
Reflections on Cerebellar Neuropathology in Classical Scrapie

 ,
,  and
and
Abstract
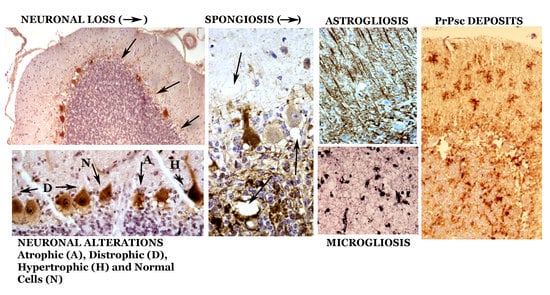
1. Introduction
2. Historical Background
2.1. Cerebellum
2.2. Scrapie as Natural Prion Disease Model
3. Neuropathologic Alterations of the Cerebellum in Classical Scrapie
3.1. Neuronal Alterations
3.2. Neuroglial Alterations
3.3. Spongiform Alterations
3.4. Deposits of Aberrant Prion Protein
4. Considerations for Studies on Cerebellar Neuropathological Alterations in Classical Scrapie
4.1. Comparison of the Cerebellar Neuropathological Aspects of Classical Natural Scrapie with Other Neuropathological Alterations in Selected Brain Regions of Prion/Prion-Like Diseases
4.2. Sequence of the Temporal Appearance of Neuropathological Markers
4.3. Initiation and Progression of Cerebellar Neuropathology
4.4. Extra-and Intraneuronal Vacuolization: A Differential Feature Not Well Explained
4.4.1. Extraneuronal Vacuoles
4.4.2. Intraneuronal Vesicles
4.5. Alterations and Neuronal Loss: Neuronal Specifity
- (1)
- Alterations in neuronal synaptic regions. Studies of the localization of normal cellular prion molecules seem to indicate that they are homogeneously localized on all surfaces of neurons, including the soma, dendrites and axons. However, several studies have indicated that the greatest neuropathological alterations are observed in dendritic areas with high synaptic functions or in axons of these neurons. The initial accumulations of neurotoxic prions (external and/or induced in the host) could be focused on areas of high neurotransmission. The evolution of areas of high synapse concentration in scrapie has been reported in several experimental studies.
- (2)
- In different neuronal types, especially in Purkinje neurons, various types of morphological alterations have been shown (as mentioned above), especially at the level of EM, and they have been interpreted in different ways over the years [76,81,82,83,84,85,86,87,88,89]. Some alterations (especially the so-called “tubulovesicular structures”) were primarily considered to be specific for prion diseases but were later “declassified” as pathognomonic markers of the disease [84,85,86,87]. Axons and dendrites of PCs have shown a large number of pathological structures with a very high variety of shapes and sizes. However, these structures have been observed in many other situations (aging and neurodegenerative diseases), and their interpretation is primarily hindered by the lack of a clear definition and a lack of information on the corresponding cellular or subcellular alterations. Some of these structures correspond to neoformations of the smooth or rough endoplasmic reticulum, while others correspond to lysosomal formations or accumulations of products and/or cytoplasmic areas of cell degradation. In different studies on experimental models of neurodegenerative diseases (especially transgenic mice), many of these structures have been observed; however, whether they are markers of specific neurotoxic changes or consequences of cell involution has not been clarified.
