
Current Approaches and Tools Used in Drug Development against Parkinson’s Disease

 , , , ,
, , , ,  and
and 
Abstract

1. Introduction
2. In Vitro Models for Parkinson’s Disease Studies
3. In Vivo Models for Parkinson’s Disease Studies
4. Computational Approaches Used in the Development of Novel Drugs against Parkinson’s Disease
5. Recent Reports of Novel Agents against Parkinson’s Disease
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| (±)-PPHT.HCl | (±)-2-(N-phenethyl-N-propyl)amino-5-hydroxytetralin hydrochloride |
| 6-OHDA | 6-hydroxydopamine |
| AChE | Acetylcholinesterase |
| AR | Adenosine receptor |
| AI | Artificial intelligence |
| BDNF | Brain-derived neurotrophic factor |
| cAMP | Cyclic adenosine monophosphate |
| Ki | Dissociation constant |
| DR | Dopamine receptor |
| GDNF | Glial cell line-derived neurotrophic factor |
| EC50 | Half maximal effective concentration |
| IC50 | Half maximal inhibitory concentration |
| H3R | Histamine H3 receptor |
| iPSC | Induced pluripotent stem cells |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAO | Monoamine oxidase |
| MAO-B | Monoamine oxidase B |
| MAPT | Microtubule-associated protein tau |
| NGF | Nerve growth factor |
| NO | Nitric oxide |
| NOR | Nociceptin opioid receptor |
| NP | Network pharmacology |
| NSCs | Neural stem cells |
| NSE | Neuron-specific enolase |
| PD | Parkinson’s disease |
| PDE4 | Phosphodiesterase-4 |
| QSAR | Quantitative structure-activity relationship |
| SI | Selectivity index |
| SAR | Structure-activity relationship |
| SVZ | Subventricular zone |
| SN | Substantia nigra |
| SVM | Support vector machine models |
| SYN | Synpatophysin protein |
| THT | Thioflavin T |
| TH | Tyrosine hydroxylase |
References
- Rozpędek-Kamińska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef]
- Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis—Parkinson’s Disease—NCBI Bookshelf. Available online: Https://www.ncbi.nlm.nih.gov/books/NBK536722/ (accessed on 16 April 2021).
- Ikeda, K.; Kurokawa, M.; Aoyama, S.; Kuwana, Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J. Neurochem. 2002, 80, 262–270. [Google Scholar] [CrossRef]
- Aryati, W.D.; Salamah, N.N.; Syahdi, R.R.; Yanuar, A. The Role and Development of the Antagonist of Adenosine A2A in Parkinson’s Disease. In Neurodegeneration; Chuen-Chung Chang, R., Ho, Y.-S., Eds.; IntechOpen: London, UK, 2019; ISBN 978-1-78984-737-6. [Google Scholar] [CrossRef]
- Dezsi, L.; Vecsei, L. Monoamine Oxidase B Inhibitors in Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2017, 16, 425–439. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nat. Cell Biol. 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Saridaki, T.; Dinter, E. Cellular models for Parkinson’s disease. J. Neurochem. 2016, 139, 121–130. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. In Neuronal Cell Culture; Amini, S., White, M.K., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1078, pp. 9–21. ISBN 978-1-62703-639-9. [Google Scholar]
- Jämsä, A.; Hasslund, K.; Cowburn, R.F.; Bäckström, A.; Vasänge, M. The retinoic acid and brain-derived neurotrophic factor differentiated SH-SY5Y cell line as a model for Alzheimer’s disease-like tau phosphorylation. Biochem. Biophys. Res. Commun. 2004, 319, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 53193, e53193. [Google Scholar] [CrossRef]
