YD0251 at Doses at Which It Exerts Anticancer Effects Does Not Induce Significant Organ Injury
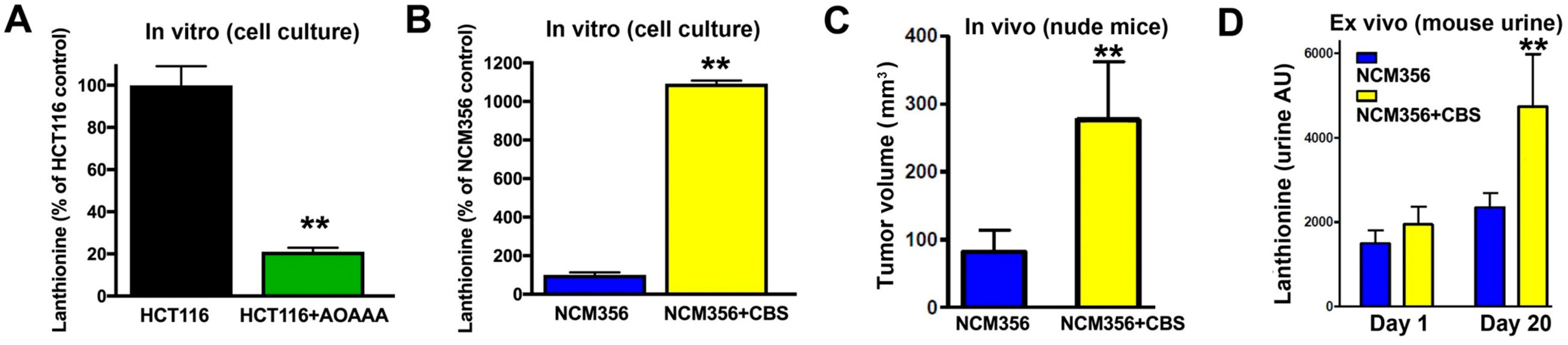
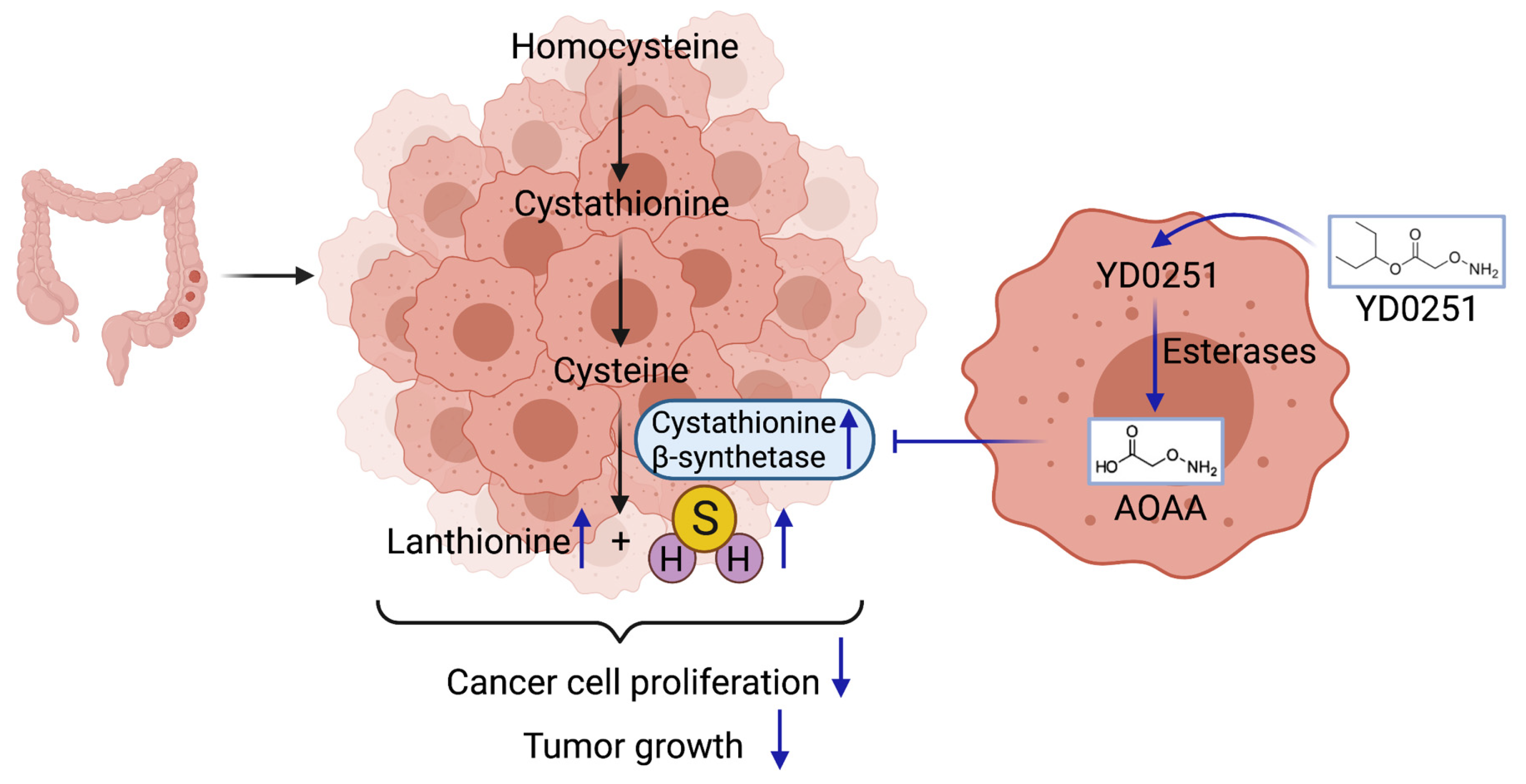
3.9. Identification of Lanthionine as a Potential Urinary Biomarker of High CBS-Expressor Tumors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen Sulfide as a Neuromodulator. Mol. Neurobiol. 2002, 26, 013–020. [Google Scholar] [CrossRef]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen Sulfide and Cell Signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef]
- Wang, R. Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. H 2 S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Panthi, S.; Chung, H.–J.; Jung, J.; Jeong, N.Y. Physiological Importance of Hydrogen Sulfide: Emerging Potent Neuroprotector and Neuromodulator. Oxidative Med. Cell. Longev. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef]
- Zezos, P.; Papaioannou, G.; Nikolaidis, N.; Vasiliadis, T.; Giouleme, O.; Evgenidis, N. Hyperhomocysteinemia in ulcerative colitis is related to folate levels. World J. Gastroenterol. 2005, 11, 6038–6042. [Google Scholar] [CrossRef]
- Kim, J.; Hong, S.J.; Park, J.H.; Park, S.Y.; Kim, S.; Cho, E.Y.; Do, I.–G.; Joh, J.–W.; Kim, D.S. Expression of cystathionine β–synthase is downregulated in hepatocellular carcinoma and associated with poor prognosis. Oncol. Rep. 2009, 21, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, S. Increased levels of homocysteine in patients with ulcerative colitis. World J. Gastroenterol. 2010, 16, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Bull, R.; Rains, J.L.; Bass, P.F.; Levine, S.N.; Reddy, S.; McVie, R.; Bocchini, J.A. Low Levels of Hydrogen Sulfide in the Blood of Diabetes Patients and Streptozotocin–Treated Rats Causes Vascular Inflammation? Antioxid. Redox Signal. 2010, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Alshorafa, A.K.; Guo, Q.; Zeng, F.; Chen, M.; Tan, G.; Tang, Z.; Yin, R. Psoriasis Is Associated with Low Serum Levels of Hydrogen Sulfide, a Potential Anti–inflammatory Molecule. Tohoku J. Exp. Med. 2012, 228, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Yang, W.; Wu, C.; Lin, D.; Ding, R.; Sun, L.; Jiang, L. Association of ulcerative colitis with transcobalamin II gene polymorphisms and serum homocysteine, vitamin B12, and folate levels in Chinese patients. Immunogenetics 2017, 69, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. The re-emerging pathophysiological