
Solvent Exposure and Ionic Condensation Drive Fuzzy Dimerization of Disordered Heterochromatin Protein Sequence



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
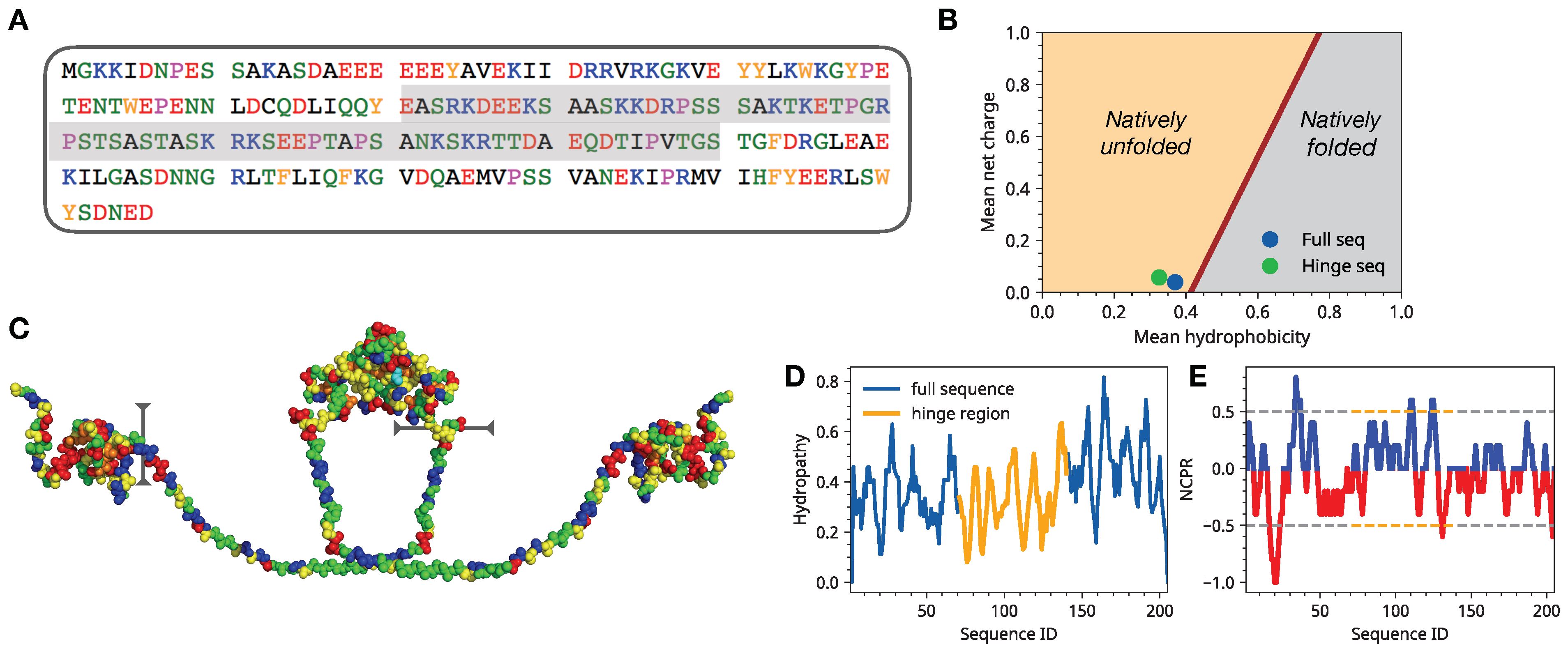
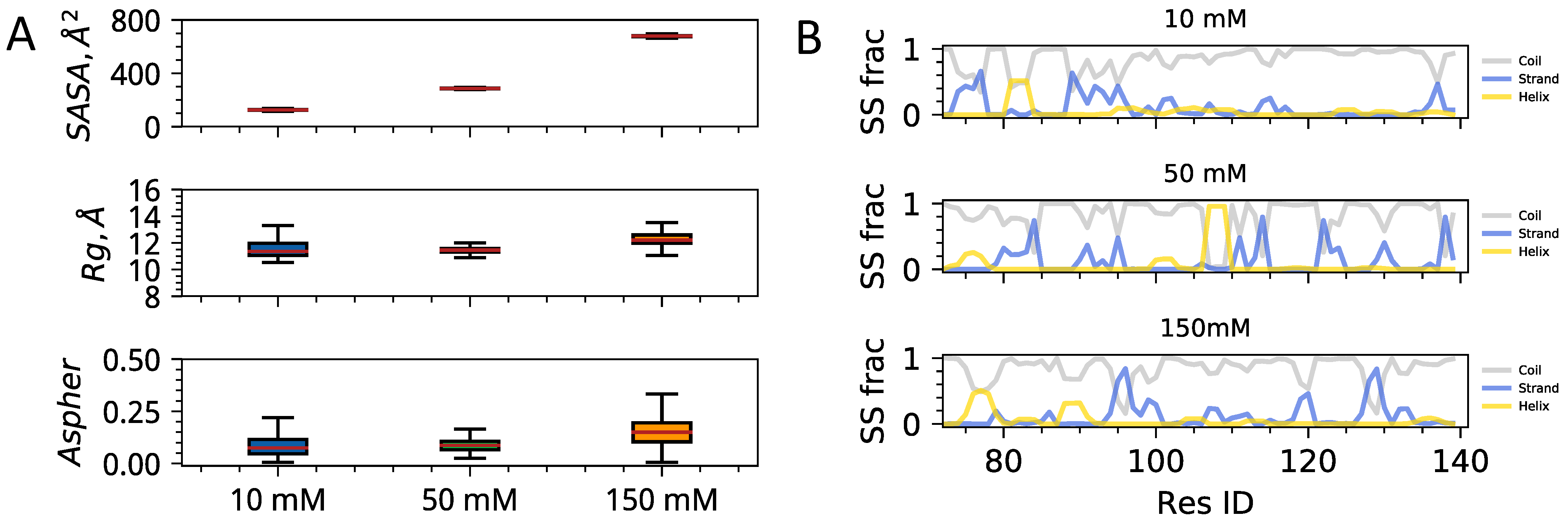
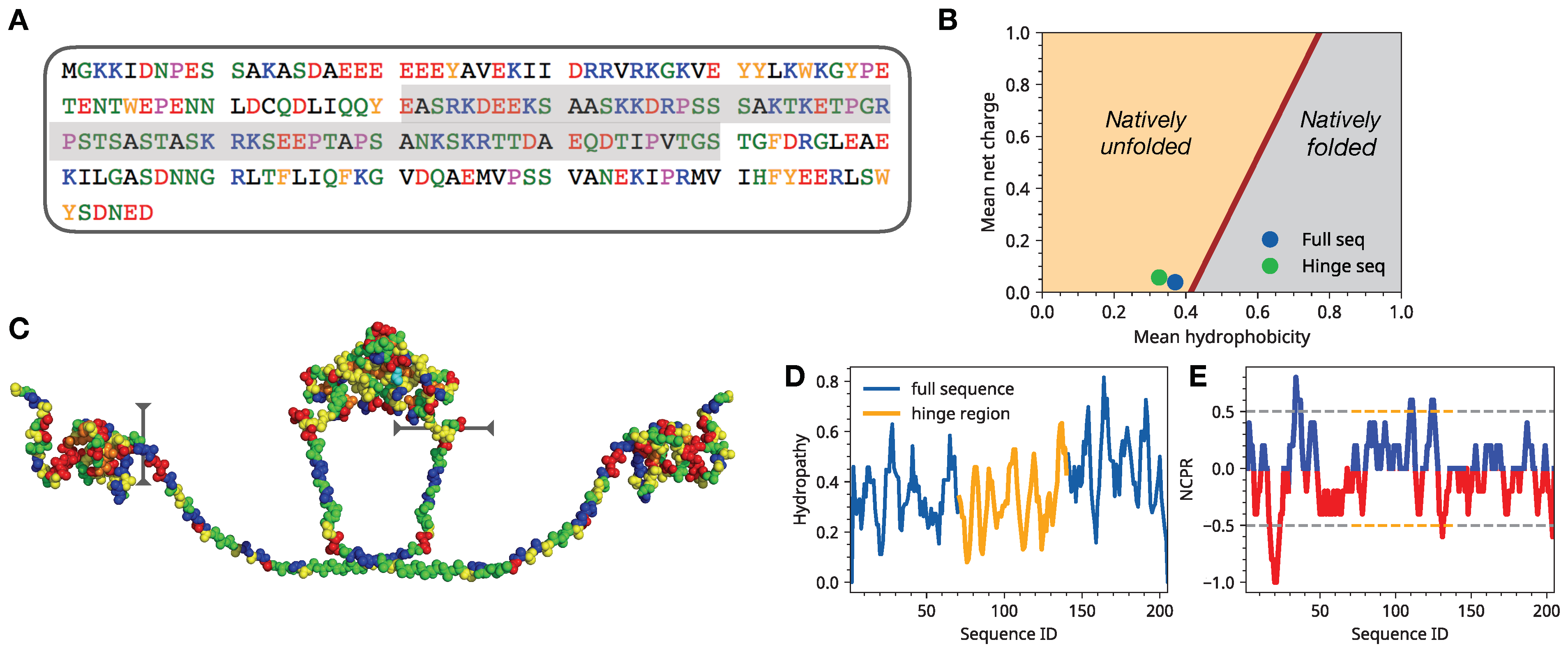
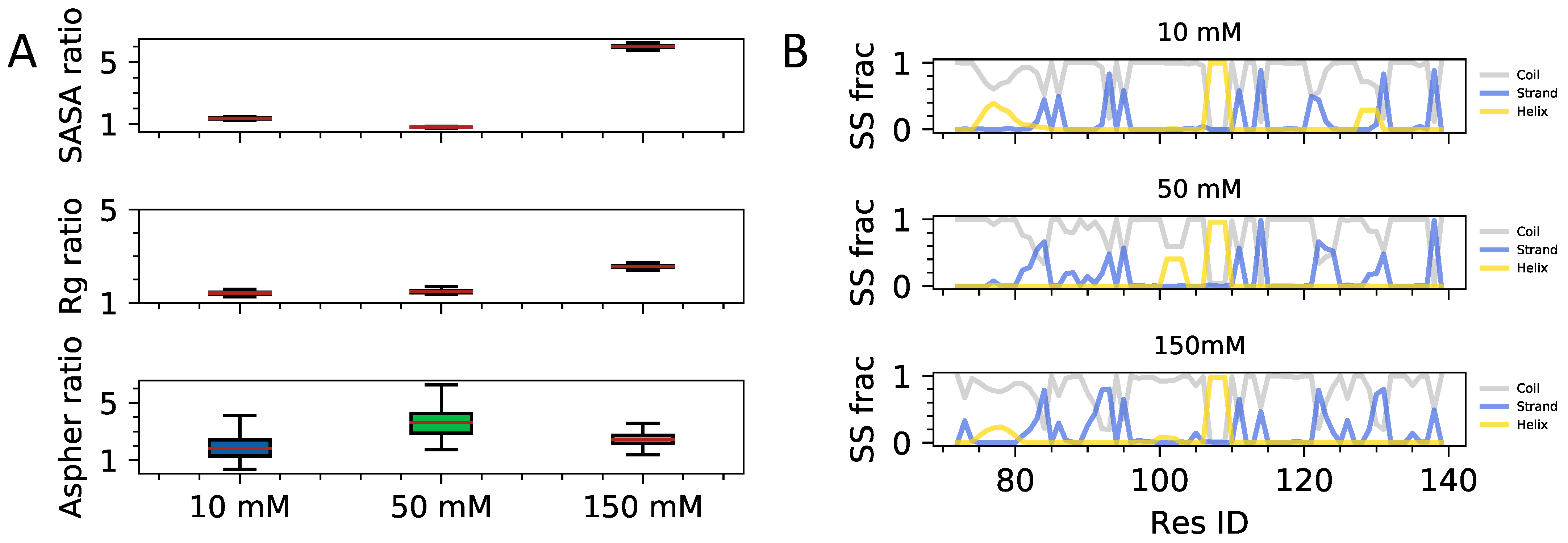
3.1. Single Chain Conformational Preferences of HP1 Hinge Regions as a Function of Ionic Strength
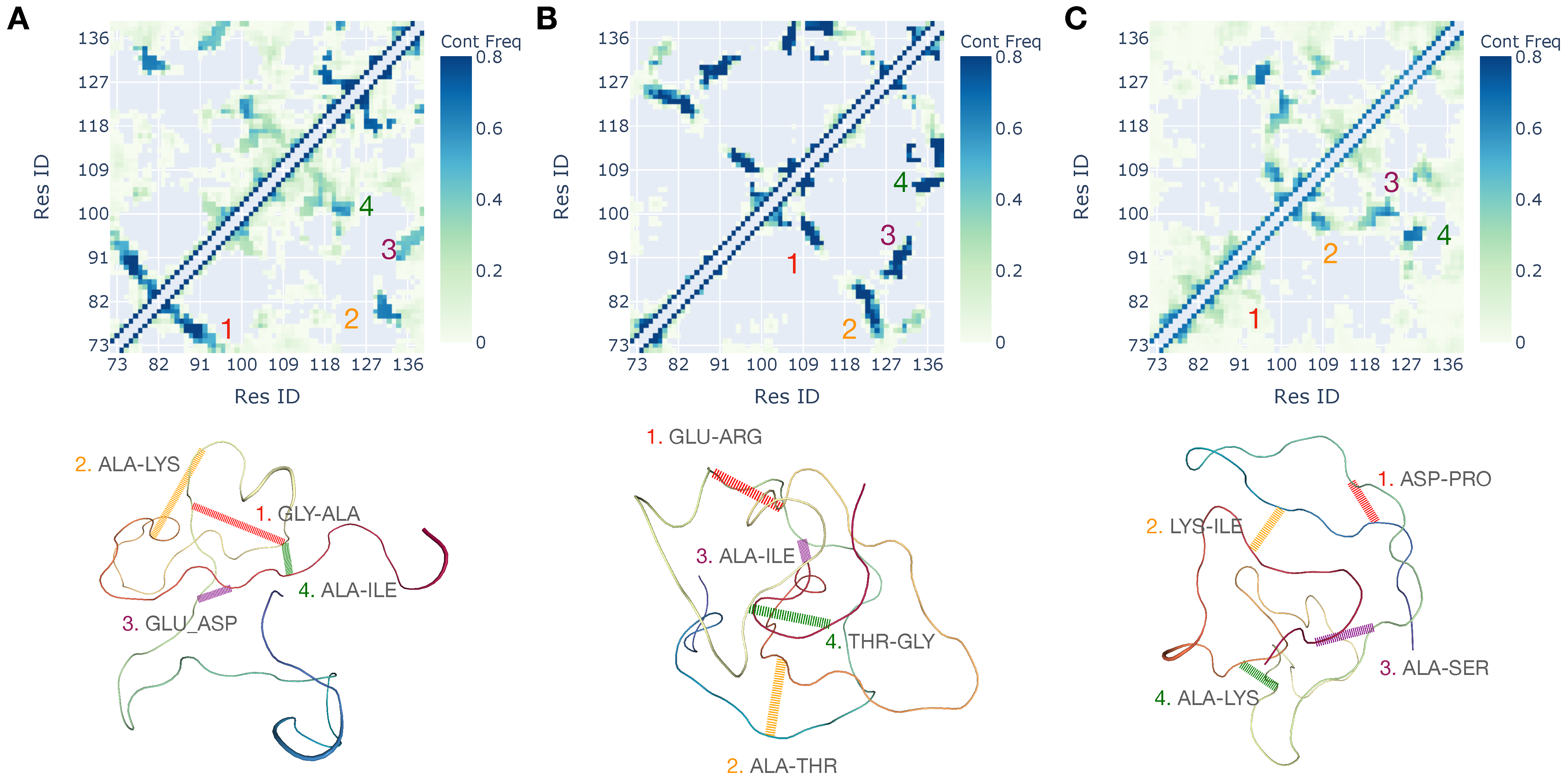
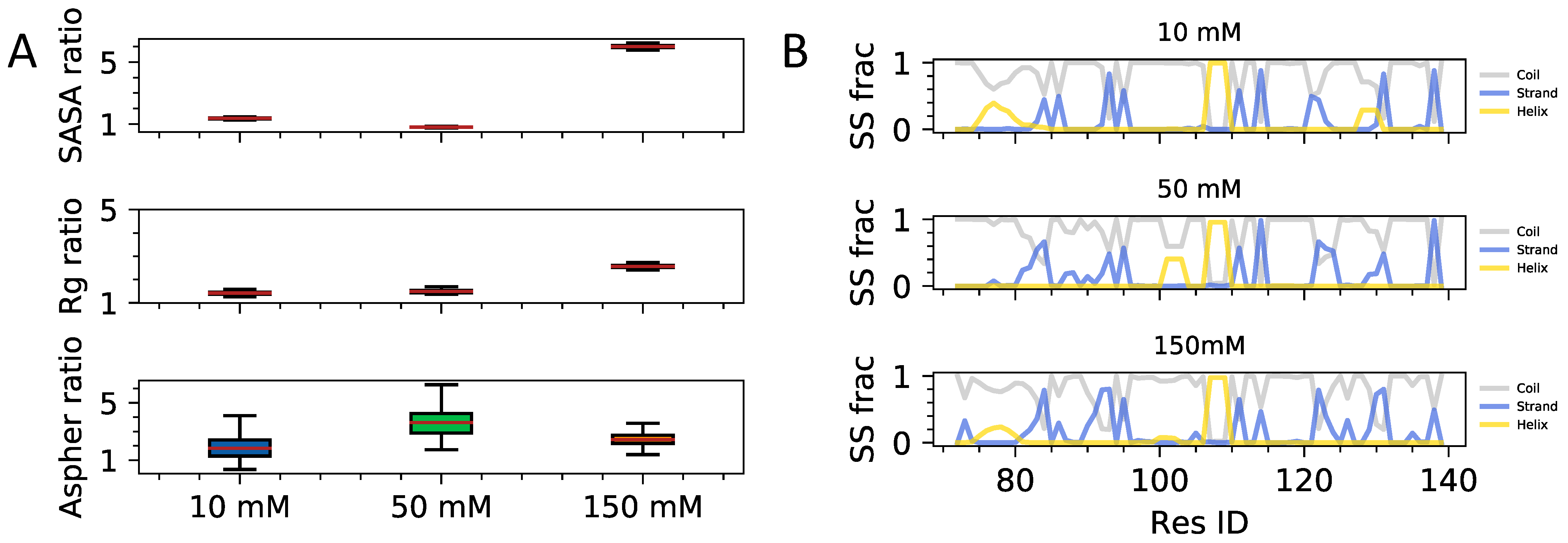
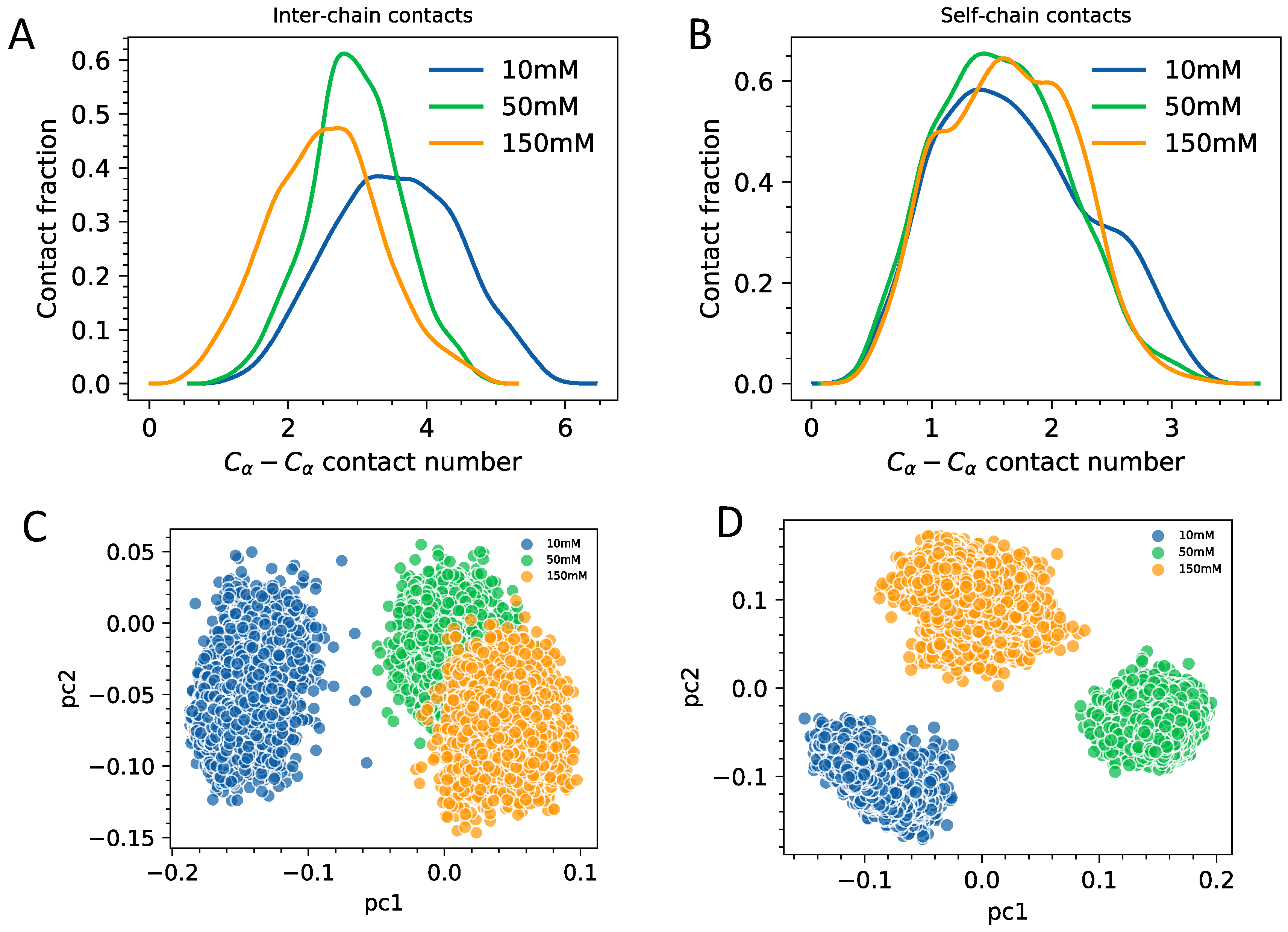
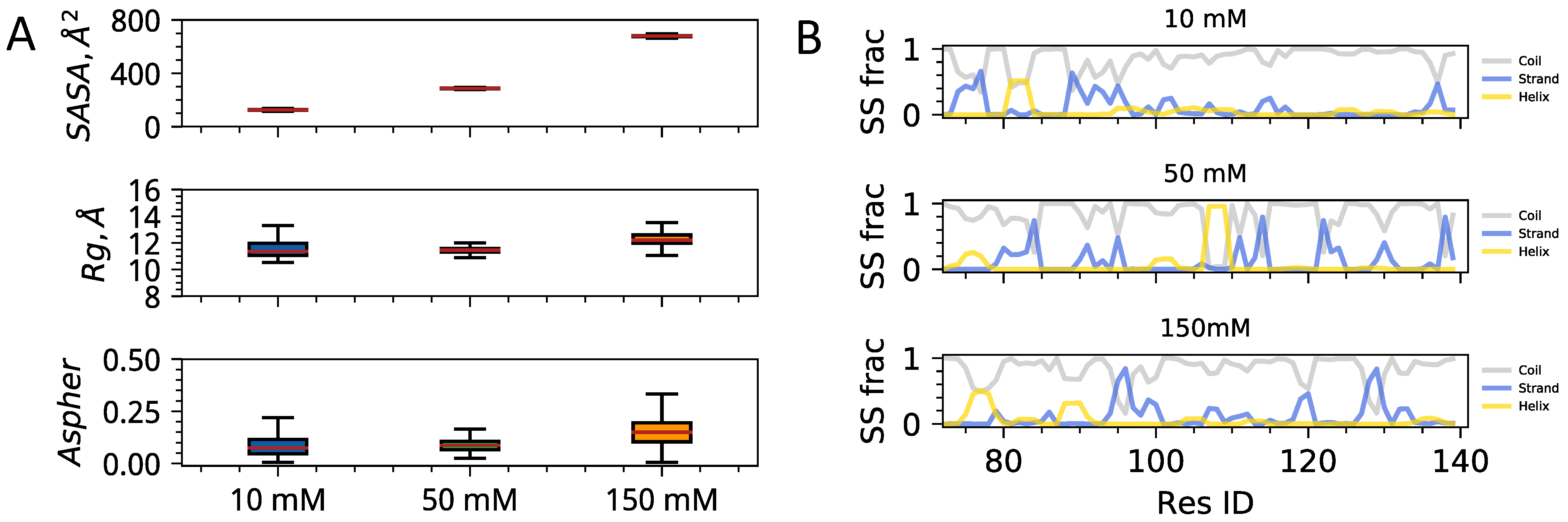
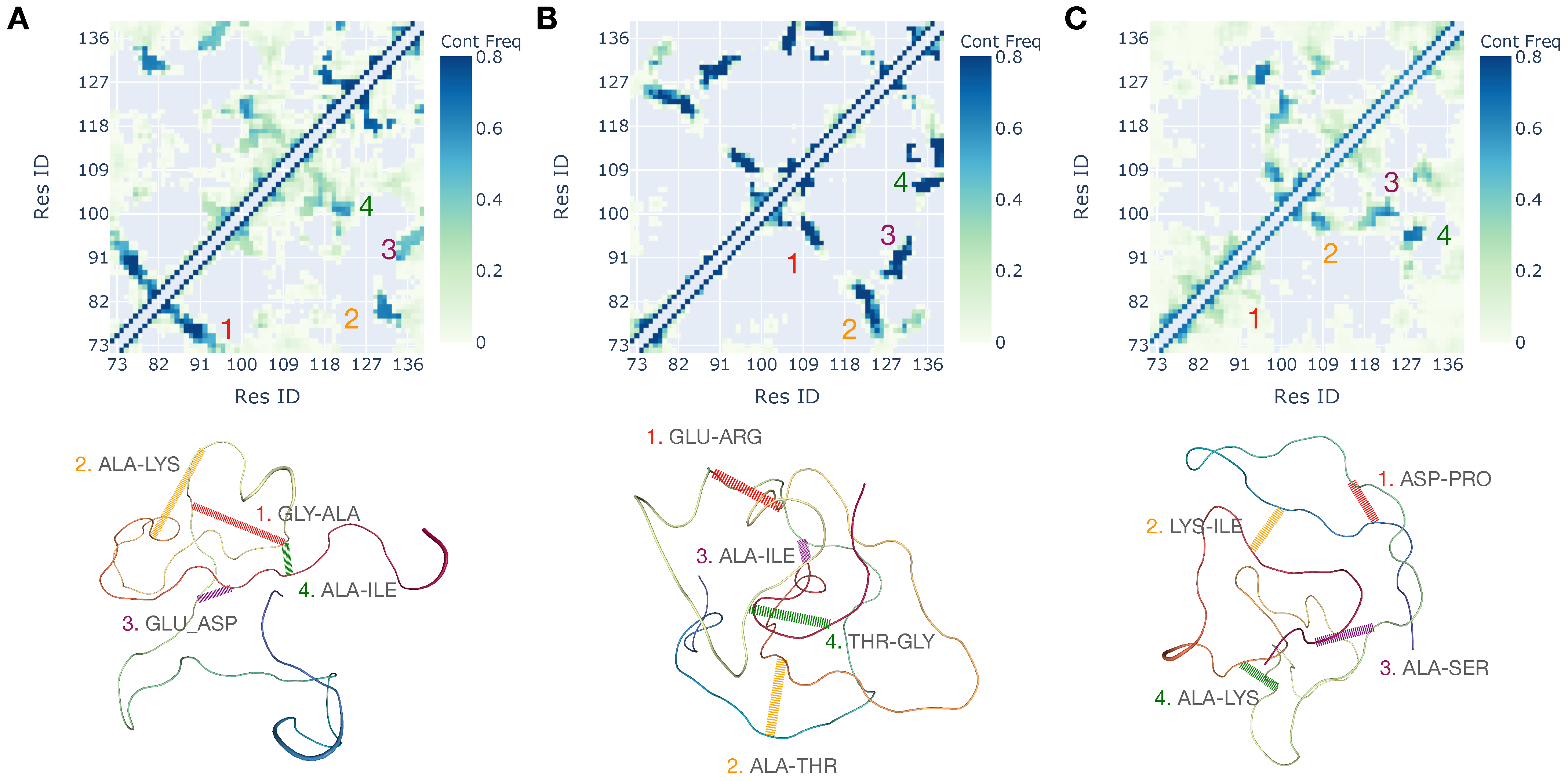
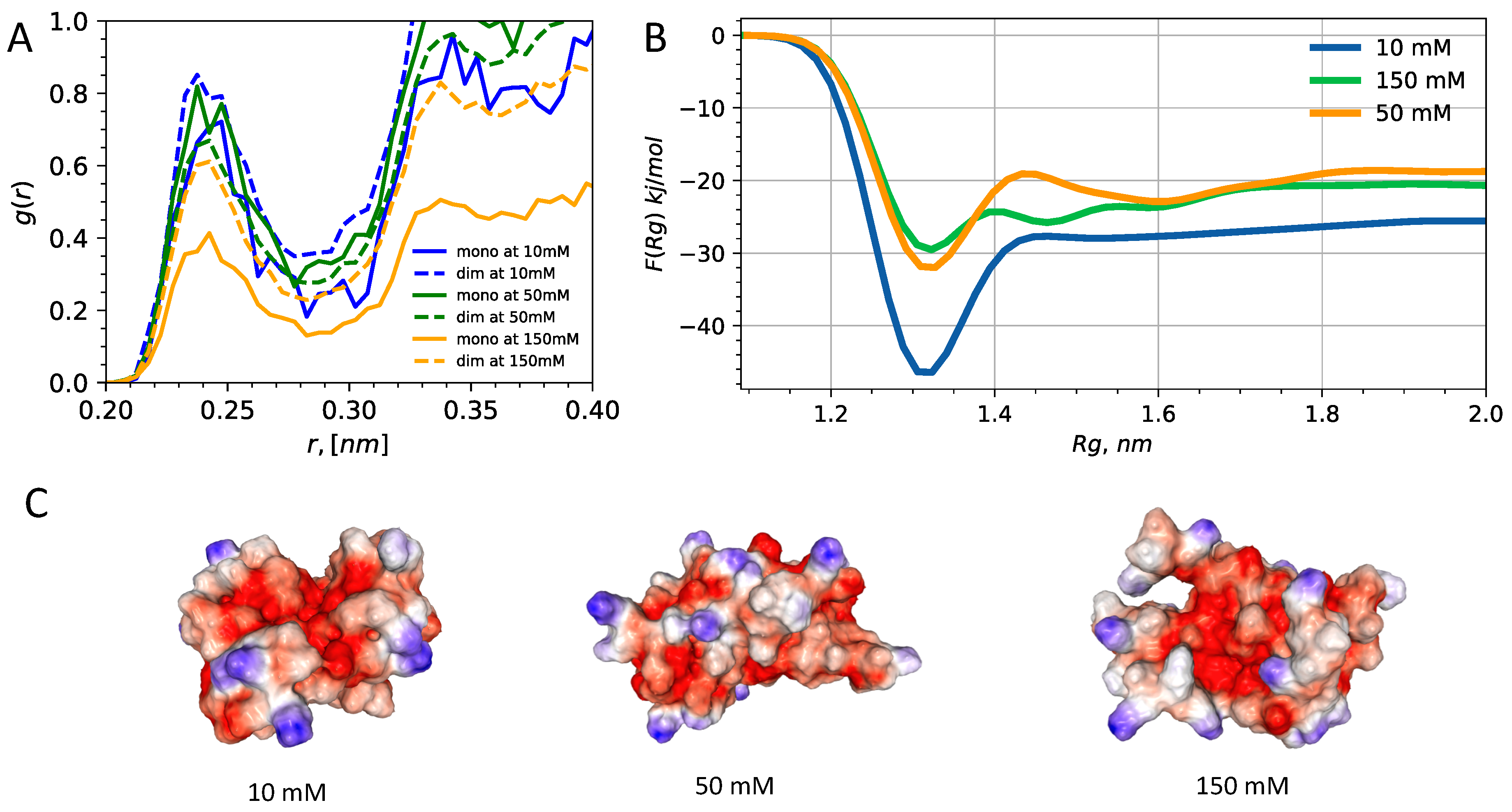
3.2. Interplay of Inter- and Intra-Chain Contacts in a Fuzzy Dimer Formed by Disordered HP1 Hinge Regions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LLPS | Liquid–liquid phase separation |
| IDP | Intrinsically disordered protein |