- Påhlman, S.; Ruusala, A.-I.; Abrahamsson, L.; Mattsson, M.E.; Esscher, T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: A comparison with phorbolester-induced differentiation. Cell Differ. 1984, 14, 135–144. [Google Scholar] [CrossRef]
- Alberio, T.; Mammucari, C.; D’Agostino, G.; Rizzuto, R.; Fasano, M. Altered dopamine homeostasis differentially affects mitochondrial voltage-dependent anion channels turnover. Biochim. Biophys. Acta 2014, 1842, 1816–1822. [Google Scholar] [CrossRef]
- Grau, C.M.; Greene, L.A. Use of PC12 Cells and Rat Superior Cervical Ganglion Sympathetic Neurons as Models for Neuroprotective Assays Relevant to Parkinson’s Disease. In Neurotrophic Factors; Skaper, S.D., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 846, pp. 201–211. ISBN 978-1-61779-535-0. [Google Scholar]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- Kim, A.R.; Ugryumov, M.V. Changes in plasma catecholamines levels as preclinical biomarkers in experimental models of Parkinson’s disease. Dokl. Biochem. Biophys. 2015, 464, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Cao, Q.; Sun, Z.; Chen, J.; Zheng, Q.; Xiao, F. A novel method of neural differentiation of PC12 cells by using Opti-MEM as a basic induction medium. Int. J. Mol. Med. 2017, 41, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, S.; Lee, C.H.; Gilyazova, N.; Ibeanu, G.C. Analysis of Gene Expression and Neuronal Phenotype in Neuroscreen-1 (NS-1) Cells. Int. J. Biomed. Investig. 2018, 1, 115. [Google Scholar]
- Nie, B.-M.; Lu, Y.; Li, W.-P.; Ma, K.; Jiang, X.-Y.; Yang, R.; Lu, P. Molecular mechanism of panaxydol on promoting axonal growth in PC12 cells. Neural Regen. Res. 2018, 13, 1927–1936. [Google Scholar] [CrossRef]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s Disease Patient-Derived Induced Pluripotent Stem Cells Free of Viral Reprogramming Factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef]
- Li, H.; Jiang, H.; Zhang, B.; Feng, J. Modeling Parkinson’s Disease Using Patient-specific Induced Pluripotent Stem Cells. J. Park. Dis. 2018, 8, 479–493. [Google Scholar] [CrossRef]
- Hu, X.; Mao, C.; Fan, L.; Luo, H.; Hu, Z.; Zhang, S.; Yang, Z.; Zheng, H.; Sun, H.; Fan, Y.; et al. Modeling Parkinson’s Disease Using Induced Pluripotent Stem Cells. Stem Cells Int. 2020, 2020, 1061470. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Bloom, D.C. Lund Human Mesencephalic (LUHMES) Neuronal Cell Line Supports Herpes Simplex Virus 1 Latency In Vitro. J. Virol. 2019, 93, 02210-18. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Yin, M.; Zhang, M.-H. Cell-based assays for Parkinson’s disease using differentiated human LUHMES cells. Acta Pharmacol. Sin. 2014, 35, 945–956. [Google Scholar] [CrossRef]
- Tüshaus, J.; Kataka, E.S.; Zaucha, J.; Frishman, D.; Müller, S.A.; Lichtenthaler, S.F. Neuronal Differentiation of LUHMES Cells Induces Substantial Changes of the Proteome. Proteomics 2021, 21, 2000174. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.M.; Schwamborn, J.C. Midbrain Organoids: A New Tool to Investigate Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 359. [Google Scholar] [CrossRef]
- Chlebanowska, P.; Tejchman, A.; Sułkowski, M.; Skrzypek, K.; Majka, M. Use of 3D Organoids as a Model to Study Idiopathic Form of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 694. [Google Scholar] [CrossRef]
- Kaushik, G.; Ponnusamy, M.P.; Batra, S.K. Concise Review: Current Status of Three-Dimensional Organoids as Preclinical Models: 3D Organoid Culture as a Tool for Research. Stem Cells 2018, 36, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Curtius, H.; Wolfensberger, M.; Steinmann, B.; Redweik, U.; Siegfried, J. Mass fragmentography of dopamine and 6-hydroxydopamine. J. Chromatogr. A 1974, 99, 529–540. [Google Scholar] [CrossRef]