4.6. Neurogliosis: A Main Feature
4.7. Abnormal Prion Protein Deposits: Diversity and Controversial Cell Association
5. Conclusions and Future Perspectives in the Research
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Liberski, P. Historical overview of prion diseases: A view from afar. Folia Neuropathol. 2012, 50, 1–12. [Google Scholar]
- Acín, C.; Martín-Burriel, I.; Goldmann, W.; Lyahyai, J.; Monzón, M.; Bolea, R.; Smith, A.; Rodellar, C.; Badiola, J.J.; Zaragoza, P. Prion protein gene polymorphisms in healthy and scrapie-affected Spanish sheep. J. Gen. Virol. 2004, 85, 2103–2110. [Google Scholar] [CrossRef]
- Baringer, J.R.; Prusiner, S.B. Experimental scrapie in mice: Ultrastructural observations. Ann. Neurol. 1978, 4, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.L. Ultrastructural pathology of scrapie in the mouse: An electron microscopic study of spinal cord and cerebellar areas. Br. J. Exp. Pathol. 1968, 49, 52–59. [Google Scholar]
- Fraser, H.; Dickinson, A.G. Distribution of experimentally induced scrapie lesions in the brain. Nature 1967, 216, 1310–1311. [Google Scholar] [CrossRef] [PubMed]
- Field, E.J.; Raine, C.S.; Joyce, G. Scrapie in the rat: An electron-microscopic study. II. Glial inclusions. Acta Neuropathol. 1967, 8, 9305–9315. [Google Scholar] [CrossRef]
- Beck, E.; Daniel, P.M.; Parry, H.B. Degeneration of the cerebellar and hypothalamo neurohypophysial systems in sheep with scrapie; and its relationship to human system degenerations. Brain 1964, 87, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.J.; Carlson, E.S. Resistance, vulnerability and resilience: A review of the cognitive cerebellum in aging and neurodegenerative diseases. Neurobiol. Learn Mem. 2020, 170, 106981. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.I.; Rivas, L.; LaCruz, C.; Toledano, A. Astroglial cell subtypes in the cerebella of normal adults, elderly adults, and patients with Alzheimer’s disease: A histological and immunohistochemical comparison. Glia 2015, 63, 287–312. [Google Scholar] [CrossRef]
- Vidal, E.; Acín, C.; Foradada, L.; Monzón, M.; Márquez, M.; Monleón, E.; Pumarola, M.; Badiola, J.J.; Bolea, R. Immunohistochemical characterisation of classical scrapie neuropathology in sheep. J. Comp. Pathol. 2009, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Alvarez, M.I.; Monleón, E.; Toledano-Díaz, A.; Badiola, J.J.; Monzón, M. Changes induced by natural scrapie in the calretinin-immunopositive cells and fibres of the sheep cerebellar cortex. Cerebellum 2012, 11, 593–604. [Google Scholar] [CrossRef]
- Monzón, M.; Hernández, R.S.; Garcés, M.; Sarasa, R.; Badiola, J.J. Glial alterations in human prion diseases: A correlative study of astroglia, reactive microglia, protein deposition, and neuropathological lesions. Medicine (Baltimore) 2018, 97, e0320. [Google Scholar] [CrossRef] [PubMed]
- VoŽeh, F. Jan Evangelista Purkyně and the cerebellum then and now. Physiol. Res. 2015, 64, S567–S584. [Google Scholar] [CrossRef] [PubMed]
- Ramón y Cajal, S. Sobre las fibras nerviosas de la capa molecular del cerebelo. Rev. Trim. Histol. 1888, 2, 33–41. [Google Scholar]
- Ramón y Cajal, S. Sur l’origine et la direction des prolongations nerveuses de la couche moléculaire du cervelet. Int. Mschr. Anat. Physiol. 1889, 6, 158–174. [Google Scholar]
- Ramón y Cajal, S. Histologie du Systeme Nerveox de L’homme et des Vertébrés; Paris Maloine; Reprinted 1952 and 1955; Consejo Superior de Investigaciones Científicas: Madrid, Spain, 1911; Volume 2.
