
Fecal Metaproteomics Reveals Reduced Gut Inflammation and Changed Microbial Metabolism Following Lifestyle-Induced Weight Loss

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
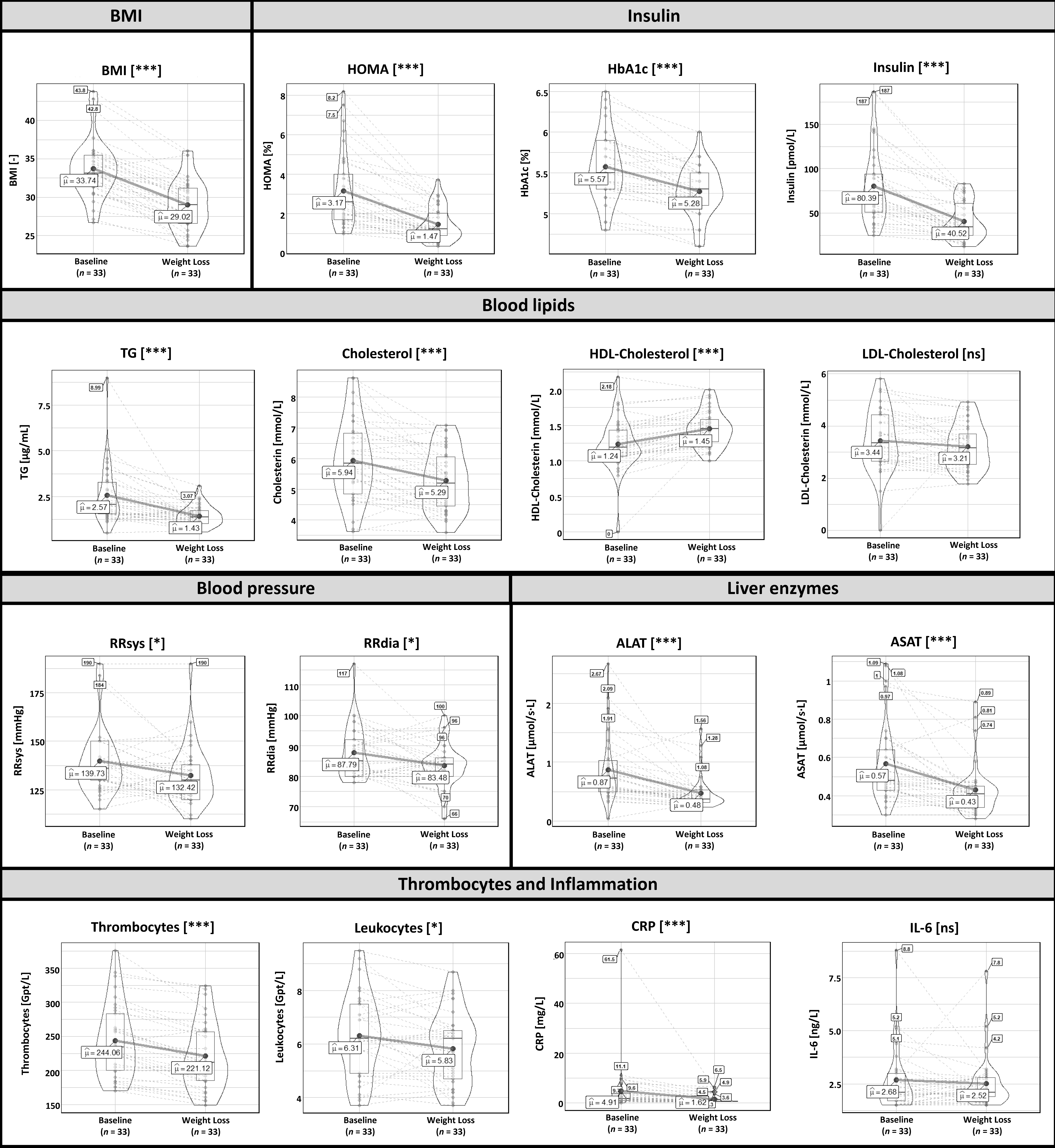
3.1. Clinical and Laboratory Parameters
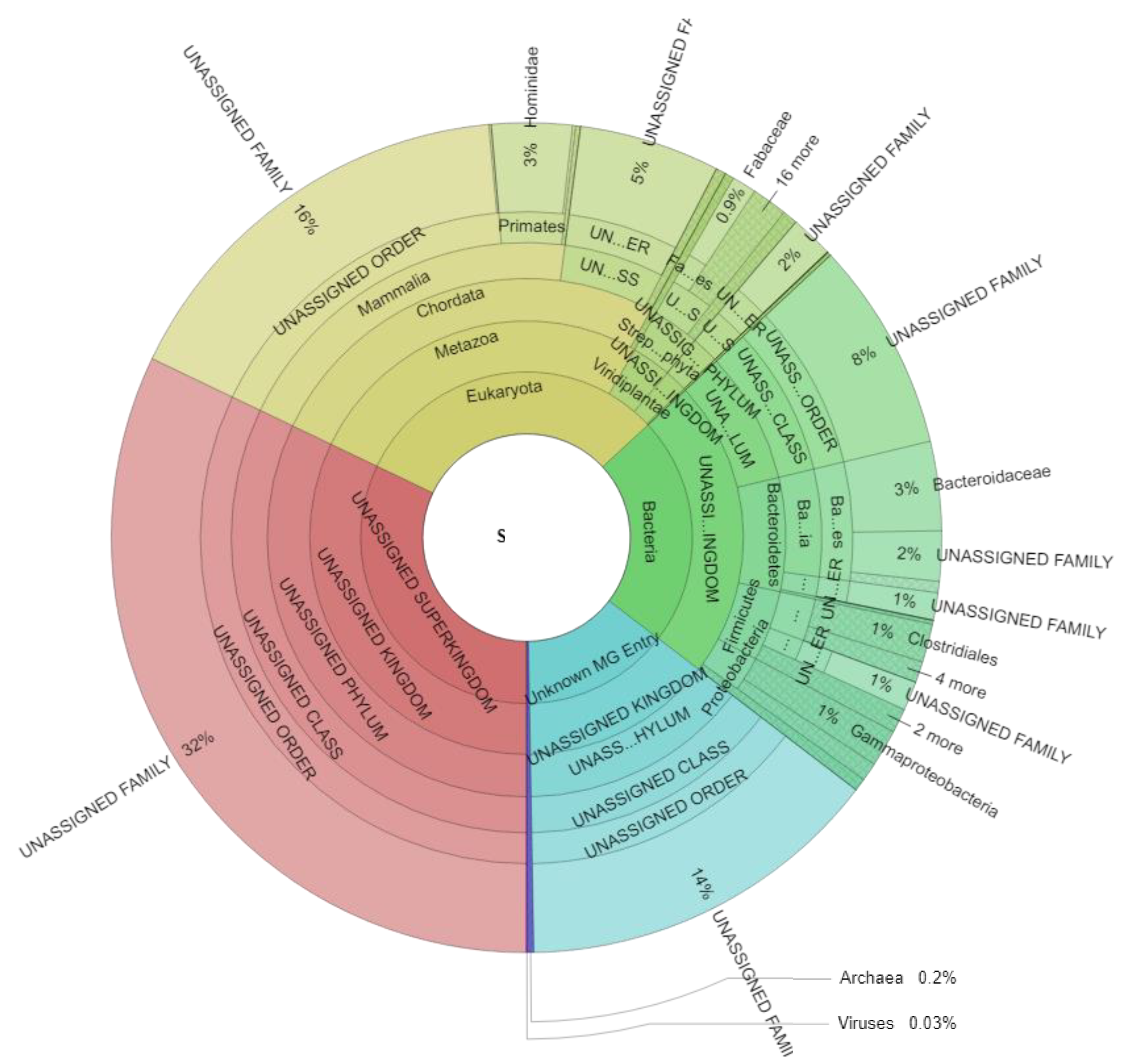
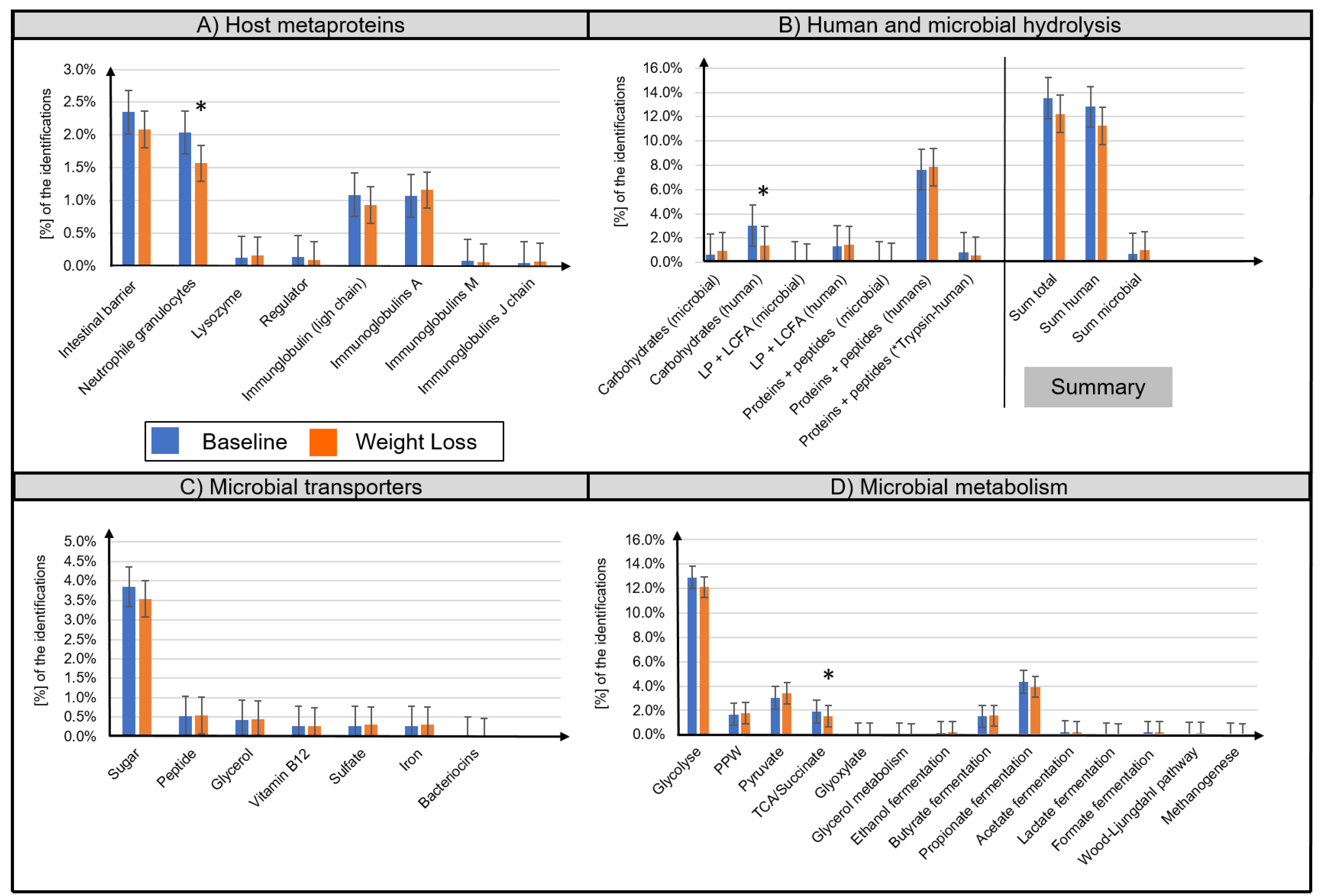
3.2. Metaproteome Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cani, P.D.; Jordan, B.F. Gut microbiota-mediated inflammation in obesity: A link with gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, T.; Schallert, K.; Vilchez-Vargas, R.; Benndorf, D.; Püttker, S.; Sydor, S.; Schulz, C.; Bechmann, L.; Canbay, A.; Heidrich, B.; et al. Metaproteomics of fecal samples of Crohn’s disease and Ulcerative Colitis. J. Proteom. 2019, 201, 93–103. [Google Scholar] [CrossRef]
- Gavin, P.G.; Mullaney, J.A.; Loo, D.; Cao, K.-A.L.; Gottlieb, P.A.; Hill, M.M.; Zipris, D.; Hamilton-Williams, E.E. Intestinal Metaproteomics Reveals Host-Microbiota Interactions in Subjects at Risk for Type 1 Diabetes. Diabetes Care 2018, 41, 2178–2186. [Google Scholar] [CrossRef]
- Ferrer, M.; Ruiz, A.; Lanza, F.; Haange, S.-B.; Oberbach, A.; Till, H.; Bargiela, R.; Campoy, C.; Segura, M.T.; Richter, M.; et al. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 2013, 15, 211–226. [Google Scholar] [CrossRef]
- Kolmeder, C.A.; Ritari, J.; Verdam, F.J.; Muth, T.; Keskitalo, S.; Varjosalo, M.; Fuentes, S.; Greve, J.M.; Buurman, W.A.; Reichl, U.; et al. Colonic metaproteomic signatures of active bacteria and the host in obesity. Proteomics 2015, 15, 3544–3552. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef]