- Khan, M.; Ahmad, A.; Ishrat, T.; Hoda, N.; Khuwaja, G.; Raza, S.S.; Khan, A.; Javed, H.; Vaibhav, K.; Islam, F. Resveratrol attenuates 6-hydroxydopamine-induced oxidative damage and dopamine depletion in rat model of Parkinson’s disease. Brain Res. 2010, 1328, 139–151. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Park. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and New Animal Models of Parkinson’s Disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Dawson, T.M.; Dawson, V.L. Models of LRRK2-Associated Parkinson’s Disease. In Leucine-Rich Repeat Kinase 2 (LRRK2); Rideout, H.J., Ed.; Advances in Neurobiology; Springer International Publishing: Cham, Switzerland, 2017; Volume 14, pp. 163–191. ISBN 978-3-319-49967-3. [Google Scholar]
- Xu, Q.; Shenoy, S.; Li, C. Mouse models for LRRK2 Parkinson’s disease. Park. Relat. Disord. 2012, 18, S186–S189. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic Animal Models of Parkinson’s Disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From Magic Bullets to Designed Multiple Ligands. Drug Discov Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Makhouri, F.R.; Ghasemi, J.B. In Silico Studies in Drug Research Against Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 664–725. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Ma, X.H.; Shi, Z.; Tan, C.; Jiang, Y.; Go, M.L.; Low, B.C.; Chen, Y.Z. In-Silico Approaches to Multi-target Drug Discovery. Pharm. Res. 2010, 27, 739–749. [Google Scholar] [CrossRef]
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1290–1308. [Google Scholar] [CrossRef]
- Del Bello, F.; Giannella, M.; Giorgioni, G.; Piergentili, A.; Quaglia, W. Receptor Ligands as Helping Hands to L-DOPA in the Treatment of Parkinson’s Disease. Biomolecules 2019, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.; Gmeiner, P. Dopamine D3 receptor ligands—Recent advances in the control of subtype selectivity and intrinsic activity. Biochim. Biophys. Acta Biomembr. 2007, 1768, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.E.; Fox, S.H. Nondopaminergic treatments for Parkinson’s disease: Current and future prospects. Neurodegener. Dis. Manag. 2016, 6, 249–268. [Google Scholar] [CrossRef] [PubMed]
- VanLe, B.; Olcott, W.; Jiménez, J.; Bashmi, L.; Danovitch, I.; Ishak, W.W. NMDA antagonists for treating the non-motor symptoms in Parkinson’s disease. Transl. Psychiatry 2018, 8, 117. [Google Scholar] [CrossRef]
- Agis-Torres, A.; Sollhuber, M.; Fernandez, M.; Sanchez-Montero, J. Multi-Target-Directed Ligands and other Therapeutic Strategies in the Search of a Real Solution for Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef] [PubMed]
- de Lera Ruiz, M.; Lim, Y.-H.; Zheng, J. Adenosine A2AReceptor as a Drug Discovery Target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef]
- Ishiki, H.M.; Filho, J.M.B.; Da Silva, M.S.; Scotti, M.T.; Scotti, L. Computer-aided Drug Design Applied to Parkinson Targets. Curr. Neuropharmacol. 2018, 16, 865–880. [Google Scholar] [CrossRef]
- Zhang, H.; Jia, H.; Liu, J.; Ao, N.; Yan, B.; Shen, W.; Wang, X.; Li, X.; Luo, C.; Liu, J. Combined R-α-lipoic acid and acetyl-L-carnitine exerts efficient preventative effects in a cellular model of Parkinson’s disease. J. Cell. Mol. Med. 2008, 14, 215–225. [Google Scholar] [CrossRef]