- Ramón y Cajal, S. Contribución al conocimiento de la neuroglía del cerebro humano. Trab. Lab. Invest. Biol. 1913, 6, 255–315. [Google Scholar]
- Ramón y Cajal, S. Contribution á la connaissance de la névroglie cérébrale et cérébelleuse dans la paralysie générale progressive. Trav. Lab. Recher. Biol. Univ. Madrid. 1925, 23, 157–216. [Google Scholar]
- Del Río Hortega, P. El “tercer elemento” de los centros nerviosos. Poder fagocitario y movilidad de la microglia. Bol. Soc. Esp. Biol. 1919, 8, 68–82. [Google Scholar]
- Del Río Hortega, P. Histogénesis y evolución normal, éxodo y distribución regional de la microglia. Mem. Real. Soci. Esp. Hist. Nat. 1921, 11, 213–268. [Google Scholar]
- Achúcarro, N.; Gayarre, M. Contribución al estudio de la neuroglía en la corteza de la demencia senil y su participación en la alteración celular de Alzheimer. Trab. Lab. Invest. Biol. Univ. Madrid. 1914, 12, 68–83. [Google Scholar]
- Eccles, J.C.; Ito, M.; Szentagothar, J. The Cerebellum as a Neuronal Machine; Springer-Verlag: New York, NY, USA, 1967. [Google Scholar]
- De Zeeuw, C.I.; Hoogland, T.M. Reappraisal of Bergmann glial cells as modulators of cerebellar circuit function. Front. Cell. Neurosci. 2015, 9, 246. [Google Scholar] [CrossRef]
- Reeber, S.L.; Otis, T.S.; Sillitoe, R.V. New roles for the cerebellum in health and disease. Front. Syst. Neurosci. 2013, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Roostaei, T.; Nazeri, A.; Sahraian, M.A.; Minagar, A. The human cerebellum: A review of physiologic neuroanatomy. Neurol. Clin. 2014, 32, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Bostan, A.C.; Dum, R.P.; Strick, P.L. Cerebellar networks with the cerebral cortex and basal ganglia. Trends. Cogn. Sci. 2013, 17, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Peterburs, J.; Desmond, J.E. The role of the human cerebellum in performance monitoring. Curr. Opin. Neurobiol. 2016, 40, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Leisman, G.; Moustafa, A.A.; Shafir, T. Thinking, Walking, Talking: Integratory Motor and Cognitive Brain Function. Front Public Health 2016, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Kasanova, Z.; Goossens, L.; Leibold, N.; De Zeeuw, C.I.; van Amelsvoort, T.; Schruers, K. The anatomy of fear learning in the cerebellum: A systematic meta-analysis. Neurosci. Biobehav. Rev. 2015, 59, 83–91. [Google Scholar] [CrossRef]
- Van Overwalle, F.; D’aes, T.; Mariën, P. Social cognition and the cerebellum: A meta-analytic connectivity analysis. Hum. Brain Mapp. 2015, 36, 5137–5154. [Google Scholar] [CrossRef] [PubMed]
- Miquel, M.; Vazquez-Sanroman, D.; Carbo-Gas, M.; Gil-Miravet, I.; Sanchis-Segura, C.; Carulli, D.; Manzo, J.; Coria-Avila, G.A. Have we been ignoring the elephant in the room? Seven arguments for considering the cerebellum as part of addiction circuitry. Neurosci. Biobehav. Rev. 2016, 60, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Crippa, A.; Del Vecchio, G.; Busti Ceccarelli, S.; Nobile, M.; Arrigoni, F.; Brambilla, P. Cortico-Cerebellar Connectivity in Autism Spectrum Disorder: What Do We Know So Far? Front. Psychiatry 2016, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Mariën, P.; Borgatti, R. Language and the cerebellum. Handb. Clin. Neurol. 2018, 154, 181–202. [Google Scholar] [CrossRef]
- Bodranghien, F.; Bastian, A.; Casali, C.; Hallett, M.; Louis, E.D.; Manto, M.; Mariën, P.; Nowak, D.A.; Schmahmann, J.D.; Serrao, M.; et al. Consensus Paper: Revisiting the Symptoms and Signs of Cerebellar Syndrome. Cerebellum 2016, 15, 369–391. [Google Scholar] [CrossRef]
