
Neuropathology of Animal Prion Diseases

 , , , ,
, , , , 
 ,
,  , and
, and
Abstract
1. Introduction
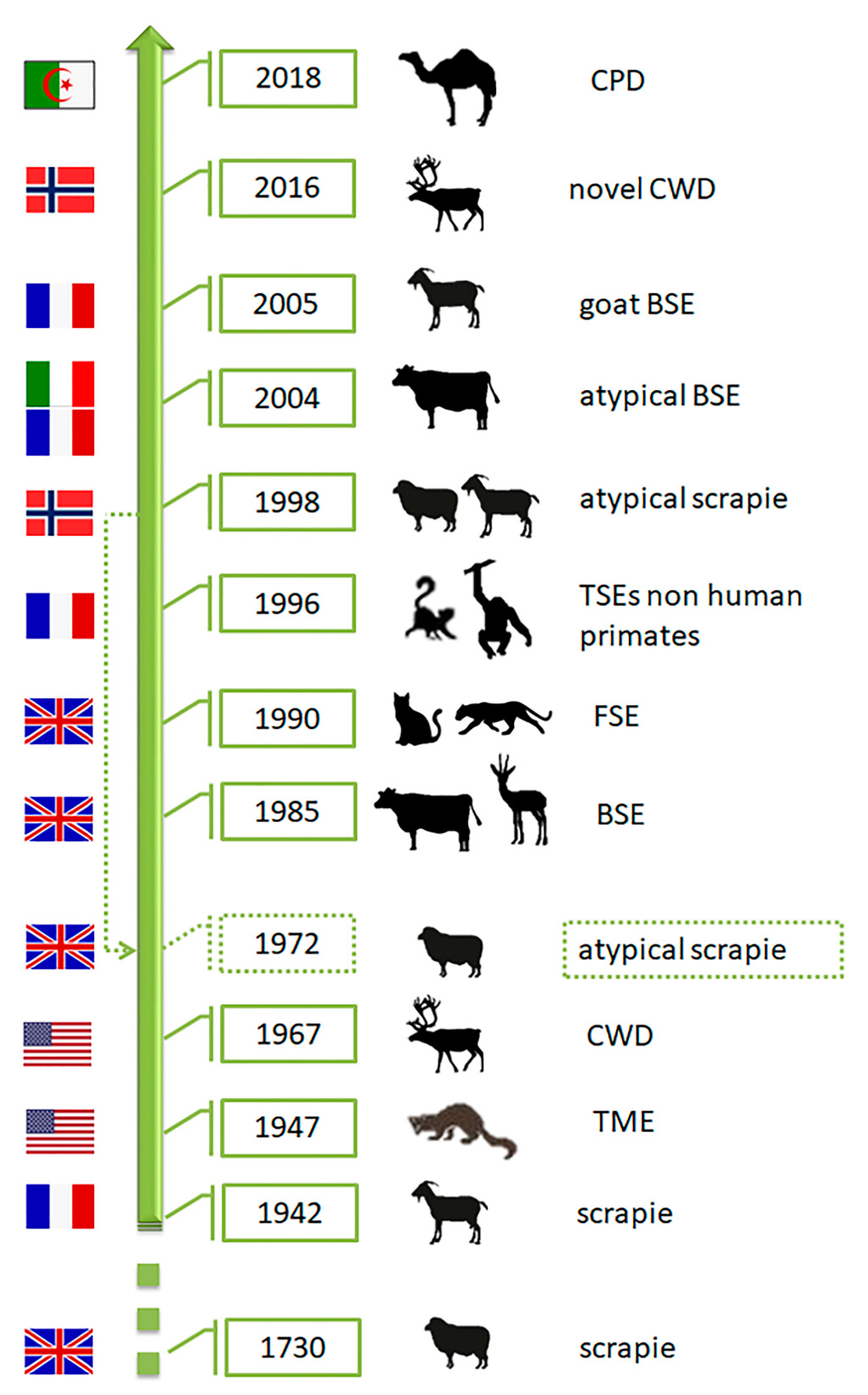
2. Prions and Animal Prion Diseases
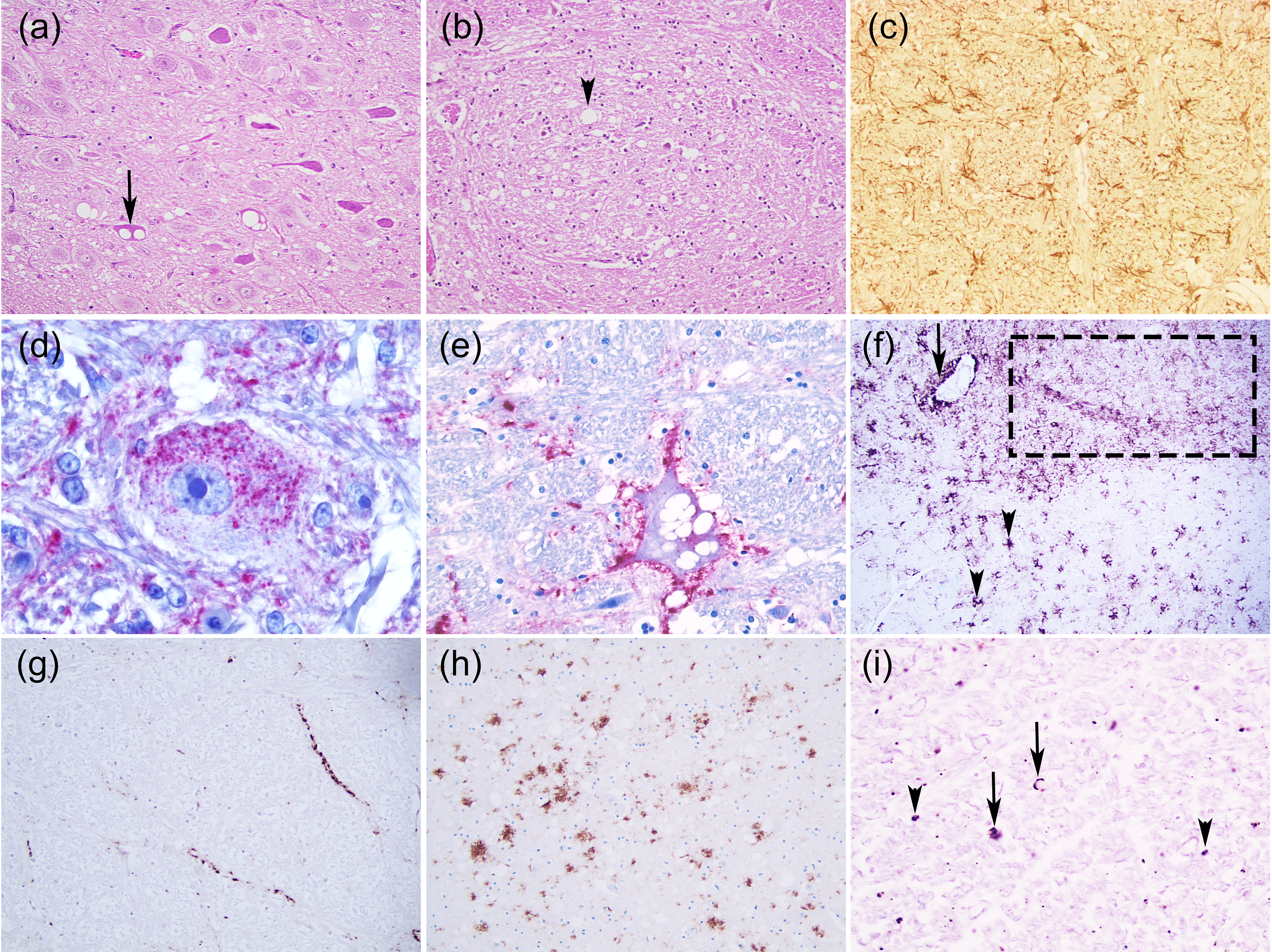
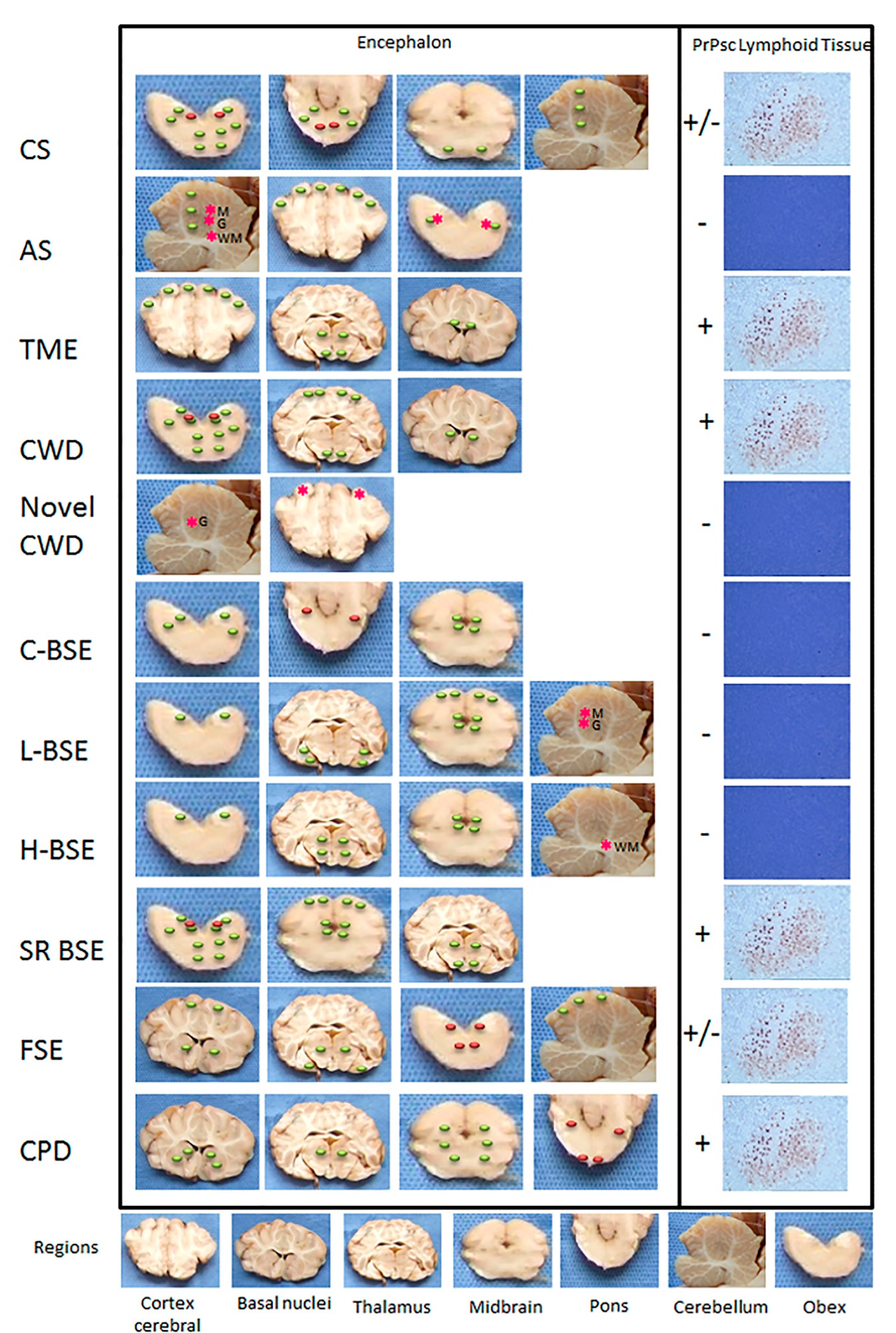
3. Neuropathology
3.1. Scrapie
3.1.1. Classical Scrapie