- Butini, S.; Nikolic, K.; Kassel, S.; Brückmann, H.; Filipic, S.; Agbaba, D.; Gemma, S.; Brogi, S.; Brindisi, M.; Campiani, G.; et al. Polypharmacology of dopamine receptor ligands. Prog. Neurobiol. 2016, 142, 68–103. [Google Scholar] [CrossRef]
- Na, Y.; Song, Z.; Zhang, A. Dual Ligands Targeting Dopamine D2 and Serotonin 5-HT1A Receptors as New Antipsychotical or Anti-Parkinsonian Agents. Curr. Med. Chem. 2014, 21, 437–457. [Google Scholar] [CrossRef]
- Blair, H.A.; Dhillon, S. Safinamide: A Review in Parkinson’s Disease. CNS Drugs 2017, 31, 169–176. [Google Scholar] [CrossRef]
- Wang, M.; Hou, S.; Wei, Y.; Li, D.; Lin, J. Discovery of Novel Dual Adenosine A1/A2A Receptor Antagonists Using Deep Learning, Pharmacophore Modeling and Molecular Docking. PLoS Comput. Biol. 2021, 17, e1008821. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Benchekroun, M.; Maramai, S. Multitarget-Directed Ligands for Neurodegenerative Diseases: Real Opportunity or Blurry Mirage? Future Med. Chem. 2019, 11, 261–263. [Google Scholar] [CrossRef]
- Mathew, B.; Oh, J.M.; Baty, R.S.; Batiha, G.E.-S.; Parambi, D.G.T.; Gambacorta, N.; Nicolotti, O.; Kim, H. Piperazine-substituted chalcones: A new class of MAO-B, AChE, and BACE-1 inhibitors for the treatment of neurological disorders. Environ. Sci. Pollut. Res. 2021, 1–12. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; Pruccoli, L.; Bisi, A.; Gobbi, S.; Rampa, A.; Martinez, A.; Pérez, C.; Martinez-Gonzalez, L.; Paglione, M.; Di Schiavi, E.; et al. Novel Curcumin-Diethyl Fumarate Hybrid as a Dualistic GSK-3β Inhibitor/Nrf2 Inducer for the Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 2728–2740. [Google Scholar] [CrossRef]
- Więckowska, A.; Szałaj, N.; Góral, I.; Bucki, A.; Latacz, G.; Kiec-Kononowicz, K.; Bautista-Aguilera, Ò.M.; Romero, A.; Ramos, E.; Egea, J.; et al. In Vitro and In Silico ADME-Tox Profiling and Safety Significance of Multifunctional Monoamine Oxidase Inhibitors Targeting Neurodegenerative Diseases. ACS Chem. Neurosci. 2020, 11, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Zondagh, L.S.; Malan, S.F.; Joubert, J. Design, synthesis and biological evaluation of edaravone derivatives bearing the N-benzyl pyridinium moiety as multifunctional anti-Alzheimer’s agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Gontijo, V.S.; Viegas, F.P.D.; Ortiz, C.J.C.; Silva, M.D.F.; Damasio, C.M.; Rosa, M.C.; Campos, T.G.; Couto, D.S.; Dias, K.S.T.; Viegas, C.; et al. Molecular Hybridization as a Tool in the Design of Multi-target Directed Drug Candidates for Neurodegenerative Diseases. Curr. Neuropharmacol. 2020, 18, 348–407. [Google Scholar] [CrossRef] [PubMed]
- Van Der Schyf, C.J. The use of multi-target drugs in the treatment of neurodegenerative diseases. Expert Rev. Clin. Pharmacol. 2011, 4, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-D.; Zhao, L.; Chen, H.-Y.; Gong, J.-N.; Chen, X.; Chen, C.Y.-C. A novel artificial intelligence protocol to investigate potential leads for Parkinson’s disease. RSC Adv. 2020, 10, 22939–22958. [Google Scholar] [CrossRef]
- Vatansever, S.; Schlessinger, A.; Wacker, D.; Kaniskan, H.Ü.; Jin, J.; Zhou, M.; Zhang, B. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: State-of-the-arts and future directions. Med. Res. Rev. 2021, 41, 1427–1473. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Disrupting Drug Discovery: How Artificial Intelligence Is Transforming Drug Research; IBM Corporation: Armonk, NY, USA, 2018.