- Bindels, L.B.; Delzenne, N.M.; Cani, P.; Walter, J. Towards a more comprehensive concept for prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 303–310. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nat. Cell Biol. 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Li, H.; Wang, R.; Tang, J.; Huang, T.; et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: A 6-month randomised controlled-feeding trial. Gut 2019, 68, 1417–1429. [Google Scholar] [CrossRef]
- Massier, L.; Chakaroun, R.; Tabei, S.; Crane, A.; Didt, K.D.; Fallmann, J.; Von Bergen, M.; Haange, S.-B.; Heyne, H.; Stumvoll, M.; et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut 2020, 69. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.B.; Mather, K. Targeting the Consequences of the Metabolic Syndrome in the Diabetes Prevention Program. Arter. Thromb. Vasc. Biol. 2012, 32, 2077–2090. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Büdel, A.; Zoun, R.; Dorl, S.; Behne, A.; Kohrs, F.; Püttker, S.; Siewert, C.; Benndorf, D.; et al. A Robust and Universal Metaproteomics Workflow for Research Studies and Routine Diagnostics within 24 h Using Phenol Extraction, FASP Digest, and the MetaProteomeAnalyzer. Front Microbiol. 2019, 10, 1883. [Google Scholar] [CrossRef] [PubMed]
- Rechenberger, J.; Samaras, P.; Jarzab, A.; Behr, J.; Frejno, M.; Djukovic, A.; Kohrs, F.; Püttker, S.; Siewert, C.; Muth, T.; et al. Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae. Proteomes 2019, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Biemann, R.; Penner, M.; Borucki, K.; Westphal, S.; Luley, C.; Rönicke, R.; Biemann, K.; Weikert, C.; Lux, A.; Goncharenko, N.; et al. Serum bile acids and GLP-1 decrease following telemetric induced weight loss: Results of a randomized controlled trial. Sci. Rep. 2016, 6, 30173. [Google Scholar] [CrossRef]