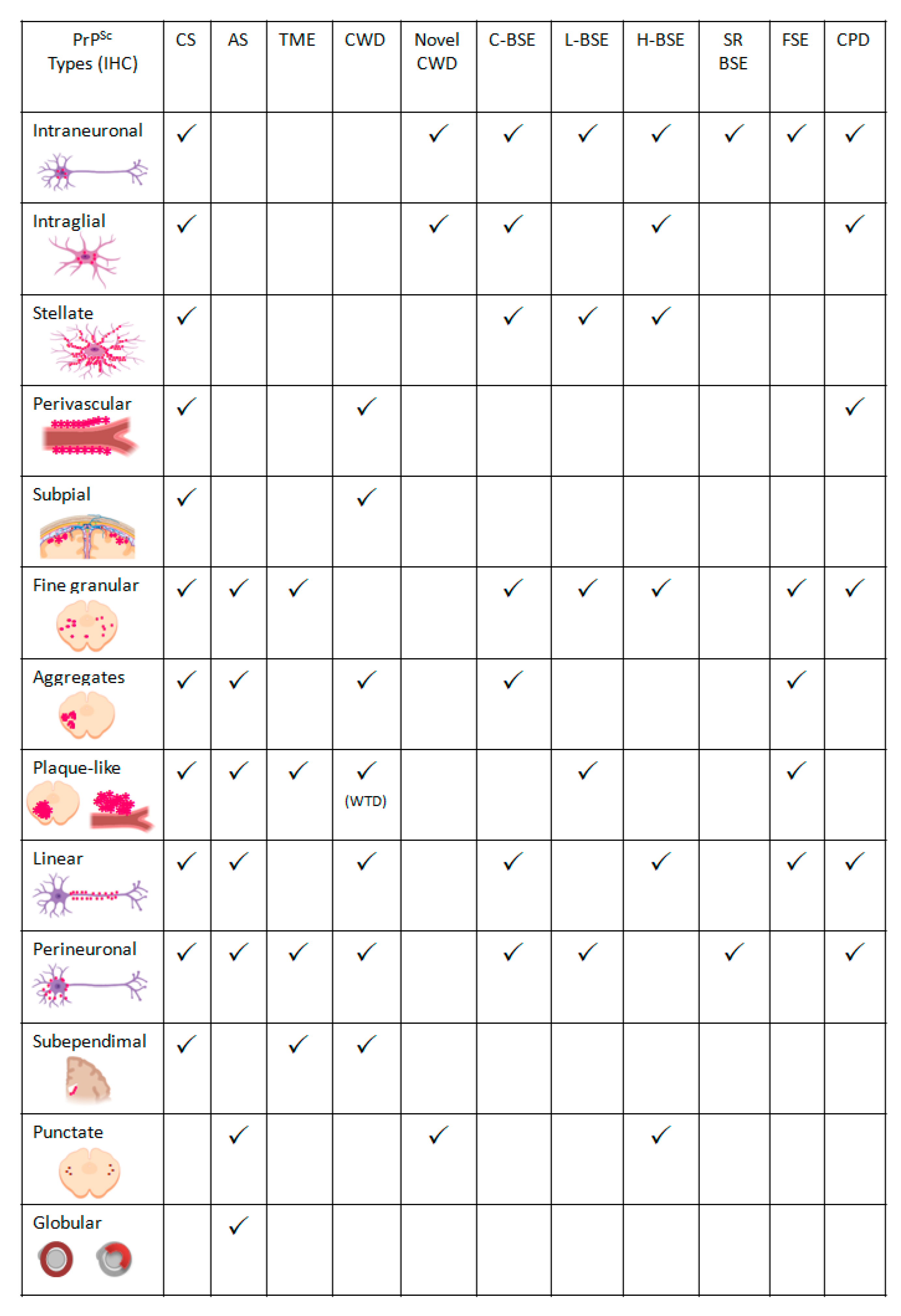
- Intraneuronal—fine PrPSc granular deposits to coarse, sometimes confluent scattered in the cytoplasm of the perikarya of neurons surrounding the nucleus;
- Intraglial—intense granular or ovoid deposits of PrPSc, slightly larger than those observed in neurons, in close proximity to the glial cell nucleus.