- Koeppen, A.H. The neuropathology of the adult cerebellum. Handb. Clin. Neurol. 2018, 154, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Álvarez, M.I.; Toledano-Díaz, A. Peculiaridades de la Involución Normal y Patológica en Cerebelo Humano. Características del Cerebelo Senil Normal y Patológico (Alzheimer), y su Singularidad Involutiva Frente al Resto del SNC; Editorial Académica Española: Chisinau, Moldova; Elsevier: Amsterdam, The Netherlands; Omni Scriptum GmbH &Co. KG.: Saarbrücken, Germany, 2017. [Google Scholar]
- Toledano, A.; Álvarez, M.I.; Toledano-Díaz, A.; Merino, J.J.; Rodríguez, J.J. Brain local and regional neuroglial alterations in Alzheimer’s Disease: Cell types, responses and implications. Curr. Alzheimer Res. 2016, 13, 321–342. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Alvarez, M.I.; Rivas, L.; Lacruz, C.; Martínez-Rodríguez, R. Amyloid precursor proteins in the cerebellar cortex of Alzheimer’s disease patients devoid of cerebellar beta-amyloid deposits: Immunocytochemical study of five cases. J. Neural. Transm. 1999, 106, 1151–1169. [Google Scholar] [CrossRef]
- Azevedo, F.A.; Carvalho, L.R.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.; Leite, R.E.; Jacob Filho, W.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef]
- Galliano, E.; Gao, Z.; Schonewille, M.; Todorov, B.; Simons, E.; Pop, A.S.; D’Angelo, E.; van den Maagdenberg, A.M.; Hoebeek, F.E.; De Zeeuw, C.I. Silencing of the majority of cerebellar granule cells uncovers their essential role in motor learning and consolidation. Cell. Rep. 2013, 3, 1239–1251. [Google Scholar] [CrossRef]
- Hawkes, R. The Ferdinando Rossi Memorial Lecture: Zones and Stripes—Pattern Formation in the Cerebellum. Cerebellum 2018, 17, 12–16. [Google Scholar] [CrossRef]
- Schmahmann, J. Cerebellum and cognition. Neurosci. Lett. 2019, 688, 62–75. [Google Scholar] [CrossRef]
- Hadlow, W.J.; Kennedy, R.C.; Race, R.E.; Eklund, C.M. Virologic and neurohistologic findings in dairy goats affected with natural scrapie. Vet. Pathol. 1980, 17, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, J.J. Review: Update on Classical and Atypical Scrapie in Sheep and Goats. Vet. Pathol. 2019, 56, 6–16. [Google Scholar] [CrossRef]
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559. [Google Scholar] [CrossRef]
- Wood, J.L.; McGill, I.S.; Done, S.H.; Bradley, R. Neuropathology of scrapie: A study of the distribution patterns of brain lesions in 222 cases of natural scrapie in sheep, 1982–1991. Vet. Rec. 1997, 140, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Hashizume, Y.; Yoshida, M.; Wang, Y. Neuropathological study of cerebellar degeneration in prion disease. Neuropathology 1999, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Sarasa, R.; Martínez, A.; Monleón, E.; Bolea, R.; Vargas, A.; Badiola, J.J.; Monzón, M. Involvement of astrocytes in transmissible spongiform encephalopathies: A confocal microscopy study. Cell. Tissue. Res. 2012, 350, 127–134. [Google Scholar] [CrossRef]
- Sarasa, R.; Junquera, C.; Toledano, A.; Badiola, J.J.; Monzón, M. Ultrastructural changes in the progress of natural Scrapie regardless fixation protocol. Histochem. Cell. Biol. 2015, 144, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.P.; Serrano, J.; Rodrigo, J.; Monleón, E.; Monzón, M.; Vargas, A.; Badiola, J.J.; Martínez-Murillo, R.; Martínez, A. Changes in the expression pattern of the nitrergic system of ovine cerebellum affected by scrapie. J. Neuropathol. Exp. Neurol. 2007, 66, 196–207. [Google Scholar] [CrossRef]