- Luley, C.; Blaik, A.; Götz, A.; Kicherer, F.; Kropf, S.; Isermann, B.; Stumm, G.; Westphal, S. Weight loss by telemonitoring of nutrition and physical activity in patients with metabolic syndrome for 1 year. J. Am. Coll. Nutr. 2014, 33, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Verdam, F.J.; Fuentes, S.; Jonge, C.; de Zoetendal, E.G.; Erbil, R.; Greve, J.W.; Buurman, W.A.; de Vos, W.M.; Rensen, S.S.; Rensen, S.S.; et al. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity 2013, 21, 15. [Google Scholar] [CrossRef]
- Mortensen, O.H.; Nielsen, A.R.; Erikstrup, C.; Plomgaard, P.; Fischer, C.P.; Krogh-Madsen, R.; Lindegaard, B.; Petersen, A.M.; Taudorf, S.; Pedersen, B.K.; et al. Calprotectin—A novel marker of obesity. PLoS ONE 2009, 4, e7419. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.B.; Zheng, W.L.; Wang, L.H.; Lu, L.Y. Alpha 1-antitrypsin. A novel biomarker for obesity in humans. Saudi Med. J. 2013, 34, 34–39. [Google Scholar]
- Kristinsson, J.; Armbruster, C.H.; Ugstad, M.; Kriwanek, S.; Nygaard, K.; Tøn, H.; Fuglerud, P. Fecal excretion of calprotectin in colorectal cancer: Relationship to tumor characteristics. Scand J. Gastroenterol. 2001, 36, 202–207. [Google Scholar] [CrossRef]
- LeDuc, Y.A.; Prasad, L.; Laivenieks, M.; Zeikus, J.G.; Delbaere, L.T.J. Structure of PEP carboxykinase from the succinate-producingActinobacillus succinogenes: A new conserved active-site motif. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Veledo, S.; Vendrell, J. Gut microbiota-derived succinate: Friend or foe in human metabolic diseases? Rev. Endocr. Metab. Disord. 2019, 20, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Serena, C.; Ceperuelo-Mallafré, V.; Keiran, N.; Queipo-Ortuño, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Pérez-Brocal, V.; Andrés-Lacueva, C.; et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef] [PubMed]