| HP1 | Heterochromatin protein-1 |
| HR | Hinge region |
References
- Nishibuchi, G.; Nakayama, J.I. Biochemical and structural properties of heterochromatin protein 1: Understanding its role in chromatin assembly. J. Biochem. 2014, 156, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.H.; Workman, J.L. The heterochromatin protein 1 (HP1) family: Put away a bias toward HP1. Mol. Cells 2008, 26, 217–227. [Google Scholar] [PubMed]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Fanti, L.; Pimpinelli, S. HP1: A functionally multifaceted protein. Curr. Opin. Genet. Dev. 2008, 18, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, J.C.; Elgin, S.C. HP1a: A structural chromosomal protein regulating transcription. Trends Genet. 2014, 30, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Ryu, H.W.; Kim, G.W.; Kwon, S.H. Comparison of three heterochromatin protein 1 homologs in Drosophila. J. Cell Sci. 2019, 132, jcs222729. [Google Scholar] [CrossRef] [Green Version]
- Maison, C.; Almouzni, G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004, 5, 296. [Google Scholar] [CrossRef] [PubMed]
- Assland, R.; Stewart, F. The chromo shadow domain, a second chromo domain in heterochromatin-binding protein 1, HP1. Nucleic Acids Res. 1995, 23, 3168–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, L.C.; Weilandt, D.R.; Bachmann, A.L.; Kilic, S.; Lechner, C.C.; Odermatt, P.D.; Fantner, G.E.; Georgeon, S.; Hantschel, O.; Hatzimanikatis, V.; et al. Single-molecule kinetic analysis of HP1-chromatin binding reveals a dynamic network of histone modification and DNA interactions. Nucleic Acids Res. 2017, 45, 10504–10517. [Google Scholar] [CrossRef]
- Nielsen, P.R.; Nietlispach, D.; Mott, H.R.; Callaghan, J.; Bannister, A.; Kouzarides, T.; Murzin, A.G.; Murzina, N.V.; Laue, E.D. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 2002, 416, 103. [Google Scholar] [CrossRef]