- Stellate (also named glial)—radiating, branching PrPSc deposits centred on a visible glial-type nucleus, conferring a star-like appearance;
- Perivascular—PrPSc deposits located around the blood vessels in the white matter;
- Subpial—loose-mesh to more amorphous, multi-located continuous PrPSc accumulations occurred beneath the pia mater;
- Subependimal—similar to the perivascular PrPSc type but in the glial layer underneath the ependymal lining of the ventricular system deposits, usually discontinuous and seen mainly around the lateral ventricles;
- Linear—thread-like deposits of PrPSc located in the neuropil;
- Fine granular (fine punctate or fine particulate)—Numerous, small PrPSc granules observed in the neuropil;
- Aggregates (or coarse granular, coarse particulate to coalescing and moss-like)—appear as large amorphous PrPSc accumulations scattered in the neuropil;
- Perineuronal—thin deposits of PrPSc around an individual, scattered neuronal perikarya and neurites;
- Plaques-Like (or plaques, vascular plaques)—fibrillar, radiate relatively large accumulations of PrPSc often distributed around blood vessels of a different calibre.
3.1.2. Atypical Scrapie
3.2. Transmissible Mink Encephalopathy (TME)
3.3. Chronic Wasting Disease (CWD)
Novel CWD (Nor16CWD)
3.4. Bovine Spongiform Encephalopathy (BSE)
3.4.1. Cattle
- Intraneuronal—often observed in the dorsal motor nucleus of the vagus nerve (DMV), reticular formation, olivary nuclei, vestibular complex, pontine and thalamic nuclei, and hypothalamus;
- Intraglial;
- Stellate—predominantly present in the central grey matter, the cerebral lamina, and within the medial pontine nuclei in the cerebral cortex, thalamus, and obex;
- Fine granular—seen in the neuropil of the DMV, NST and the thalamic nuclei;
- Aggregates;
- Linear—noticed particularly at the level of the reticular formation of the brainstem;
- Perineuronal—detected in the caudate and putamen nuclei of the basal ganglia and in the DMV;
Atypical BSE (L-BSE and H-BSE)
3.4.2. Zoo Ruminants and Non-Human Primates
3.4.3. Small Ruminants
3.5. Feline Spongiform Encephalopathy (FSE)
3.6. Prion Diseases in Dromedary Camels (CPD)
4. Neuroinflammation
Author Contributions
Funding
Conflicts of Interest
References
- Budka, H. Neuropathology of Prion Diseases. Br. Med. Bull. 2003, 66, 121–130. [Google Scholar] [CrossRef]
- VanDeVelde, M.; Higgins, R.J.; Oevermann, A. Veterinary Neuropathology: Essentials of Theory and Practice; Wiley-Blackwell: Oxford, UK, 2012; ISBN 978-0-470-67056-9. [Google Scholar]
- Spiropoulos, J.; Simmons, M.M. Pathology of Animal Transmissible Spongiform Encephalopathies (TSEs). Food Saf. 2017, 5, 1–9. [Google Scholar] [CrossRef][Green Version]
- Soto, C.; Satani, N. The Intricate Mechanisms of Neurodegeneration in Prion Diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef]
- Prusiner, S. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-Propagation of Pathogenic Protein Aggregates in Neurodegenerative Diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment 1. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Nicholas, K.B.; Nicholas, H.B.J. Deerfield D.W.GeneDoc Analysis and Visualization of Genetic Variation. EMBnet News 1997, 4, 1–4. [Google Scholar]
- Vaccari, G.; D’Agostino, C.; Nonno, R.; Rosone, F.; Conte, M.; Di Bari, M.A.; Chiappini, B.; Esposito, E.; De Grossi, L.; Giordani, F.; et al. Prion Protein Alleles Showing a Protective Effect on the Susceptibility of Sheep to Scrapie and Bovine Spongiform Encephalopathy. J. Virol. JVI 2007, 81, 7306–7309. [Google Scholar] [CrossRef] [PubMed]
- Laegreid, W.W.; Clawson, M.L.; Heaton, M.P.; Green, B.T.; O’Rourke, K.I.; Knowles, D.P. Scrapie Resistance in ARQ Sheep. J. Virol. 2008, 82, 10318–10320. [Google Scholar] [CrossRef]
- Greenlee, J.J. Review: Update on Classical and Atypical Scrapie in Sheep and Goats. Vet. Pathol. 2019, 56, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Cassmann, E.D.; Moore, S.J.; Smith, J.D.; Greenlee, J.J. Sheep With the Homozygous Lysine-171 Prion Protein Genotype Are Resistant to Classical Scrapie After Experimental Oronasal Inoculation. Vet. Pathol. 2019, 56, 409–417. [Google Scholar] [CrossRef]
- DeSilva, U.; Guo, X.; Kupfer, D.M.; Fernando, S.C.; Pillai, A.T.V.; Najar, F.Z.; So, S.; Fitch, G.Q.; Roe, B.A. Allelic Variants of Ovine Prion Protein Gene (PRNP) in Oklahoma Sheep. Cytogenet. Genome Res. 2003, 102, 89–94. [Google Scholar] [CrossRef]
- Kdidi, S.; Yahyaoui, M.H.; Conte, M.; Chiappini, B.; Zaccaria, G.; Ben Sassi, M.; Ben Ammar El Gaaied, A.; Khorchani, T.; Vaccari, G. PRNP Polymorphisms in Tunisian Sheep Breeds. Livest. Sci. 2014, 167, 100–103. [Google Scholar] [CrossRef]
- Kim, S.-K.; Kim, Y.-C.; Won, S.-Y.; Jeong, B.-H. Potential Scrapie-Associated Polymorphisms of the Prion Protein Gene (PRNP) in Korean Native Black Goats. Sci. Rep. 2019, 9, 15293. [Google Scholar] [CrossRef] [PubMed]
- Teferedegn, E.Y.; Yaman, Y.; Ün, C. Novel Variations in Native Ethiopian Goat Breeds PRNP Gene and Their Potential Effect on Prion Protein Stability. Sci. Rep. 2020, 10, 6953. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Jeffrey, M.; Dagleish, M.P.; Goldmann, W.; Sisó, S.; Eaton, S.L.; Martin, S.; Finlayson, J.; Stewart, P.; Steele, P.; et al. Susceptibility to Scrapie and Disease Phenotype in Sheep: Cross-PRNP Genotype Experimental Transmissions with Natural Sources. Vet. Res. 2012, 43, 55. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, J.J.; Zhang, X.; Nicholson, E.M.; Kunkle, R.A.; Hamir, A.N. Prolonged Incubation Time in Sheep with Prion Protein Containing Lysine at Position 171. J. Vet. Diagn. Investig. 2012, 24, 554–558. [Google Scholar] [CrossRef]
- Torricelli, M.; Sebastiani, C.; Ciullo, M.; Ceccobelli, S.; Chiappini, B.; Vaccari, G.; Capocefalo, A.; Conte, M.; Giovannini, S.; Lasagna, E.; et al. PRNP Polymorphisms in Eight Local Goat Populations/Breeds from Central and Southern Italy. Animals 2021, 11, 333. [Google Scholar] [CrossRef]