- Lumppio, H.L.; Shenvi, N.V.; Summers, A.O.; Voordouw, G.; Kurtz, D.M. Rubrerythrin and rubredoxin oxidoreductase in Desulfovibrio vulgaris: A novel oxidative stress protection system. J. Bacteriol. 2001, 183, 101–108. [Google Scholar] [CrossRef]
- Cullender, T.C.; Chassaing, B.; Janzon, A.; Kumar, K.; Muller, C.E.; Werner, J.J.; Angenent, L.T.; Bell, M.E.; Hay, A.G.; Peterson, D.A.; et al. Innate and Adaptive Immunity Interact to Quench Microbiome Flagellar Motility in the Gut. Cell Host Microbe 2013, 14, 571–581. [Google Scholar] [CrossRef]
- Tran, H.Q.; Mills, R.H.; Peters, N.V.; Holder, M.K.; de Vries, G.J.; Knight, R.; Chassaing, B.; Gonzalez, D.J.; Gewirtz, A.T. Associations of the Fecal Microbial Proteome Composition and Proneness to Diet-induced Obesity. Mol. Cell Proteom. 2019, 18, 1864–1879. [Google Scholar] [CrossRef]
- Söling, H.D.; Unger, K.O. The Role of Insulin in the Regulation of α-Amylase Synthesis in the Rat Pancreas. Eur. J. Clin. Investig. 1972, 2, 199–212. [Google Scholar] [CrossRef]
- Afsartala, Z.; Savabkar, S.; Mojarad, E.N.; Assadollahi, V.; Tanha, S.; Bijangi, K.; Gholami, M. Expression of liver alpha-amylase in obese mouse hepatocytes. Gastroenterol. Hepatol. Bed Bench 2016, 9, 278–285. [Google Scholar] [PubMed]
- Azzout-Marniche, D.; Chaumontet, C.; Piedcoq, J.; Khodorova, N.; Fromentin, G.; Tomé, D.; Gaudichon, C.; Even, P.C. High Pancreatic Amylase Expression Promotes Adiposity in Obesity-Prone Carbohydrate-Sensitive Rats. J. Nutr. 2019, 149, 270–279. [Google Scholar] [CrossRef]
- Ahuja, M.; Schwartz, D.M.; Tandon, M.; Son, A.; Zeng, M.; Swaim, W.; Xiao, B.; Cui, Y.; Worley, P.F.; Muallem, S. Orai1-Mediated Antimicrobial Secretion from Pancreatic Acini Shapes the Gut Microbiome and Regulates Gut Innate Immunity. Cell Metab. 2017, 25, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar]