- Voigtländer, T.; Unterberger, U.; Guentchev, M.; Schwaller, B.; Celio, M.R.; Meyer, M.; Budka, H. The role of parvalbumin and calbindin D28k in experimental scrapie. Neuropathol. Appl. Neurobiol. 2008, 34, 435–445. [Google Scholar] [CrossRef]
- Alvarez, M.I.; Lacruz, C.; Toledano-Díaz, A.; Monleón, E.; Monzón, M.; Badiola, J.J.; Toledano, A. Calretinin-immunopositive cells and fibers in the cerebellar cortex of normal sheep. Cerebellum 2008, 7, 417–729. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.M.; Jeung, E.B. Calbindin-D28k in the Brain Influences the Expression of Cellular Prion Protein. Oxid. Med. Cell. Longev. 2018, 4670210. [Google Scholar] [CrossRef]
- Liberski, P.; Gajos, A.; Sikorska, B. Dystrophic neurites accumulating autophagic vacuoles show early stages of neuritic destruction. Folia Neuropathol. 2018, 56, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Bolea, R.; Lyahyai, J.; Filali, H.; Varona, L.; Marcos-Carcavilla, A.; Acín, C.; Calvo, J.H.; Serrano, M.; Badiola, J.J.; et al. Changes in HSP gene and protein expression in natural scrapie with brain damage. Vet. Res. 2011, 42, 13. [Google Scholar] [CrossRef]
- Mander, P.; Brown, G.C. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflammation 2005, 2, 20. [Google Scholar] [CrossRef]
- Guentchev, M.; Groschup, M.H.; Kordek, R.; Liberski, P.P.; Budka, H. Severe, early and selective loss of a subpopulation of GABAergic inhibitory neurons in experimental transmissible spongiform encephalopathies. Brain Pathol. 1998, 8, 615–623. [Google Scholar] [CrossRef]
- Campeau, J.L.; Wu, G.; Bell, J.R.; Rasmussen, J.; Sim, V.L. Early Increase and Late Decrease of Purkinje Cell Dendritic Spine Density in Prion-Infected Organotypic Mouse Cerebellar Cultures. PLoS ONE 2013, 8, e81776. [Google Scholar] [CrossRef]
- Domich, L.; Delhaye-Bouchaud, N.; Laget, P. Alterations of the functional properties of the parallel fibers in the cerebellum of the “scrapie mouse”. Arch. Ital. Biol. 1986, 124, 27–41. [Google Scholar] [PubMed]
- Ersdal, C.; Ulvund, M.J.; Benestad, S.L.; Tranulis, M.A. Accumulation of pathogenic prion protein (PrPSc) in nervous and lymphoid tissues of sheep with subclinical scrapie. Vet. Pathol. 2003, 40, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Hernández, R.S.; Sarasa, R.; Toledano, A.; Badiola, J.J.; Monzón, M. Morphological approach to assess the involvement of astrocytes in prion propagation. Cell. Tissue. Res. 2014, 358, 57–63. [Google Scholar] [CrossRef]
- Garcés, M.; Guijarro, M.I.; Vargas, A.; Badiola, J.J.; Monzón, M. Neuroglial patterns are shared by cerebella from prion and prion-like disorder affected patients. Mech. Ageing Dev. 2019, 184, 111176. [Google Scholar] [CrossRef]
- Mironov, A., Jr.; Latawiec, D.; Wille, H.; Bouzamondo-Bernstein, E.; Legname, G.; Williamson, R.A.; Burton, D.; DeArmond, S.J.; Prusiner, S.B.; Peters, P.J. Cytosolic prion protein in neurons. J. Neurosci. 2003, 23, 7183–7193. [Google Scholar] [CrossRef]
- Laine, J.; Marc, M.E.; Sy, M.S.; Axelrad, H. Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur. J. Neurosci. 2001, 14, 47–56. [Google Scholar] [CrossRef]
- Kretzschmar, H.A.; Prusiner, S.B.; Stowring, L.E.; DeArmond, S.J. Scrapie prion proteins are synthesized in neurons. Am. J. Pathol. 1986, 122, 1–5. [Google Scholar]
- Jeffrey, M.; McGovern, G.; Sisó, S.; González, L. Cellular and sub-cellular pathology of animal prion diseases: Relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 2011, 121, 113–134. [Google Scholar] [CrossRef]