- Zhang, R.; Zhu, X.; Bai, H.; Ning, K. Network Pharmacology Databases for Traditional Chinese Medicine: Review and Assessment. Front. Pharmacol. 2019, 10, 123. [Google Scholar] [CrossRef]
- Fan, Q.; Guo, L.; Guan, J.; Chen, J.; Fan, Y.; Chen, Z.; Li, H. Network Pharmacology-Based Study on the Mechanism of Gegen Qinlian Decoction against Colorectal Cancer. Evid.-Based Complement. Altern. Med. 2020, 2020, 8897879. [Google Scholar] [CrossRef]
- Du, W.; Liang, X.; Wang, S.; Lee, P.; Zhang, Y. The Underlying Mechanism of Paeonia lactiflora Pall. in Parkinson’s Disease Based on a Network Pharmacology Approach. Front. Pharmacol. 2020, 11, 581984. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qiu, H.; Liu, M.; Cai, Y. A Network Pharmacology-Based Study of the Molecular Mechanisms of Shaoyao-Gancao Decoction in Treating Parkinson’s Disease. Interdiscip. Sci. Comput. Life Sci. 2020, 12, 131–144. [Google Scholar] [CrossRef]
- Li, J.; Qi, X.; Sun, Y.; Zhang, Y.; Chen, J. Network Pharmacology Analysis on Zhichan Powder in the Treatment of Parkinson’s Disease. Comb. Chem. High Throughput Screen. 2020, 23, 28–40. [Google Scholar] [CrossRef]
- Masih, A.; Singh, S.; Agnihotri, A.K.; Giri, S.; Shrivastava, J.K.; Pandey, N.; Bhat, H.R.; Singh, U.P. Design and development of 1,3,5-triazine-thiadiazole hybrids as potent adenosine A2A receptor (A2AR) antagonist for benefit in Parkinson’s disease. Neurosci. Lett. 2020, 735, 135222. [Google Scholar] [CrossRef] [PubMed]
- Masih, A.; Agnihotri, A.K.; Srivastava, J.K.; Pandey, N.; Bhat, H.R.; Singh, U.P. Discovery of novel 1,3,5-triazine as adenosine A 2A receptor antagonist for benefit in Parkinson’s disease. J. Biochem. Mol. Toxicol. 2021, 35, e22659. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Barawkar, D.A.; Ramdas, V.; Naykodi, M.; Shejul, Y.D.; Patel, M.; Thorat, S.; Panmand, A.; Kashinath, K.; Bonagiri, R.; et al. Discovery of Potent and Selective A2A Antagonists with Efficacy in Animal Models of Parkinson’s Disease and Depression. ACS Med. Chem. Lett. 2017, 8, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, H.J.; Legoabe, L.; Terre’Blanche, G.; Van Der Walt, M. 2-Benzylidene-1-Indanone Analogues as Dual Adenosine A1/A2a Receptor Antagonists for the Potential Treatment of Neurological Conditions. Drug Res. 2019, 69, 382–391. [Google Scholar] [CrossRef]