- Grembi, J.A.; Nguyen, L.H.; Haggerty, T.D.; Gardner, C.D.; Holmes, S.P.; Parsonnet, J. Gut microbiota plasticity is correlated with sustained weight loss on a low-carb or low-fat dietary intervention. Sci. Rep. 2020, 10, 1405. [Google Scholar] [CrossRef] [PubMed]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.; Sokol, H.; Doré, J.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Cani, P.D.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Campbell, C.L.; Yu, R.; Li, F.; Zhou, Q.; Chen, D.; Qi, C.; Yin, Y.; Sun, J. Modulation of fat metabolism and gut microbiota by resveratrol on high-fat diet-induced obese mice. Diabetes Metab. Syndr. Obes. Targets 2019, 12, 97–107. [Google Scholar] [CrossRef]
- Sonoyama, K.; Fujiwara, R.; Takemura, N.; Ogasawara, T.; Watanabe, J.; Ito, H.; Morita, T. Response of Gut Microbiota to Fasting and Hibernation in Syrian Hamsters. Appl. Environ. Microbiol. 2009, 75, 6451–6456. [Google Scholar] [CrossRef]
- Derrien, M.; van Passel, M.W.; van de Bovenkamp, J.H.; Schipper, R.G.; Vos, W.M.; de Dekker, J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Family | Abundance Baseline | Abundance after WL | Fold Change | p-Value |
|---|---|---|---|---|
| Thermotogaceae | 3.1 × 10−4 ± 4.3 × 10−4 | 8.0 × 10−4 ± 1.3 × 10−3 | 2.6 | 0.01 |
| Desulfovibrionaceae | 1.1 × 10−4 ± 2.1 × 10−4 | 3.4 × 10−4 ± 4.8 × 10−4 | 3.2 | 0.01 |
| Leptospiraceae | 7.0 × 10−5 ± 2.1 × 10−4 | 1.5 × 10−4 ± 2.6 × 10−4 | 2.1 | 0.04 |
| Syntrophomonadaceae | 1.0 × 10−5 ± 4.0 × 10−5 | 7.5 × 10−5 ± 1.6 × 10−4 | 7.5 | 0.03 |
| Siphoviridae | 7.4 × 10−6 ± 3.0 × 10−5 | 3.4 × 10−5 ± 6.3 × 10−5 | 4.6 | 0.03 |
| Rutaceae | 4.4 × 10−6 ± 2.5 × 10−5 | 2.6 × 10−5 ± 5.8 × 10−5 | 5.9 | 0.05 |