- Papasavva-Stylianou, P.; Kleanthous, M.; Toumazos, P.; Mavrikiou, P.; Loucaides, P. Novel Polymorphisms at Codons 146 and 151 in the Prion Protein Gene of Cyprus Goats, and Their Association with Natural Scrapie. Vet. J. 2007, 173, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Colussi, S.; Vaccari, G.; Maurella, C.; Bona, C.; Lorenzetti, R.; Troiano, P.; Casalinuovo, F.; Di Sarno, A.; Maniaci, M.G.; Zuccon, F.; et al. Histidine at Codon 154 of the Prion Protein Gene Is a Risk Factor for Nor98 Scrapie in Goats. J. Gen. Virol. 2008, 89, 3173–3176. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.I.; Spraker, T.R.; Zhuang, D.; Greenlee, J.J.; Gidlewski, T.E.; Hamir, A.N. Elk with a Long Incubation Prion Disease Phenotype Have a Unique PrPd Profile. NeuroReport 2007, 18, 1935–1938. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, W. PrP Genetics in Ruminant Transmissible Spongiform Encephalopathies. Vet. Res. 2008, 39, 30. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.E.; Conner, M.M.; Wolfe, L.L.; Miller, M.W.; Williams, E.S. Low Frequency of PrP Genotype 225SF among Free-Ranging Mule Deer (Odocoileus Hemionus) with Chronic Wasting Disease. J. Gen. Virol. 2005, 86, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Sander, P.; Hamann, H.; Pfeiffer, I.; Wemheuer, W.; Brenig, B.; Groschup, M.H.; Ziegler, U.; Distl, O.; Leeb, T. Analysis of Sequence Variability of the Bovine Prion Protein Gene (PRNP) in German Cattle Breeds. Neurogenetics 2004, 5, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Juling, K.; Schwarzenbacher, H.; Williams, J.L.; Fries, R. A Major Genetic Component of BSE Susceptibility. BMC Biol. 2006, 4, 33. [Google Scholar] [CrossRef]
- Richt, J.A.; Hall, S.M. BSE Case Associated with Prion Protein Gene Mutation. PLoS Pathog. 2008, 4, e1000156. [Google Scholar] [CrossRef]
- Gurgul, A.; Polak, M.P.; Larska, M.; Słota, E. PRNP and SPRN Genes Polymorphism in Atypical Bovine Spongiform Encephalopathy Cases Diagnosed in Polish Cattle. J. Appl. Genet. 2012, 53, 337–342. [Google Scholar] [CrossRef]
- Zoubeyda, K.; Imane, M.; Youcef, C.; Baaissa, B.; Suheil, G.S.B.; Michela, C.; Antonio, C.; Umberto, A.; Barbara, C.; Gabriele, V. Variability of the Prion Protein Gene (PRNP) in Algerian Dromedary Populations. Anim. Gene 2020, 17–18, 200106. [Google Scholar] [CrossRef]
- Babelhadj, B.; Di Bari, M.A.; Pirisinu, L.; Chiappini, B.; Gaouar, S.B.S.; Riccardi, G.; Marcon, S.; Agrimi, U.; Nonno, R.; Vaccari, G. Prion Disease in Dromedary Camels, Algeria. Emerg. Infect. Dis. 2018, 24, 1029–1036. [Google Scholar] [CrossRef]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational Variations in an Infectious Protein Determine Prion Strain Differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Abid, K.; Soto, C. The Prion Strain Phenomenon: Molecular Basis and Unprecedented Features. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 681–691. [Google Scholar] [CrossRef]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef]
- Morales, R. Prion Strains in Mammals: Different Conformations Leading to Disease. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Nonno, R.; Marin-Moreno, A.; Carlos Espinosa, J.; Fast, C.; Van Keulen, L.; Spiropoulos, J.; Lantier, I.; Andreoletti, O.; Pirisinu, L.; Di Bari, M.A.; et al. Characterization of Goat Prions Demonstrates Geographical Variation of Scrapie Strains in Europe and Reveals the Composite Nature of Prion Strains. Sci. Rep. 2020, 10, 19. [Google Scholar] [CrossRef]
- Aguilar-Calvo, P.; García, C.; Espinosa, J.C.; Andreoletti, O.; Torres, J.M. Prion and Prion-like Diseases in Animals. Virus Res. 2015, 207, 82–93. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Miller, M.W. Other Animal Prion Diseases. Br. Med. Bull. 2003, 66, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Chong, A.; Kennedy, I.; Goldmann, W.; Green, A.; González, L.; Jeffrey, M.; Hunter, N. Archival Search for Historical Atypical Scrapie in Sheep Reveals Evidence for Mixed Infections. J. Gen. Virol. 2015, 96, 3165–3178. [Google Scholar] [CrossRef][Green Version]
- Benestad, S.L.; Sarradin, P.; Thu, B.; Schonheit, J.; Tranulis, M.A.; Bratberg, B. Cases of Scrapie with Unusual Features in Norway and Designation of a New Type, Nor98. Vet. Rec. 2003, 153, 202–208. [Google Scholar] [CrossRef]
- Benestad, S.L.; Arsac, J.-N.; Goldmann, W.; Nöremark, M. Atypical/Nor98 Scrapie: Properties of the Agent, Genetics, and Epidemiology. Vet. Res. 2008, 39, 19. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L.N.; McGill, I.S.; Done, S.H.; Bradley, R. Neuropathology of Scrapie: A Study of the Distribution Patterns of Brain Lesions in 222 Cases of Natural Scrapie in Sheep, 1982–1991. Vet. Rec. 1997, 140, 167–174. [Google Scholar] [CrossRef]
- Spiropoulos, J.; Casalone, C.; Caramelli, M.; Simmons, M.M. Immunohistochemistry for PrPSc in Natural Scrapie Reveals Patterns Which Are Associated with the PrP Genotype. Neuropathol. Appl. Neurobiol. 2007, 33, 398–409. [Google Scholar] [CrossRef]
- Jeffrey, M.; González, L. Pathology and Pathogenesis of Bovine Spongiform Encephalopathy and Scrapie. Curr. Top. Microbiol. Immunol. 2004, 284, 65–97. [Google Scholar] [CrossRef]
- Goldmann, W.; Hunter, N.; Foster, J.D.; Salbaum, J.M.; Beyreuther, K.; Hope, J. Two Alleles of a Neural Protein Gene Linked to Scrapie in Sheep. Proc. Natl. Acad. Sci. USA 1990, 87, 2476–2480. [Google Scholar] [CrossRef]
- Hunter, N.; Goldmann, W.; Smith, G.; Hope, J. The Association of a Codon 136 PrP Gene Variant with the Occurrence of Natural Scrapie. Arch. Virol. 1994, 137, 171–177. [Google Scholar] [CrossRef]
- Hunter, N.; Foster, J.D.; Goldmann, W.; Stear, M.J.; Hope, J.; Bostock, C. Natural Scrapie in a Closed Flock of Cheviot Sheep Occurs Only in Specific PrP Genotypes. Arch. Virol. 1996, 141, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Laplanche, J.L.; Chatelain, J.; Westaway, D.; Thomas, S.; Dussaucy, M.; Brugere-Picoux, J.; Launay, J.M. PrP Polymorphisms Associated with Natural Scrapie Discovered by Denaturing Gradient Gel Electrophoresis. Genomics 1993, 15, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Westaway, D.; Zuliani, V.; Cooper, C.M.; Da Costa, M.; Neuman, S.; Jenny, A.L.; Detwiler, L.; Prusiner, S.B. Homozygosity for Prion Protein Alleles Encoding Glutamine-171 Renders Sheep Susceptible to Natural Scrapie. Genes Dev. 1994, 8, 959–969. [Google Scholar] [CrossRef]
- Groschup, M.H.; Lacroux, C.; Buschmann, A.; Lühken, G.; Mathey, J.; Eiden, M.; Lugan, S.; Hoffmann, C.; Espinosa, J.C.; Baron, T.; et al. Classic Scrapie in Sheep with the ARR/ARR Prion Genotype in Germany and France. Emerg. Infect. Dis. 2007, 13, 1201–1207. [Google Scholar] [CrossRef]
- Guo, X.; Kupfer, D.M.; Fitch, G.Q.; Roe, B.A.; DeSilva, U. Identification of a Novel Lysine-171 Allele in the Ovine Prion Protein (PRNP) Gene: Brief Notes. Anim. Genet. 2003, 34, 303–305. [Google Scholar] [CrossRef]