- Canzio, D.; Chang, E.Y.; Shankar, S.; Kuchenbecker, K.M.; Simon, M.D.; Madhani, H.D.; Narlikar, G.J.; Al-Sady, B. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell 2011, 41, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, P.J.; Koslover, E.F.; Spakowitz, A.J. Thermodynamic model of heterochromatin formation through epigenetic regulation. J. Phys. Cond. Matter 2015, 27, 064109. [Google Scholar] [CrossRef]
- Teif, V.B.; Kepper, N.; Yserentant, K.; Wedemann, G.; Rippe, K. Affinity, stoichiometry and cooperativity of heterochromatin protein 1 (HP1) binding to nucleosomal arrays. J. Phys. Cond. Matter 2015, 27, 064110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacPherson, Q.; Beltran, B.; Spakowitz, A.J. Bottom—Up modeling of chromatin segregation due to epigenetic modifications. Proc. Nat. Acad. Sci. USA 2018, 115, 12739–12744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdel, F.; Rippe, K. Formation of Chromatin Subcompartments by Phase Separation. Biophys. J. 2018, 114, 2262–2270. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Stengl, A.; Ugur, E.; Leidescher, S.; Ryan, J.; Cardoso, M.C.; Leonhardt, H. HP1β carries an acidic linker domain and requires H3K9me3 for phase separation. Nucleus 2021, 12, 44–57. [Google Scholar] [CrossRef]
- Sales-Gil, R.; Vagnarelli, P. How HP1 Post-Translational Modifications Regulate Heterochromatin Formation and Maintenance. Cells 2020, 9, 1460. [Google Scholar] [CrossRef]
- Sanulli, S.; Trnka, M.; Dharmarajan, V.; Tibble, R.; Pascal, B.; Burlingame, A.; Griffin, P.; Gross, J.; Narlikar, G. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 2019, 575, 390–394. [Google Scholar] [CrossRef]