| Verrucomicrobiaceae | Not Detectable | 1.0 × 10−4 ± 3.7 × 10−4 | Only in WL | 0.04 |
| Microbial Metaproteins (ID) | Description | Taxonomy | Average % Abundance | p-Value | Fold Change |
|---|---|---|---|---|---|
| 87 | Phosphoenolpyruvate carboxykinase (ATP) | Superkingdom: Bacteria | 1.5 × 10−2 | 0.03 | 0.8 |
| 1176 | 5-keto-D-gluconate 5-reductase | Unknown Superkingdom | 4.6 × 10−4 | 0.04 | 1.6 |
| 3789 | Endoglucanase A | Order: Clostridiales | 3.3 × 10−4 | 0.00 | Only in WL |
| 346 | Galactokinase | Unknown Superkingdom | 2.3 × 10−4 | 0.00 | 2.5 |
| 8627 | Endoglucanase A | Species: Clostridium thermocellum | 1.3 × 10−4 | 0.04 | Only in WL |
| 1745 | Flagellin | Superkingdom: Bacteria | 9.0 × 10−5 | 0.01 | 7.7 |
| 6585 | Rubredoxin | Species: Clostridium acetobutylicum | 4.5 × 10−5 | 0.01 | Only in WL |
| 1838 | Beta-1,4-mannooligosaccharide phosphorylase | Species: Ruminococcus albus | 4.5 × 10−5 | 0.04 | Only in WL |
| 6265 | Phosphate propanoyltransferase | Species: Thermotoga maritima | 1.4 × 10−5 | 0.04 | Only in WL |
| Human Metaproteins (ID) | Description | Taxonomy | Average % Abundance | p-Value | Fold Change |
| 33 | Pancreatic alpha-amylase | Phylum: Chordata | 1.9 × 10−2 | 0.00 | 0.4 |
| 67 | Calcium-activated chloride channel regulator 1 | Class: Mammalia | 3.8 × 10−3 | 0.01 | 0.7 |
| 159 | Protein S100-A9 | Species: Homo sapiens | 2.4 × 10−4 | 0.01 | 0.6 |
| 197 | Alpha-1-antichymotrypsin | Family: Hominidae | 1.7 × 10−4 | 0.02 | 0.6 |
| 212 | Immunoglobulin kappa variable 1–33 | Species: Homo sapiens | 8.5 × 10−4 | 0.03 | 0.6 |
| 370 | Cadherin-1 | Phylum: Chordata | 6.1 × 10−4 | 0.03 | 0.5 |
| 1571 | Immunoglobulin J chain | Species: Homo sapiens | 6.1 × 10−4 | 0.02 | 1.7 |