role of the cystathionine-β-synthase—Hydrogen sulfide system in Down syndrome. FEBS J. 2020, 287, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor–derived hydrogen sulfide, produced by cystathionine–β–synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.; Nair, K.S.; Jennings, N.B.; Rodriguez–Aguayo, C.; Lopez–Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine Beta–Synthase (CBS) Contributes to Advanced Ovarian Cancer Progression and Drug Resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Guo, H.; Gai, J.–W.; Wang, Y.; Jin, H.–F.; Du, J.–B.; Jin, J. Characterization of Hydrogen Sulfide and Its Synthases, Cystathionine β–Synthase and Cystathionine γ–Lyase, in Human Prostatic Tissue and Cells. Urology 2012, 79, 483.e1–483.e5. [Google Scholar] [CrossRef]
- Sen, S.; Kawahara, B.; Gupta, D.; Tsai, R.; Khachatryan, M.; Roy–Chowdhuri, S.; Bose, S.; Yoon, A.; Faull, K.; Farias–Eisner, R.; et al. Role of cystathionine β–synthase in human breast Cancer. Free. Radic. Biol. Med. 2015, 86, 228–238. [Google Scholar] [CrossRef]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of Cystathionine–β–Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef]
- Untereiner, A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2S–producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Coletta, C.; Chao, C.; Szabo, C. The Therapeutic Potential of Cystathionine β–Synthetase/Hydrogen Sulfide Inhibition in Cancer. Antioxid. Redox Signal. 2015, 22, 424–448. [Google Scholar] [CrossRef]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2016, 15, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen Sulfide, an Endogenous Stimulator of Mitochondrial Function in Cancer Cells. Cells 2021, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Augsburger, F.; Szabo, C. Potential role of the 3–mercaptopyruvate sulfurtransferase (3–MST)—Hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2020, 154, 104083. [Google Scholar] [CrossRef]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine–β–synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Druzhyna, N.; Szczesny, B.; Olah, G.; Módis, K.; Asimakopoulou, A.; Pavlidou, A.; Szoleczky, P.; Gerö, D.; Yanagi, K.; Törö, G.; et al. Screening of a composite library of clinically used drugs and well–characterized pharmacological compounds for cystathionine β–synthase inhibition identifies benserazide as a drug potentially suitable for repurposing for the experimental therapy of colon cancer. Pharmacol. Res. 