- Van Rensburg, H.D.J.; Legoabe, L.J.; Terre’Blanche, G. Synthesis and Structure Activity Relationships of Chalcone based Benzocycloalkanone Derivatives as Adenosine A1 and/or A2A Receptor Antagonists. Drug Res. 2020, 70, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.-M.; Ma, X.; Paira, P.; Tan, A.; Herr, D.R.; Lim, K.L.; Ng, C.H.; Venkatesan, G.; Klotz, K.-N.; Federico, S.; et al. Discovery of indolylpiperazinylpyrimidines with dual-target profiles at adenosine A2A and dopamine D2 receptors for Parkinson’s disease treatment. PLoS ONE 2018, 13, e0188212. [Google Scholar] [CrossRef]
- Olanow, C.W.; Rascol, O.; Hauser, R.; Feigin, P.D.; Jankovic, J.; Lang, A.; Langston, W.; Melamed, E.; Poewe, W.; Stocchi, F.; et al. A Double-Blind, Delayed-Start Trial of Rasagiline in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1268–1278. [Google Scholar] [CrossRef]
- Kavully, F.S.; Oh, J.M.; Dev, S.; Kaipakasseri, S.; Palakkathondi, A.; Vengamthodi, A.; Azeez, R.F.A.; Tondo, A.R.; Nicolotti, O.; Kim, H.; et al. Design of enamides as new selective monoamine oxidase-B inhibitors. J. Pharm. Pharmacol. 2020, 72, 916–926. [Google Scholar] [CrossRef]
- Chavarria, D.; Fernandes, C.; Silva, V.; Silva, C.; Gil-Martins, E.; Soares, P.; Silva, T.; Silva, R.; Remião, F.; Oliveira, P.J.; et al. Design of novel monoamine oxidase-B inhibitors based on piperine scaffold: Structure-activity-toxicity, drug-likeness and efflux transport studies. Eur. J. Med. Chem. 2020, 185, 111770. [Google Scholar] [CrossRef]
- Tao, D.; Wang, Y.; Bao, X.-Q.; Yang, B.-B.; Gao, F.; Wang, L.; Zhang, D.; Li, L. Discovery of coumarin Mannich base derivatives as multifunctional agents against monoamine oxidase B and neuroinflammation for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2019, 173, 203–212. [Google Scholar] [CrossRef]
- Jismy, B.; El Qami, A.; Pišlar, A.; Frlan, R.; Kos, J.; Gobec, S.; Knez, D.; Abarbri, M. Pyrimido[1,2-b]indazole derivatives: Selective inhibitors of human monoamine oxidase B with neuroprotective activity. Eur. J. Med. Chem. 2021, 209, 112911. [Google Scholar] [CrossRef]
- Carradori, S.; Ortuso, F.; Petzer, A.; Bagetta, D.; De Monte, C.; Secci, D.; de Vita, D.; Guglielmi, P.; Zengin, G.; Aktumsek, A.; et al. Design, synthesis and biochemical evaluation of novel multi-target inhibitors as potential anti-Parkinson agents. Eur. J. Med. Chem. 2018, 143, 1543–1552. [Google Scholar] [CrossRef]
- Affini, A.; Hagenow, S.; Zivkovic, A.; Marco-Contelles, J.; Stark, H. Novel indanone derivatives as MAO B/H3R dual-targeting ligands for treatment of Parkinson’s disease. Eur. J. Med. Chem. 2018, 148, 487–497. [Google Scholar] [CrossRef]
- Osborne, J.; Birchall, K.; Tsagris, D.J.; Lewis, S.J.; Smiljanic-Hurley, E.; Taylor, D.L.; Levy, A.; Alessi, D.; McIver, E.G. Discovery of potent and selective 5-azaindazole inhibitors of leucine-rich repeat kinase 2 (LRRK2)—Part 1. Bioorg. Med. Chem. Lett. 2019, 29, 668–673. [Google Scholar] [CrossRef]