- Boukouvala, E.; Gelasakis, A.I.; Kanata, E.; Fragkiadaki, E.; Giadinis, N.D.; Palaska, V.; Christoforidou, S.; Sklaviadis, T.; Ekateriniadou, L.V. The Association between 171 K Polymorphism and Resistance against Scrapie Affection in Greek Dairy Sheep. Small Rumin. Res. 2018, 161, 51–56. [Google Scholar] [CrossRef]
- Cassmann, E.D.; Greenlee, J.J. Pathogenesis, Detection, and Control of Scrapie in Sheep. Am. J. Vet. Res. 2020, 81, 600–614. [Google Scholar] [CrossRef] [PubMed]
- OIE. Terrestrial Manual 2018, Chapter 3.4.5.-Bovine Spongiform Encephalopathy; OIE: Paris, France, 2018. [Google Scholar]
- Van Keulen, L.J.M.; Schreuder, B.E.C.; Meloen, R.H.; Poelen-van den Berg, M.; Mooij-Harkes, G.; Vromans, M.E.W.; Langeveld, J.P.M. Immumohistochemical Detection and Localization of Prion Protein in Brain Tissue of Sheep With Natural Scrapie. Vet. Pathol. 1995, 32, 299–308. [Google Scholar] [CrossRef]
- Ryder, S.J.; Spencer, Y.I.; Bellerby, P.J.; March, S.A. Immunohistochemical Detection of PrP in the Medulla Oblongata of Sheep: The Spectrum of Staining in Normal and Scrapie-Affected Sheep. Vet. Rec. 2001, 148, 7–13. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Martin, S.; Begara-McGorum, I.; Hunter, N.; Houston, F.; Simmons, M.; Jeffrey, M. Effects of Agent Strain and Host Genotype on PrP Accumulation in the Brain of Sheep Naturally and Experimentally Affected with Scrapie. J. Comp. Pathol. 2002, 126, 17–29. [Google Scholar] [CrossRef]
- Gonzalez, L.; Siso, S.; Monleon, E.; Casalone, C.; van Keulen, L.J.M.; Balkema-Buschmann, A.; Ortiz-Pelaez, A.; Iulini, B.; Langeveld, J.P.M.; Hoffmann, C.; et al. Variability in Disease Phenotypes within a Single PRNP Genotype Suggests the Existence of Multiple Natural Sheep Scrapie Strains within Europe. J. Gen. Virol. 2010, 91, 2630–2641. [Google Scholar] [CrossRef]
- Moore, S.J.; Simmons, M.; Chaplin, M.; Spiropoulos, J. Neuroanatomical Distribution of Abnormal Prion Protein in Naturally Occurring Atypical Scrapie Cases in Great Britain. Acta Neuropathol. 2008, 116, 547–559. [Google Scholar] [CrossRef]
- Sisó, S.; Jeffrey, M.; Martin, S.; Chianini, F.; Dagleish, M.P.; González, L. Characterization of Strains of Ovine Transmissible Spongiform Encephalopathy with a Short PrPd Profiling Method. J. Comp. Pathol. 2010, 142, 300–310. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Biological Hazards (BIOHAZ). Scientific Opinion on the Scrapie Situation in the EU after 10 Years of Monitoring and Control in Sheep and Goats. EFSA J. 2014, 12. [Google Scholar] [CrossRef]
- Andréoletti, O.; Berthon, P.; Marc, D.; Sarradin, P.; Grosclaude, J.; van Keulen, L.; Schelcher, F.; Elsen, J.-M.; Lantier, F. Early Accumulation of PrPSc in Gut-Associated Lymphoid and Nervous Tissues of Susceptible Sheep from a Romanov Flock with Natural Scrapie. J. Gen. Virol. 2000, 81, 3115–3126. [Google Scholar] [CrossRef]
- Hadlow, W.J.; Kennedy, R.C.; Race, R.E. Natural Infection of Suffolk Sheep with Scrapie Virus. J. Infect. Dis. 1982, 146, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, L.J.M.; Bossers, A.; van Zijderveld, F. TSE Pathogenesis in Cattle and Sheep. Vet. Res. 2008, 39, 24. [Google Scholar] [CrossRef] [PubMed]
- Andréoletti, O.; Lacroux, C.; Chabert, A.; Monnereau, L.; Tabouret, G.; Lantier, F.; Berthon, P.; Eychenne, F.; Lafond-Benestad, S.; Elsen, J.-M.; et al. PrPSc Accumulation in Placentas of Ewes Exposed to Natural Scrapie: Influence of Foetal PrP Genotype and Effect on Ewe-to-Lamb Transmission. J. Gen. Virol. 2002, 83, 2607–2616. [Google Scholar] [CrossRef] [PubMed]
- Garza, M.C.; Monzón, M.; Marín, B.; Badiola, J.J.; Monleón, E. Distribution of Peripheral PrPSc in Sheep with Naturally Acquired Scrapie. PLoS ONE 2014, 9, e97768. [Google Scholar] [CrossRef] [PubMed]
- Thomzig, A.; Schulz-Schaeffer, W.; Wrede, A.; Wemheuer, W.; Brenig, B.; Kratzel, C.; Lemmer, K.; Beekes, M. Accumulation of Pathological Prion Protein PrPSc in the Skin of Animals with Experimental and Natural Scrapie. PLoS Pathog. 2007, 3, e66. [Google Scholar] [CrossRef] [PubMed]
- Ligios, C.; Sigurdson, C.J.; Santucciu, C.; Carcassola, G.; Manco, G.; Basagni, M.; Maestrale, C.; Cancedda, M.G.; Madau, L.; Aguzzi, A. PrPSc in Mammary Glands of Sheep Affected by Scrapie and Mastitis. Nat. Med. 2005, 11, 1137–1138. [Google Scholar] [CrossRef]
- Curcio, L.; Sebastiani, C.; Di Lorenzo, P.; Lasagna, E.; Biagetti, M. Review: A Review on Classical and Atypical Scrapie in Caprine: Prion Protein Gene Polymorphisms and Their Role in the Disease. Animal 2016, 10, 1585–1593. [Google Scholar] [CrossRef]
- Lacroux, C.; Perrin-Chauvineau, C.; Corbiere, F.; Aron, N.; Aguilar-Calvo, P.; Torres, J.M.; Costes, P.; Bremaud, I.; Lugan, S.; Schelcher, F.; et al. Genetic Resistance to Scrapie Infection in Experimentally Challenged Goats. J. Virol. 2014, 88, 2406–2413. [Google Scholar] [CrossRef]
- Papasavva-Stylianou, P.; Simmons, M.M.; Ortiz-Pelaez, A.; Windl, O.; Spiropoulos, J.; Georgiadou, S. Effect of Polymorphisms at Codon 146 of the Goat PRNP Gene on Susceptibility to Challenge with Classical Scrapie by Different Routes. J. Virol. 2017, 91, e01142-17. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (BIOHAZ); Ricci, A.; Allende, A.; Bolton, D.; Chemaly, M.; Davies, R.; Fernández Escámez, P.S.; Gironés, R.; Herman, L.; Koutsoumanis, K.; et al. Bovine Spongiform Encephalopathy (BSE) Cases Born after the Total Feed Ban. EFSA J. 2017, 15. [Google Scholar] [CrossRef]
- Migliore, S.; Puleio, R.; Loria, G.R. Scrapie Control in EU Goat Population: Has the Last Gap Been Overcome? Front. Vet. Sci. 2020, 7, 581969. [Google Scholar] [CrossRef]
- Sofianidis, G.; Psychas, V.; Billinis, C.; Spyrou, V.; Argyroudis, S.; Papaioannou, N.; Vlemmas, I. Histopathological and Immunohistochemical Features of Natural Goat Scrapie. J. Comp. Pathol. 2006, 135, 116–129. [Google Scholar] [CrossRef]
- Dustan, B.H.; Spencer, Y.I.; Casalone, C.; Brownlie, J.; Simmons, M.M. A Histopathologic and Immunohistochemical Review of Archived UK Caprine Scrapie Cases. Vet. Pathol. 2008, 45, 443–454. [Google Scholar] [CrossRef]