2016, 113, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Zatarain, J.R.; Ding, Y.; Coletta, C.; Mrazek, A.A.; Druzhyna, N.; Johnson, P.; Chen, H.; Hellmich, J.L.; Asimakopoulou, A.; et al. Cystathionine–β–Synthase Inhibition for Colon Cancer: Enhancement of the Efficacy of Aminooxyacetic Acid via the Prodrug Approach. Mol. Med. 2016, 22, 361–379. [Google Scholar] [CrossRef]
- Perna, A.F.; Pane, F.; Sepe, N.; Fontanarosa, C.; Pinto, G.; Zacchia, M.; Trepiccione, F.; Anishchenko, E.; Ingrosso, D.; Pucci, P.; et al. Lanthionine and Other Relevant Sulfur Amino Acid Metabolites: Detection of Prospective Uremic Toxins in Serum by Multiple Reaction Monitoring Tandem Mass Spectrometry. Breast Cancer 2019, 2007, 9–17. [Google Scholar] [CrossRef]
- Bahar, F.G.; Ohura, K.; Ogihara, T.; Imai, T. Species Difference of Esterase Expression and Hydrolase Activity in Plasma. J. Pharm. Sci. 2012, 101, 3979–3988. [Google Scholar] [CrossRef] [PubMed]
- Yoshigae, Y.; Imai, T.; Horita, A.; Otagiri, M. Species differences for stereoselective hydrolysis of propranolol prodrugs in plasma and liver. Chirality 1997, 9, 661–666. [Google Scholar] [CrossRef]
- Berry, L.M.; Wollenberg, L.; Zhao, Z. Esterase activities in the blood, liver and intestine of several preclinical species and humans. Drug Metab. Lett. 2009, 3, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Widen, S.G.; Wood, T.G.; Zatarain, J.R.; Johnson, P.; Gajjar, A.; Gomez, G.; Qiu, S.; Thompson, J.; Spratt, H.; et al. Patient–derived Xenografts from Colorectal Carcinoma: A Temporal and Hierarchical Study of Murine Stromal Cell Replacement. Anticancer Res. 2017, 37, 3405–3412. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative Contributions of Cystathionine β–Synthase and γ–Cystathionase to H2S Biogenesis via Alternative Trans–sulfuration Reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed]
- Majtan, T.; Krijt, J.; Sokolová, J.; Křížková, M.; Ralat, M.A.; Kent, J.; Gregory, J.F.; Kožich, V.; Kraus, J.P. Biogenesis of Hydrogen Sulfide and Thioethers by Cystathionine Beta–Synthase. Antioxid. Redox Signal. 2018, 28, 311–323. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Category | Structure | CBS Enzyme (% Inhibition at 100 µM) | HCT116 Proliferation (% Inhibition at 10, 30, 100, 300 µM for 48 h) | Tumor Homogen. CBS Activity (% Inhibition) | Liver Homogen. CBS Activity (% Inhibition) |
|---|---|---|---|---|---|---|
| AOAA | Reference |  | 92% | 0, 0, 0, 36% | 0% | 36% |