- Safar, J.; Ceroni, M.; Piccardo, P.; Gajdusek, D.C.; Gibbs, C.J., Jr. Scrapie-associated precursor proteins: Antigenic relationship between species and immunocytochemical localization in normal, scrapie, and Creutzfeldt-Jakob disease brains. Neurology 1990, 40, 513–517. [Google Scholar] [CrossRef]
- Diedrich, J.F.; Bendheim, P.E.; Kim, Y.S.; Carp, R.I.; Haase, A.T. Scrapie-associated prion protein accumulates in astrocytes during scrapie infection. Proc. Natl. Acad. Sci. USA 1991, 88, 375–379. [Google Scholar] [CrossRef]
- González, L.; Martin, S.; Jeffrey, M. Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: Implications for differential cell targeting and PrP processing. J. Gen. Virol. 2003, 84, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Martin, S.; Begara-McGorum, I.; Hunter, N.; Houston, F.; Simmons, M.; Jeffrey, M. Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J. Comp. Pathol. 2002, 126, 17–29. [Google Scholar] [CrossRef]
- DeArmond, S.J.; Sánchez, H.; Yehiely, F.; Qiu, Y.; Ninchak-Casey, A.; Daggett, V.; Camerino, A.P.; Cayetano, J.; Rogers, M.; Groth, D.; et al. Selective neuronal targeting in prion disease. Neuron 1997, 19, 1337–1348. [Google Scholar] [CrossRef]
- Jeffrey, M.; McGovern, G.; Goodsir, C.M.; Síso, S.; González, L. Strain-associated variations in abnormal PrP trafficking of sheep scrapie. Brain Pathol. 2009, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Simmons, M.; Chaplin, M.; Spiropoulos, J. Neuroanatomical distribution of abnormal prion protein in naturally occurring atypical scrapie cases in Great Britain. Acta Neuropathol. 2008, 116, 547–559. [Google Scholar] [CrossRef]
- Kim, Y.S.; Carp, R.I.; Callahan, S.M.; Wisniewski, H.M. Pathogenesis and pathology of scrapie after stereotactic injection of strain 22 in intact and bisected cerebella. J. Neuropathol. Exp. Neurol. 1990, 49, 114–121. [Google Scholar] [CrossRef]
- Liberski, P. Axonal changes in experimental prion diseases recapitulate those following constriction of postganglionic branches of the superior cervical ganglion: A comparison 40 years later. Prion 2019, 13, 83–93. [Google Scholar] [CrossRef]
- Bignami, A.; Parry, H.B. Electron microscopic studies of the brain of sheep with natural scrapie. I. The fine structure of neuronal vacuolation. Brain 1972, 95, 319–326. [Google Scholar] [CrossRef]
- Christensen, H.M.; Dikranian, K.; Li, A.; Baysac, K.C.; Walls, K.C.; Olney, J.W.; Roth, K.A.; Harris, D.A. A highly toxic cellular prion protein induces a novel, nonapoptotic form of neuronal death. Am. J. Pathol. 2010, 176, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, O.; Otero, A.; Filali, H.; Sanz-Rubio, D.; Toivonen, J.M.; Zaragoza, P.; Badiola, j.j.; Bolea, R.; Martín-Burriel, I. Dysregulation of autophagy in the central nervous system of sheep naturally infected with classical scrapie. Sci. Rep. 2019, 9, 1911. [Google Scholar] [CrossRef]
- Basant, A.; Abdulrahman, A.; Dalia, H.; Abdelaziz, A.; Hermann, M.; Schatz, J. Autophagy regulates exosomal release of prions in neuronal cells. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef]
- Xu, Y.; Tian, C.; Wang, S.B.; Xie, W.L.; Guo, Y.; Zhang, J.; Shi, Q.; Chen, C.; Dong, X.P. Activation of the macroautophagic system in scrapie-infected experimental animals and human genetic prion diseases. Autophagy 2012, 8, 1604–1620. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, Ó.; Badiola, J.J.; Bolea, R.; Ferrer, I.; Llorens, F.; Martín-Burriel, I. An update on autophagy in Prion Diseases. Front. Bioeng. Biotechnol. 2020, 8, 975. [Google Scholar] [CrossRef]