- González, L.; Martin, S.; Sisó, S.; Konold, T.; Ortiz-Peláez, A.; Phelan, L.; Goldmann, W.; Stewart, P.; Saunders, G.; Windl, O.; et al. High Prevalence of Scrapie in a Dairy Goat Herd: Tissue Distribution of Disease-Associated PrP and Effect of PRNP Genotype and Age. Vet. Res. 2009, 40, 65. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, J.P.M.; Pirisinu, L.; Jacobs, J.G.; Mazza, M.; Lantier, I.; Simon, S.; Andréoletti, O.; Acin, C.; Esposito, E.; Fast, C.; et al. Four Types of Scrapie in Goats Differentiated from Each Other and Bovine Spongiform Encephalopathy by Biochemical Methods. Vet. Res. 2019, 50, 97. [Google Scholar] [CrossRef] [PubMed]
- Le Dur, A.; Beringue, V.; Andreoletti, O.; Reine, F.; Lai, T.L.; Baron, T.; Bratberg, B.; Vilotte, J.-L.; Sarradin, P.; Benestad, S.L.; et al. A Newly Identified Type of Scrapie Agent Can Naturally Infect Sheep with Resistant PrP Genotypes. Proc. Natl. Acad. Sci. USA 2005, 102, 16031–16036. [Google Scholar] [CrossRef] [PubMed]
- De Bosschere, H.; Roels, S.; Dechamps, P.; Vanopdenbosch, E. TSE Detected in a Belgian ARR-Homozygous Sheep via Active Surveillance. Vet. J. 2007, 173, 449–451. [Google Scholar] [CrossRef]
- Saunders, G.C.; Cawthraw, S.; Mountjoy, S.J.; Hope, J.; Windl, O. PrP Genotypes of Atypical Scrapie Cases in Great Britain. J. Gen. Virol. 2006, 87, 3141–3149. [Google Scholar] [CrossRef]
- Orge, L.; Galo, A.; Machado, C.; Lima, C.; Ochoa, C.; Silva, J.; Ramos, M.; Simas, J.P. Identification of Putative Atypical Scrapie in Sheep in Portugal. J. Gen. Virol. 2004, 85, 3487–3491. [Google Scholar] [CrossRef]
- Orge, L.; Oliveira, A.; Machado, C.; Lima, C.; Ochoa, C.; Silva, J.; Carvalho, R.; Tavares, P.; Almeida, P.; Ramos, M.; et al. Putative Emergence of Classical Scrapie in a Background of Enzootic Atypical Scrapie. J. Gen. Virol. 2010, 91, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Andréoletti, O.; Orge, L.; Benestad, S.L.; Beringue, V.; Litaise, C.; Simon, S.; Le Dur, A.; Laude, H.; Simmons, H.; Lugan, S.; et al. Atypical/Nor98 Scrapie Infectivity in Sheep Peripheral Tissues. PLoS Pathog. 2011, 7, e1001285. [Google Scholar] [CrossRef]
- Simmons, M.M.; Moore, S.J.; Konold, T.; Thurston, L.; Terry, L.A.; Thorne, L.; Lockey, R.; Vickery, C.; Hawkins, S.A.C.; Chaplin, M.J.; et al. Experimental Oral Transmission of Atypical Scrapie to Sheep. Emerg. Infect. Dis. 2011, 17, 848–854. [Google Scholar] [CrossRef]
- Seuberlich, T.; Botteron, C.; Benestad, S.L.; Brünisholz, H.; Wyss, R.; Kihm, U.; Schwermer, H.; Friess, M.; Nicolier, A.; Heim, D.; et al. Atypical Scrapie in a Swiss Goat and Implications for Transmissible Spongiform Encephalopathy Surveillance. J. Vet. Diagn. Investig. 2007, 19, 2–8. [Google Scholar] [CrossRef]
- Mink|Description, Habitat, Diet, & Facts. Available online: https://www.britannica.com/animal/mink (accessed on 29 January 2021).
- Barlow, R.M. Transmissible Mink Encephalopathy: Pathogenesis and Nature of the Aetiological Agent. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 1972, 6, 102–109. [Google Scholar] [CrossRef]
- Liberski, P.P.; Sikorska, B.; Guiroy, D.; Bessen, R.A. Transmissible Mink Encephalopathy—Review of the Etiology of a Rare Prion Disease. Folia Neuropathol. 2009, 47, 195–204. [Google Scholar]
- Spickler, A.R. Transmissible Mink Encephalopathy. Available online: https://www.cfsph.iastate.edu/Factsheets/pdfs/transmissible_mink_encephalopathy.pdf (accessed on 30 December 2020).
- Baron, T.; Bencsik, A.; Biacabe, A.-G.; Morignat, E.; Bessen, R.A. Phenotypic Similarity of Transmissible Mink Encephalopathy in Cattle and L-Type Bovine Spongiform Encephalopathy in a Mouse Model. Emerg. Infect. Dis. 2007, 13, 1887–1894. [Google Scholar] [CrossRef]
- Comoy, E.; Mikol, J.; Ruchoux, M.-M.; Durand, V.; Luccantoni-Freire, S.; Dehen, C.; Correia, E.; Casalone, C.; Richt, J.; Greenlee, J.; et al. Evaluation of the Zoonotic Potential of Transmissible Mink Encephalopathy. Pathogens 2013, 2, 520–532. [Google Scholar] [CrossRef]
- Hamir, A.N.; Miller, J.M.; O’Rourke, K.I.; Bartz, J.C.; Stack, M.J.; Chaplin, M.J. Transmission of Transmissible Mink Encephalopathy to Raccoons (Procyon lotor) by Intracerebral Inoculation. J. Vet. Diagn. Investig. 2004, 16, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Telling, G.C. Chronic wasting disease: An evolving prion disease of cervids. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 153, pp. 135–151. ISBN 978-0-444-63945-5. [Google Scholar]
- Otero, A.; Duque Velásquez, C.; Johnson, C.; Herbst, A.; Bolea, R.; Badiola, J.J.; Aiken, J.; McKenzie, D. Prion Protein Polymorphisms Associated with Reduced CWD Susceptibility Limit Peripheral PrPCWD Deposition in Orally Infected White-Tailed Deer. BMC Vet. Res. 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Aguzzi, A. Chronic Wasting Disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 610–618. [Google Scholar] [CrossRef]
- Williams, E.S.; Young, S. Neuropathology of Chronic Wasting Disease of Mule Deer (Odocoileus hemionus) and Elk (Cervus elaphus nelsoni). Vet. Pathol. 1993, 30, 36–45. [Google Scholar] [CrossRef]
- Guiroy, D.C.; Williams, E.S.; Yanagihara, R.; Gajdusek, D.C. Topographic Distribution of Scrapie Amyloid-Immunoreactive Plaques in Chronic Wasting Disease in Captive Mule Deer (Odocoileus hemionus hemionus). Acta Neuropathol. 1991, 81, 475–478. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.I.; Zhuang, D.; Lyda, A.; Gomez, G.; Williams, E.S.; Tuo, W.; Miller, M.W. Abundant PrP CWD in Tonsil from Mule Deer with Preclinical Chronic Wasting Disease. J. Vet. Diagn. Investig. 2003, 15, 320–323. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Williams, E.S.; Miller, M.W.; Spraker, T.R.; O’Rourke, K.I.; Hoover, E.A. Oral Transmission and Early Lymphoid Tropism of Chronic Wasting Disease PrPres in Mule Deer Fawns (Odocoileus hemionus). J. Gen. Virol. 1999, 80, 2757–2764. [Google Scholar] [CrossRef]
- Fox, K.A.; Jewell, J.E.; Williams, E.S.; Miller, M.W. Patterns of PrPCWD Accumulation during the Course of Chronic Wasting Disease Infection in Orally Inoculated Mule Deer (Odocoileus hemionus). J. Gen. Virol. 2006, 87, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; O’Rourke, K.I.; Dong, Z.; Jenny, A.L.; Langenberg, J.A.; Belay, E.D.; Schonberger, L.B.; Petersen, R.B.; Zou, W.; Kong, Q.; et al. Chronic Wasting Disease of Elk and Deer and Creutzfeldt-Jakob Disease. J. Biol. Chem. 2006, 281, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S. Chronic Wasting Disease. Vet. Pathol. 2005, 42, 530–549. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Spraker, T.R.; Miller, M.W.; Oesch, B.; Hoover, E.A. PrPCWD in the Myenteric Plexus, Vagosympathetic Trunk and Endocrine Glands of Deer with Chronic Wasting Disease. J. Gen. Virol. 2001, 82, 2327–2334. [Google Scholar] [CrossRef]