| YD0171 | Prodrug |  | 44% | 4, 12, 34, 55% | 68% | 15% |
| YD0222 | Prodrug |  | 17% | 0, 0, 0, 30% | 68% | 67% |
| YD0223 | Prodrug |  | 4% | 0, 0, 0, 15% | 50% | 62% |
| YD0239 | Prodrug |  | 1% | 0, 8, 21, 41% | 63% | 0% |
| YD0242 | Prodrug |  | 0% | 0, 0, 2, 65% | 0% | 60% |
| YD0246 | Prodrug |  | 0% | 0, 0, 0, 43% | 54% | 73% |
| YD0251 | Prodrug |  | 10% | 5, 5, 11, 52% | 42% | 54% |
| YD0335 | Prodrug |  | 0% | 0, 0, 16, 54% | 46% | 59% |
| YD0343 | Prodrug |  | 0% | 0, 0, 0, 24% | 0% | 0% |
| YD0381 | Prodrug |  | 1% | 0, 3, 13, 41% | 81% | 61% |
| YD0382 | Prodrug (Hypoxia-activated) |  | 20% | 0, 0, 0, 50% | 0% | 0% |
| YD0450 | Prodrug |  | 3% | 0, 0, 0, 59% | 16% | 73% |
| YD0452 | Prodrug (Hypoxia-activated) |  | 50% | 0, 0, 0, 50% | 0% | 0% |
| YD0541 | Prodrug (PEGylated) |  | 1% | 0, 0, 0, 19% | 0% | 38% |
| YD0706 | Prodrug |  | 0% | 0, 0, 0, 0% | 0% | 42% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hellmich, M.R.; Chao, C.; Módis, K.; Ding, Y.; Zatarain, J.R.; Thanki, K.; Maskey, M.; Druzhyna, N.; Untereiner, A.A.; Ahmad, A.; et al. Efficacy of Novel Aminooxyacetic Acid Prodrugs in Colon Cancer Models: Towards Clinical Translation of the Cystathionine β-Synthase Inhibition Concept. Biomolecules 2021, 11, 1073. https://doi.org/10.3390/biom11081073
Hellmich MR, Chao C, Módis K, Ding Y, Zatarain JR, Thanki K, Maskey M, Druzhyna N, Untereiner AA, Ahmad A, et al. Efficacy of Novel Aminooxyacetic Acid Prodrugs in Colon Cancer Models: Towards Clinical Translation of the Cystathionine β-Synthase Inhibition Concept. Biomolecules. 2021; 11(8):1073. https://doi.org/10.3390/biom11081073
Chicago/Turabian StyleHellmich, Mark R., Celia Chao, Katalin Módis, Ye Ding, John R. Zatarain, Ketan Thanki, Manjit Maskey, Nadiya Druzhyna, Ashley A. Untereiner, Akbar Ahmad, and et al. 2021. "Efficacy of Novel Aminooxyacetic Acid Prodrugs in Colon Cancer Models: Towards Clinical Translation of the Cystathionine β-Synthase Inhibition Concept" Biomolecules 11, no. 8: 1073. https://doi.org/10.3390/biom11081073
APA StyleHellmich, M. R., Chao, C., Módis, K., Ding, Y., Zatarain, J. R., Thanki, K., Maskey, M., Druzhyna, N., Untereiner, A. A., Ahmad, A., Xue, Y., Chen, H., Russell, W. K., Wang, J., Zhou, J., & Szabo, C. (2021). Efficacy of Novel Aminooxyacetic Acid Prodrugs in Colon Cancer Models: Towards Clinical Translation of the Cystathionine β-Synthase Inhibition Concept. Biomolecules, 11(8), 1073. https://doi.org/10.3390/biom11081073