- Salado, I.G.; Zaldivar-Diez, J.; Sebastián-Pérez, V.; Li, L.; Geiger, L.; González-Pelayo, S.; Campillo, N.E.; Gil, C.; Morales, A.V.; Perez, D.I.; et al. Leucine rich repeat kinase 2 (LRRK2) inhibitors based on indolinone scaffold: Potential pro-neurogenic agents. Eur. J. Med. Chem. 2017, 138, 328–342. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009399. [Google Scholar] [CrossRef]
- Maqbool, M.; Gadhavi, J.; Hivare, P.; Gupta, S.; Hoda, N. Diphenyl triazine hybrids inhibit α-synuclein fibrillogenesis: Design, synthesis and in vitro efficacy studies. Eur. J. Med. Chem. 2020, 207, 112705. [Google Scholar] [CrossRef]
- Li, S.; Wei, D.; Mao, Z.; Chen, L.; Yan, X.; Li, Y.; Dong, S.; Wang, D. Design, synthesis, immunocytochemistry evaluation, and molecular docking investigation of several 4-aminopyridine derivatives as potential neuroprotective agents for treating Parkinson’s disease. Bioorg. Chem. 2017, 73, 63–75. [Google Scholar] [CrossRef]
- Hu, X.-L.; Lin, J.; Lv, X.-Y.; Feng, J.-H.; Zhang, X.-Q.; Wang, H.; Ye, W.-C. Synthesis and biological evaluation of clovamide analogues as potent anti-neuroinflammatory agents in vitro and in vivo. Eur. J. Med. Chem. 2018, 151, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-Y.; Kim, I.-S.; Koppula, S.; Park, J.-Y.; Kim, B.-W.; Yoon, S.-H.; Choi, D.-K. 2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8195. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, S.; Yoo, J.S.; Kim, H.J.; Kim, H.J.; Kim, B.E.; Lee, E.H.; Lee, Y.S.; Park, J.-H.; Park, K.D. Development and optimization of halogenated vinyl sulfones as Nrf2 activators for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2021, 212, 113103. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, X.; Shi, M.; Xue, L.; Kuang, S.; Xu, R.; Qi, W.; Li, Y.; Ma, X.; Zhang, R.; et al. Identification and optimization of piperine analogues as neuroprotective agents for the treatment of Parkinson’s disease via the activation of Nrf2/keap1 pathway. Eur. J. Med. Chem. 2020, 199, 112385. [Google Scholar] [CrossRef]
- Temme, L.; Bechthold, E.; Schreiber, J.A.; Gawaskar, S.; Schepmann, D.; Robaa, D.; Sippl, W.; Seebohm, G.; Wünsch, B. Negative allosteric modulators of the GluN2B NMDA receptor with phenylethylamine structure embedded in ring-expanded and ring-contracted scaffolds. Eur. J. Med. Chem. 2020, 190, 112138. [Google Scholar] [CrossRef]