- Jeffrey, M.; Goodsir, C.M.; Bruce, M.E.; McBride, P.A.; Fraser, J.R. In vivo toxicity of prion protein in murine scrapie: Ultrastructural and immunogold studies. Neuropathol. Appl. Neurobiol. 1997, 23, 93–101. [Google Scholar] [CrossRef]
- Liberski, P.; Gajos, A.; Sikorska, B.; Moryś, J. Light and electron microscopic studies of the 139A-H strain of scrapie passaged in hamsters-. Folia Neuropathol. 2018, 56, 321–327. [Google Scholar] [CrossRef]
- Liberski, P. The tubulovesicular structures - the ultrastructural hallmark for all prion diseases. Acta Neurobiol. Exp. 2008, 68, 113–121. [Google Scholar]
- Liberski, P.; Jeffrey, M. Tubulovesicular structures: What are they really? Microsc. Res. Tech. 2000, 50, 46–57. [Google Scholar] [CrossRef]
- Liberski, P.; Jeffrey, M. Tubulovesicular structures--the ultrastructural hallmark for transmissible spongiform encephalopathies or prion diseases. Folia Neuropathol. 2004, 42 (Suppl. B), 96–108. [Google Scholar] [PubMed]
- Liberski, P.P.; Sikorska, B.; Hauw, J.J.; Kopp, N.; Streichenberger, N.; Giraud, P.; Budka, H.; Boellaard, J.W.; Brown, P. Tubulovesicular structures are a consistent (and unexplained) finding in the brains of humans with prion diseases. Virus Res. 2008, 132, 226–228. [Google Scholar] [CrossRef]
- Armstrong, R.A.; Lantos, P.L.; Ironside, J.W.; Cairns, N.J. Spatial correlation between the vacuolation, prion protein, deposition and surviving neurons in patients with Creutzfeldt-Jakob disease (vCJD). J. Neural. Transm. 2003, 110, 1303–1311. [Google Scholar] [CrossRef]
- Sisó, S.; Puig, B.; Varea, R.; Vidal, E.; Acín, C.; Prinz, M.; Montrasio, F.; Badiola, J.J.; Aguzzi, A.; Pumarola, M.; et al. Abnormal synaptic protein expression and cell death in murine scrapie. Acta Neuropathol. 2002, 103, 615–626. [Google Scholar] [CrossRef]
- Brown, D.R. Prion protein peptide neurotoxicity can be mediated by astrocytes. J. Neurochem. 1999, 73, 1105–1113. [Google Scholar] [CrossRef]
- Brown, D.R.; Mohn, C.M. Astrocytic glutamate uptake and prion protein expression. Glia 1999, 25, 282–292. [Google Scholar] [CrossRef]
- Lefrancois, T.; Fages, C.; Brugere-Picoux, J.; Tardy, M. Astroglial reactivity in natural scrapie of sheep. Microb. Pathog. 1994, 17, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Andres-Barquin, P.J.; Le Prince, G.; Fages, C.; Garcia de Jalon, J.A.; Perez-Martos, A.; Tardy, M.; Lopez-Perez, M.J. Expression of glial fibrillary acidic protein and glutamine synthetase genes in the natural scrapie of sheep. Mol. Chem. Neuropathol. 1994, 22, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia are critical in host defense against prion disease. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D. The Role of Microglia in Prion Diseases: A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207. [Google Scholar] [CrossRef]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein. Nature 1996, 380, 345–347. [Google Scholar] [CrossRef]
- Guiroy, D.C.; Wakayama, I.; Liberski, P.P.; Gajdusek, D.C. Relationship of microglia and scrapie amyloid-immunoreactive plaques in kuru, Creutzfeldt-Jakob disease and Gerstmann-Straussler syndrome. Acta Neuropathol. 1994, 87, 526–530. [Google Scholar] [CrossRef]
- Giese, A.; Brown, D.R.; Groschup, M.H.; Feldmann, C.; Haist, I.; Kretzschmar, H.A. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998, 8, 449–457. [Google Scholar] [CrossRef]