- Jewell, J.E.; Brown, J.; Kreeger, T.; Williams, E.S. Prion Protein in Cardiac Muscle of Elk (Cervus Elaphus Nelsoni) and White-Tailed Deer (Odocoileus virginianus) Infected with Chronic Wasting Disease. J. Gen. Virol. 2006, 87, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Seward, T.S.; Napier, D.; Green, M.; Hoover, E.; Spraker, T.; O’Rourke, K.; Balachandran, A.; Telling, G.C. Chronic Wasting Disease Prions in Elk Antler Velvet. Emerg. Infect. Dis. 2009, 15, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L.; Breyer, J.; Wagenfuehr, K.; Wemheuer, W.M.; Thomzig, A.; Schulz-Schaeffer, W.J.; Beekes, M. Presence and Seeding Activity of Pathological Prion Protein (PrPTSE) in Skeletal Muscles of White-Tailed Deer Infected with Chronic Wasting Disease. PLoS ONE 2011, 6, e18345. [Google Scholar] [CrossRef]
- Mitchell, G.B.; Sigurdson, C.J.; O’Rourke, K.I.; Algire, J.; Harrington, N.P.; Walther, I.; Spraker, T.R.; Balachandran, A. Experimental Oral Transmission of Chronic Wasting Disease to Reindeer (Rangifer Tarandus Tarandus). PLoS ONE 2012, 7, e39055. [Google Scholar] [CrossRef]
- Pirisinu, L.; Tran, L.; Chiappini, B.; Vanni, I.; Di Bari, M.A.; Vaccari, G.; Vikøren, T.; Madslien, K.I.; Våge, J.; Spraker, T.; et al. Novel Type of Chronic Wasting Disease Detected in Moose (Alces alces), Norway. Emerg. Infect. Dis. 2018, 24, 2210–2218. [Google Scholar] [CrossRef]
- Wells, G.; Scott, A.; Johnson, C.; Gunning, R.; Hancock, R.; Jeffrey, M.; Dawson, M.; Bradley, R. A Novel Progressive Spongiform Encephalopathy in Cattle. Vet. Rec. 1987, 121, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Wilesmith, J.W.; Wells, G.A.; Cranwell, M.P.; Ryan, J.B. Bovine Spongiform Encephalopathy: Epidemiological Studies. Vet. Rec. 1988, 123, 638–644. [Google Scholar] [CrossRef]
- Anderson, R.M.; Donnelly, C.A.; Ferguson, N.M.; Woolhouse, M.E.J.; Watt, C.J.; Udy, H.J.; MaWhinney, S.; Dunstan, S.P.; Southwood, T.R.E.; Wilesmith, J.W.; et al. Transmission Dynamics and Epidemiology of BSE in British Cattle. Nature 1996, 382, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Sidle, K.C.L.; Meads, J.; Ironside, J.; Hill, A.F. Molecular Analysis of Prion Strain Variation and the Aetiology of “new Variant” CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to Mice Indicate That ‘New Variant’ CJD Is Caused by the BSE Agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Biacabe, A.; Laplanche, J.; Ryder, S.; Baron, T. Distinct Molecular Phenotypes in Bovine Prion Diseases. EMBO Rep. 2004, 5, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Casalone, C.; Zanusso, G.; Acutis, P.; Ferrari, S.; Capucci, L.; Tagliavini, F.; Monaco, S.; Caramelli, M. Identification of a Second Bovine Amyloidotic Spongiform Encephalopathy: Molecular Similarities with Sporadic Creutzfeldt-Jakob Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3065–3070. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). The European Union Summary Report on Surveillance for the Presence of Transmissible Spongiform Encephalopathies (TSE) in 2019. EFSA J. 2020, 18. [Google Scholar] [CrossRef]
- Brunelle, B.W.; Hamir, A.N.; Baron, T.; Biacabe, A.G.; Richt, J.A.; Kunkle, R.A.; Cutlip, R.C.; Miller, J.M.; Nicholson, E.M. Polymorphisms of the Prion Gene Promoter Region That Influence Classical Bovine Spongiform Encephalopathy Susceptibility Are Not Applicable to Other Transmissible Spongiform Encephalopathies in Cattle. J. Anim. Sci. 2007, 85, 3142–3147. [Google Scholar] [CrossRef] [PubMed]
- Béringue, V.; Bencsik, A.; Le Dur, A.; Reine, F.; Laï, T.L.; Chenais, N.; Tilly, G.; Biacabé, A.-G.; Baron, T.; Vilotte, J.-L.; et al. Isolation from Cattle of a Prion Strain Distinct from That Causing Bovine Spongiform Encephalopathy. PLoS Pathog. 2006, 2, e112. [Google Scholar] [CrossRef]
- Lombardi, G.; Casalone, C.; D’Angelo, A.; Gelmetti, D.; Torcoli, G.; Barbieri, I.; Corona, C.; Fasoli, E.; Farinazzo, A.; Fiorini, M.; et al. Intraspecies Transmission of BASE Induces Clinical Dullness and Amyotrophic Changes. PLoS Pathog. 2008, 4, e1000075. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Iwamaru, Y.; Imamura, M.; Masujin, K.; Matsuura, Y.; Shimizu, Y.; Kasai, K.; Mohri, S.; Yokoyama, T.; Czub, S. Experimental H-Type Bovine Spongiform Encephalopathy Characterized by Plaques and Glial- and Stellate-Type Prion Protein Deposits. Vet. Res. 2011, 42, 79. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.H.; Wilesmith, J.W. The Neuropathology and Epidemiology of Bovine Spongiform Encephalopathy. Brain Pathol. 1995, 5, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.M.; Harris, P.; Jeffrey, M.; Meek, S.C.; Blamire, I.W.H.; Wells, G.A.H. BSE in Great Britain: Consistency of the Neurohistopathological Findings in Two Random Annual Samples of Clinically Suspect Cases. Vet. Rec. 1996, 138, 175–177. [Google Scholar] [CrossRef]
- Wells, G.A.H.; McGill, I.S. Recently Described Scrapie-like Encephalopathies of Animals: Case Definitions. Res. Vet. Sci. 1992, 53, 1–10. [Google Scholar] [CrossRef]
- Corona, C.; Vallino Costassa, E.; Iulini, B.; Caramelli, M.; Bozzetta, E.; Mazza, M.; Desiato, R.; Ru, G.; Casalone, C. Phenotypical variability in bovine spongiform encephalopathy: Epidemiology, pathogenesis, and diagnosis of classical and atypical forms. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2017; Volume 150, pp. 241–265. ISBN 978-0-12-811226-7. [Google Scholar]
- Gavier-Widén, D.; Stack, M.J.; Baron, T.; Balachandran, A.; Simmons, M. Diagnosis of Transmissible Spongiform Encephalopathies in Animals: A Review. J. Vet. Diagn. Investig. 2005, 17, 509–527. [Google Scholar] [CrossRef]
- Orge, L.; Fernandes, A.C.; Ramos, M.; Galo, A.; Simas, J.P. Similarity of the Lesion Profile of BSE in Portuguese Cattle to That Described in British Cattle. Vet. Rec. 2000, 147, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Wilesmith, J.W.; Ryan, J.B.; Atkinson, M.J. Bovine Spongiform Encephalopathy: Epidemiological Studies on the Origin. Vet. Rec. 1991, 128, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G. Review of the Evidence for the Occurrence of’BARB’BSE Cases in Cattle; Citeseer; DEFRA: London, UK, 2005. Available online: https://webarchive.nationalarchives.gov.uk/20110318144828/http://www.defra.gov.uk/foodfarm/farmanimal/diseases/atoz/bse/documents/hillreport.pdf (accessed on 21 January 2021).