- Kamakolanu, U.G.; Meyer, M.E.; Yasuda, D.; Polgar, W.E.; Marti, M.; Mercatelli, D.; Pisanò, C.A.; Brugnoli, A.; Morari, M.; Zaveri, N.T. Discovery and Structure–Activity Relationships of Nociceptin Receptor Partial Agonists That Afford Symptom Ablation in Parkinson’s Disease Models. J. Med. Chem. 2020, 63, 2688–2704. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, L.; Schwake, M.; Silverman, R.B.; Krainc, D. Design and Synthesis of Potent Quinazolines as Selective β-Glucocerebrosidase Modulators. J. Med. Chem. 2016, 59, 8508–8520. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Zhang, L.; Zhang, S.; Yao, D.; Zhao, Y.; Wang, G.; Fu, L.; Lei, P.; Liu, B. Small-Molecule Activator of UNC-51-Like Kinase 1 (ULK1) That Induces Cytoprotective Autophagy for Parkinson’s Disease Treatment. J. Med. Chem. 2018, 61, 2776–2792. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Huang, C.; Zhong, J.; He, J.; Guo, J.; Liu, M.; Xu, J.-P.; Wang, H.-T.; Zhou, Z.-Z. Discovery of Arylbenzylamines as PDE4 Inhibitors with Potential Neuroprotective Effect. Eur. J. Med. Chem. 2019, 168, 221–231. [Google Scholar] [CrossRef] [PubMed]



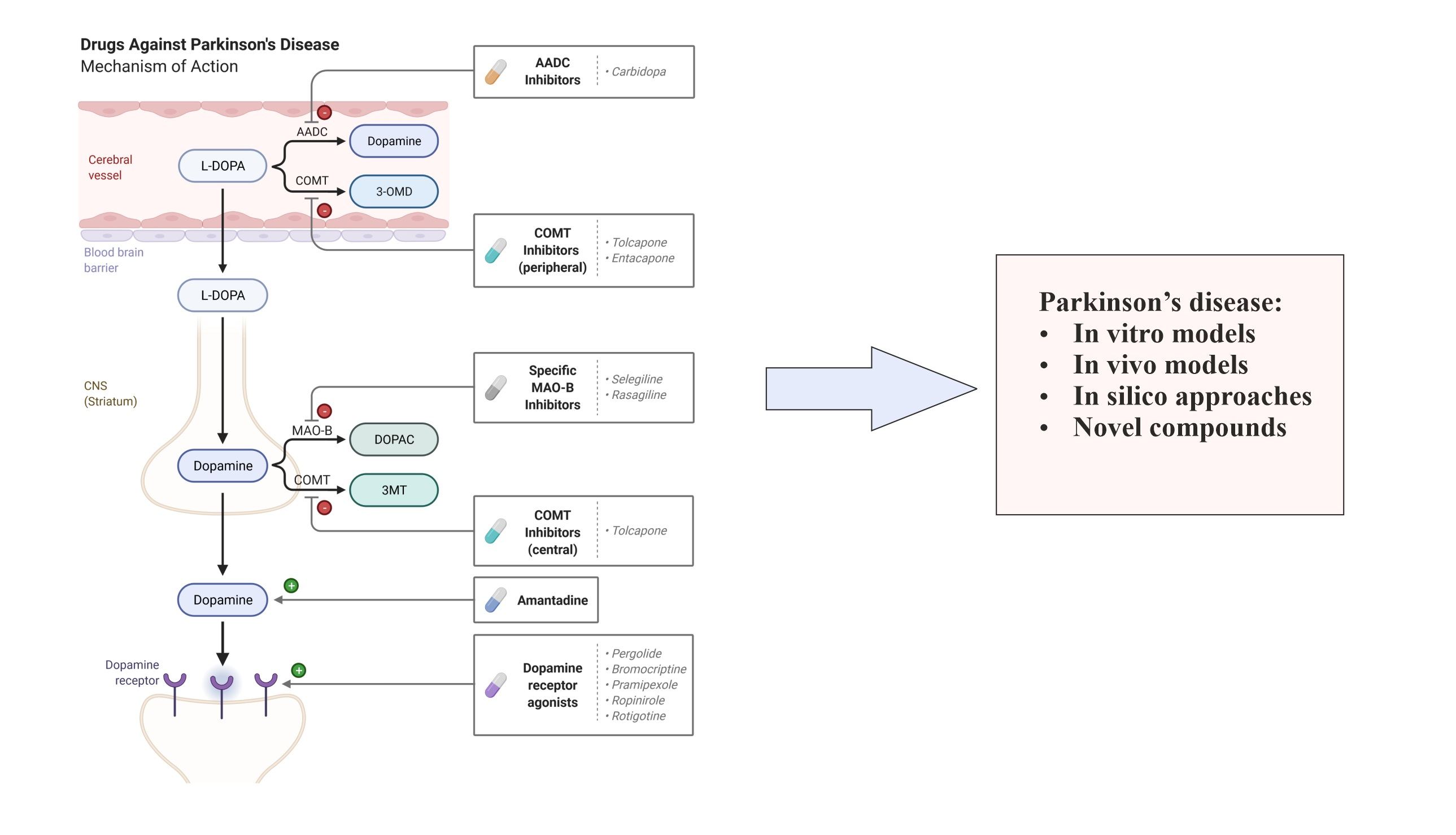{kind=link}
{kind=link}
{kind=link}
| Ki (nM) | EC50 (µM) | IC50 (µM) | % Inhibition at 3 µM | Ki (nM) | IC50 (nM) | |||
|---|---|---|---|---|---|---|---|---|
| A2AR | A1R | D2R | MAO-B | MAO SI a | AChE | H3R | LRRK2 | |
| 1 | 32 | 322 | ||||||
| 2 | 1.5 | 478 | ||||||
| 3 | 1.5 | 1700 | ||||||
| 4 | 903 | 435 | ||||||
| 5 | 434 | 792 | ||||||
| 6 | 11,200 | >100,000 | 22.5 | |||||
| 7 | 0.11 | >363 | ||||||
| 8 | 0.047 | >211 | ||||||
| 9 | 3.66 | >100 | ||||||
| 10 | 0.13 | >769 | ||||||
| 11 | 0.0053 | >501 | 44 | |||||
| 12 | 0.276 | >36 | 6.5 | |||||
| 13 | 2 | |||||||
| 14 | 10 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koszła, O.; Stępnicki, P.; Zięba, A.; Grudzińska, A.; Matosiuk, D.; Kaczor, A.A. Current Approaches and Tools Used in Drug Development against Parkinson’s Disease. Biomolecules 2021, 11, 897. https://doi.org/10.3390/biom11060897
Koszła O, Stępnicki P, Zięba A, Grudzińska A, Matosiuk D, Kaczor AA. Current Approaches and Tools Used in Drug Development against Parkinson’s Disease. Biomolecules. 2021; 11(6):897. https://doi.org/10.3390/biom11060897
Chicago/Turabian StyleKoszła, Oliwia, Piotr Stępnicki, Agata Zięba, Angelika Grudzińska, Dariusz Matosiuk, and Agnieszka A. Kaczor. 2021. "Current Approaches and Tools Used in Drug Development against Parkinson’s Disease" Biomolecules 11, no. 6: 897. https://doi.org/10.3390/biom11060897
APA StyleKoszła, O., Stępnicki, P., Zięba, A., Grudzińska, A., Matosiuk, D., & Kaczor, A. A. (2021). Current Approaches and Tools Used in Drug Development against Parkinson’s Disease. Biomolecules, 11(6), 897. https://doi.org/10.3390/biom11060897