- Priller, J.; Prinz, M.; Heikenwalder, M.; Zeller, N.; Schwarz, P.; Heppner, F.L.; Aguzzi, A. Early and Rapid Engraftment of Bone Marrow-Derived Microglia in Scrapie. J. Neurosci. 2006, 26, 11753–11762. [Google Scholar] [CrossRef] [PubMed]
- El Hachimi, K.H.; Chaunu, M.P.; Brown, P.; Foncin, J.F. Modifications of oligodendroglial cells in spongiform encephalopathies. Exp. Neurol. 1998, 154, 23–30. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Kalev, O.; Budka, H. Contribution of neuropathology to the understanding of human prion disease. Folia Neuropathol. 2004, 42, 69–76. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cerebellar Neuropathological Abnormalities in Classical Natural Scrapie | |
|---|---|
| Cerebellar Cortical Layers | Abnormalities |
| Molecular layer | Decrease in thickness |
| Purkinje Cell layer | Loss and morphohistochemical alterations of neurons |
| Granule cell layer | Decrease in thickness Loss of granule cells |
| Neurons | |
| PC neurons |
|
| Lugaro, Golgi and brush cells | Small morphohistochemical changes (dystrophy) |
| Granule cells | Decrease in cell density |
| Spongiosis (vacuolation/vacuolization) | |
| Intraneuronal vacuolization | |
| Extraneuronal vacuolization | |
| Neurogliosis | |
| Astroglia | Astrogliosis Astroglial hyperplasia |
| Microglia | Microgliosis |
| Abnormal PRP Deposits/Deposition | |
| Different types of immunodeposits Different association to cells | |
| Reflections on the Neuropathological Cerebellar Changes | |
| Comparison with other neuropathological brain patterns in selected brain regions of prion/prion-like diseases | |
| Sequence of the temporal appearance of neuropathological markers | |
| Initiation and progression of neuropathology | |
| Extra- and intra-neuronal vacuolization: a differential feature not well explained | |
| Alterations and neuronal loss: neuronal specificity | |
| Neuroglioses: a main feature | |
| Abnormal prion protein deposits: diversity and controversial cell association | |
| Pathologies | Neuronal Loss | Spongiosis | Neurogliosis | Aberrant Protein Deposition |
|---|---|---|---|---|
| CLASSICAL NATURAL SCRAPIE | ||||
| Cerebellum | +/+++ | +++ | +++ | +++ (*) |
| Frontal cortex | NE | + | +++ | ++ (*) |
| Obex | NE | +++ | +++ | +++ (*) |
| ATYPICAL SCRAPIE | ||||
| Cerebellum | +/+++ | − | ++ | ++ (*) |
| HUMAN PRION DISEASES | ||||
| Cerebellum | +/+++ | ++/+++ | +++ | +++ (*) |
| ALZHEIMER DISEASE | ||||
| Cerebellum | −/+ | − | −/+ | −/+ (**) |
| Frontal cortex and hippocampus | ++/+++ | − | ++/+++ | ++/+++ (**) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toledano-Díaz, A.; Álvarez, M.I.; Rodríguez, J.-J.; Badiola, J.J.; Monzón, M.; Toledano, A. Reflections on Cerebellar Neuropathology in Classical Scrapie. Biomolecules 2021, 11, 649. https://doi.org/10.3390/biom11050649
Toledano-Díaz A, Álvarez MI, Rodríguez J-J, Badiola JJ, Monzón M, Toledano A. Reflections on Cerebellar Neuropathology in Classical Scrapie. Biomolecules. 2021; 11(5):649. https://doi.org/10.3390/biom11050649
Chicago/Turabian StyleToledano-Díaz, Adolfo, María Isabel Álvarez, Jose-Julio Rodríguez, Juan Jose Badiola, Marta Monzón, and Adolfo Toledano. 2021. "Reflections on Cerebellar Neuropathology in Classical Scrapie" Biomolecules 11, no. 5: 649. https://doi.org/10.3390/biom11050649
APA StyleToledano-Díaz, A., Álvarez, M. I., Rodríguez, J.-J., Badiola, J. J., Monzón, M., & Toledano, A. (2021). Reflections on Cerebellar Neuropathology in Classical Scrapie. Biomolecules, 11(5), 649. https://doi.org/10.3390/biom11050649