- Laghmach, R.; Di Pierro, M.; Potoyan, D.A. The interplay of chromatin phase separation and lamina interactions in nuclear organisation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Laghmach, R.; Di Pierro, M.; Potoyan, D.A. Mesoscale liquid model of chromatin recapitulates nuclear order of eukaryotes. Biophys. J. 2019, 118, 2130–2140. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Rademacher, A.; Vlijm, R.; Tünnermann, J.; Frank, L.; Weinmann, R.; Schweigert, E.; Yserentant, K.; Hummert, J.; Bauer, C.; et al. Mouse heterochromatin adopts digital compaction states without showing hallmarks of HP1-driven liquid-liquid phase separation. Mol. Cell 2020, 78, 236–249. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Potoyan, D.A.; Papoian, G.A. Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J. Am. Chem. Soc. 2011, 133, 7405–7415. [Google Scholar] [CrossRef]
- Winogradoff, D.; Echeverria, I.; Potoyan, D.A.; Papoian, G.A. The Acetylation Landscape of the H4 Histone Tail: Disentangling the Interplay between the Specific and Cumulative Effects. J. Am. Chem. Soc. 2015, 137, 6245–6253. [Google Scholar] [CrossRef]
- Potoyan, D.A.; Papoian, G.A. Regulation of the H4 tail binding and folding landscapes via Lys-16 acetylation. Proc. Natl. Acad. Sci. USA 2012, 109, 17857–17862. [Google Scholar] [CrossRef] [Green Version]
- Roche, J.; Potoyan, D.A. Disorder Mediated Oligomerization of DISC1 Proteins Revealed by Coarse-Grained Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 9567–9575. [Google Scholar] [CrossRef] [PubMed]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 244111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenk, F.; Zhan, Y.; Kos, P.; Löser, E.; Atinbayeva, N.; Schächtle, M.; Tiana, G.; Giorgetti, L.; Iovino, N. HP1 drives de novo 3D genome reorganization in early Drosophila embryos. Nature 2021, 593, 289–293. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mueterthies, J.; Potoyan, D.A. Solvent Exposure and Ionic Condensation Drive Fuzzy Dimerization of Disordered Heterochromatin Protein Sequence. Biomolecules 2021, 11, 915. https://doi.org/10.3390/biom11060915
Mueterthies J, Potoyan DA. Solvent Exposure and Ionic Condensation Drive Fuzzy Dimerization of Disordered Heterochromatin Protein Sequence. Biomolecules. 2021; 11(6):915. https://doi.org/10.3390/biom11060915
Chicago/Turabian StyleMueterthies, Jazelli, and Davit A. Potoyan. 2021. "Solvent Exposure and Ionic Condensation Drive Fuzzy Dimerization of Disordered Heterochromatin Protein Sequence" Biomolecules 11, no. 6: 915. https://doi.org/10.3390/biom11060915
APA StyleMueterthies, J., & Potoyan, D. A. (2021). Solvent Exposure and Ionic Condensation Drive Fuzzy Dimerization of Disordered Heterochromatin Protein Sequence. Biomolecules, 11(6), 915. https://doi.org/10.3390/biom11060915