| 169 | Neutrophil gelatinase-associated lipocalin | Species: Homo sapiens | 3.3 × 10−4 | 0.05 | 0.5 |
| 600 | Immunoglobulin kappa variable 3–15 | Species: Homo sapiens | 2.3 × 10−4 | 0.04 | 0.4 |
| 3996 | Fibrillin-1 | Class: Mammalia | 6.3 × 10−5 | 0.02 | 0.0 |
| 1852 | HLA class II histocompatibility antigen, DRB1-4 beta chain | Species: Homo sapiens | 7.6 × 10−6 | 0.04 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biemann, R.; Buß, E.; Benndorf, D.; Lehmann, T.; Schallert, K.; Püttker, S.; Reichl, U.; Isermann, B.; Schneider, J.G.; Saake, G.; et al. Fecal Metaproteomics Reveals Reduced Gut Inflammation and Changed Microbial Metabolism Following Lifestyle-Induced Weight Loss. Biomolecules 2021, 11, 726. https://doi.org/10.3390/biom11050726
Biemann R, Buß E, Benndorf D, Lehmann T, Schallert K, Püttker S, Reichl U, Isermann B, Schneider JG, Saake G, et al. Fecal Metaproteomics Reveals Reduced Gut Inflammation and Changed Microbial Metabolism Following Lifestyle-Induced Weight Loss. Biomolecules. 2021; 11(5):726. https://doi.org/10.3390/biom11050726
Chicago/Turabian StyleBiemann, Ronald, Enrico Buß, Dirk Benndorf, Theresa Lehmann, Kay Schallert, Sebastian Püttker, Udo Reichl, Berend Isermann, Jochen G. Schneider, Gunter Saake, and et al. 2021. "Fecal Metaproteomics Reveals Reduced Gut Inflammation and Changed Microbial Metabolism Following Lifestyle-Induced Weight Loss" Biomolecules 11, no. 5: 726. https://doi.org/10.3390/biom11050726
APA StyleBiemann, R., Buß, E., Benndorf, D., Lehmann, T., Schallert, K., Püttker, S., Reichl, U., Isermann, B., Schneider, J. G., Saake, G., & Heyer, R. (2021). Fecal Metaproteomics Reveals Reduced Gut Inflammation and Changed Microbial Metabolism Following Lifestyle-Induced Weight Loss. Biomolecules, 11(5), 726. https://doi.org/10.3390/biom11050726