- Konold, T.; Bone, G.E.; Clifford, D.; Chaplin, M.J.; Cawthraw, S.; Stack, M.J.; Simmons, M.M. Experimental H-Type and L-Type Bovine Spongiform Encephalopathy in Cattle: Observation of Two Clinical Syndromes and Diagnostic Challenges. BMC Vet. Res. 2012, 8, 22. [Google Scholar] [CrossRef]
- Stack, M.J.; Moore, S.J.; Davis, A.; Webb, P.R.; Bradshaw, J.M.; Lee, Y.H.; Chaplin, M.; Focosi-Snyman, R.; Thurston, L.; Spencer, Y.I.; et al. Bovine Spongiform Encephalopathy: Investigation of Phenotypic Variation among Passive Surveillance Cases. J. Comp. Pathol. 2011, 144, 277–288. [Google Scholar] [CrossRef]
- Porcario, C.; Hall, S.M.; Martucci, F.; Corona, C.; Iulini, B.; Perazzini, A.Z.; Acutis, P.; Hamir, A.N.; Loiacono, C.M.; Greenlee, J.J.; et al. Evaluation of Two Sets of Immunohistochemical and Western Blot Confirmatory Methods in the Detection of Typical and Atypical BSE Cases. BMC Res. Notes 2011, 4, 376. [Google Scholar] [CrossRef]
- Guldimann, C.; Gsponer, M.; Drögemüller, C.; Oevermann, A.; Seuberlich, T. Atypical H-Type Bovine Spongiform Encephalopathy in a Cow Born after the Reinforced Feed Ban on Meat-and-Bone Meal in Europe. J. Clin. Microbiol. 2012, 50, 4171–4174. [Google Scholar] [CrossRef][Green Version]
- Orge, L.; Machado, C.G.; Ramalho, L.; Carvalho, R.; Silva, J.; Almeida, P.; Tavares, P.; Ochoa, C.; Lima, C.; Pinto, M.J.M.; et al. Identification of H-Type BSE in Portugal. Prion 2015, 9, 22–28. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cunningham, A.A.; Kirkwood, J.K.; Dawson, M.; Spencer, Y.I.; Green, R.B.; Wells, G.A.H. Bovine Spongiform Encephalopathy Infectivity in Greater Kudu (Tragelaphus strepsiceros). Emerg. Infect. Dis. 2004, 10, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.; Chree, A.; McConnell, I.; Foster, J.; Pearson, G.; Fraser, H.; Collinge, J.; Weissmann, C. Transmission of Bovine Spongiform Encephalopathy and Scrapie to Mice: Strain Variation and the Species Barrier. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1994, 343, 405–411. [Google Scholar] [CrossRef]
- Bons, N.; Mestre-Frances, N.; Belli, P.; Cathala, F.; Gajdusek, D.C.; Brown, P. Natural and Experimental Oral Infection of Nonhuman Primates by Bovine Spongiform Encephalopathy Agents. Proc. Natl. Acad. Sci. USA 1999, 96, 4046–4051. [Google Scholar] [CrossRef]
- Baker, H.F.; Ridley, R.M.; Wells, G.A.; Ironside, J.W. Spontaneous Spongiform Encephalopathy in a Monkey. Lancet 1996, 348, 955–956. [Google Scholar] [CrossRef]
- Eloit, M.; Adjou, K.; Coulpier, M.; Fontaine, J.J.; Hamel, R.; Lilin, T.; Messiaen, S.; Andreoletti, O.; Baron, T.; Bencsik, A.; et al. BSE Agent Signatures in a Goat. Vet. Rec. 2005, 156, 523–524. [Google Scholar] [CrossRef]
- Spiropoulos, J.; Lockey, R.; Sallis, R.E.; Terry, L.A.; Thorne, L.; Holder, T.M.; Beck, K.E.; Simmons, M.M. Isolation of Prion with BSE Properties from Farmed Goat. Emerg. Infect. Dis. 2011, 17, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; Parnham, D.; Chong, A.; Goldmann, W.; Hunter, N. Clinical Signs, Histopathology and Genetics of Experimental Transmission of BSE and Natural Scrapie to Sheep and Goats. Vet. Rec. 2001, 148, 165–171. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Martin, S.; Houston, F.E.; Hunter, N.; Reid, H.W.; Bellworthy, S.J.; Jeffrey, M. Phenotype of Disease-Associated PrP Accumulation in the Brain of Bovine Spongiform Encephalopathy Experimentally Infected Sheep. J. Gen. Virol. 2005, 86, 827–838. [Google Scholar] [CrossRef]
- Foster, J.; Hope, J.; Fraser, H. Transmission of Bovine Spongiform Encephalopathy to Sheep and Goats. Vet. Rec. 1993, 133, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Martin, S.; González, L.; Ryder, S.J.; Bellworthy, S.J.; Jackman, R. Differential Diagnosis of Infections with the Bovine Spongiform Encephalopathy (BSE) and Scrapie Agents in Sheep. J. Comp. Pathol. 2001, 125, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Martin, S.; González, L.; Foster, J.; Langeveld, J.P.M.; van Zijderveld, F.G.; Grassi, J.; Hunter, N. Immunohistochemical Features of PrPd Accumulation in Natural and Experimental Goat Transmissible Spongiform Encephalopathies. J. Comp. Pathol. 2006, 134, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Iulini, B.; Cantile, C.; Mandara, M.T.; Maurella, C.; Loria, G.R.; Castagnaro, M.; Salvadori, C.; Porcario, C.; Corona, C.; Perazzini, A.Z.; et al. Neuropathology of Italian Cats in Feline Spongiform Encephalopathy Surveillance. Vet. Pathol. 2008, 45, 626–633. [Google Scholar] [CrossRef]
- Kelly, D.F.; Wells, G.A.H.; Haritani, M.; Higgins, R.J.; Jeffrey, M. Neuropathological Findings in Cats with Clinically Suspect but Histologically Unconfirmed Feline Spongiform Encephalopathy. Vet. Rec. 2005, 156, 472–477. [Google Scholar] [CrossRef]
- Wyatt, J.; Pearson, G.; Smerdon, T.; Gruffydd-Jones, T.; Wells, G.; Wilesmith, J. Naturally Occurring Scrapie-like Spongiform Encephalopathy in Five Domestic Cats. Vet. Rec. 1991, 129, 233–236. [Google Scholar] [CrossRef]
- Hilbe, M.M.; Soldati, G.G.; Zlinszky, K.K.; Wunderlin, S.S.; Ehrensperger, F.F. Immunohistochemical Study of PrPSc Distribution in Neural and Extraneural Tissues of Two Cats with Feline Spongiform Encephalopathy. BMC Vet. Res. 2009, 5, 11. [Google Scholar] [CrossRef]
- Silva, J.; Correia, J.; Ribeiro, J.; Carmo, S.; Orge, L. Feline Spongiform Encephalopathy: First Confirmed Case in Portugal. Abstract Book, Poster Session-Diagnosis [DIA-45]:392. In Proceedings of the Prion 2006, Torino, Italy, 4–6 October 2006; Available online: https://www.researchgate.net/publication/216830198_Feline_spongiform_encephalopathy_first_confirmed_case_in_Portugal (accessed on 21 January 2021).
- Lezmi, S.; Bencsik, A.; Monks, E.; Petit, T.; Baron, T. First Case of Feline Spongiform Encephalopathy in a Captive Cheetah Born in France: PrPsc Analysis in Various Tissues Revealed Unexpected Targeting of Kidney and Adrenal Gland. Histochem. Cell Biol. 2003, 119, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Bencsik, A.; Debeer, S.; Petit, T.; Baron, T. Possible Case of Maternal Transmission of Feline Spongiform Encephalopathy in a Captive Cheetah. PLoS ONE 2009, 4, e6929. [Google Scholar] [CrossRef] [PubMed]
- Camel|Bactrian, Dromedary, & Facts. Available online: https://www.britannica.com/animal/camel (accessed on 29 January 2021).
- Harek, D.; Ikhlef, H.; Rachid, B.; Sahel, H.; Cherifi, Y.A.; Djallout, N.; Khelifa ChelihI, S.; El Mokhefi, M.; Boukhtala, K.; Suheil Gaouar, S.B.; et al. Genetic diversity status of Camel’s resources (Camelus dromedarius. Linnaeus, 1758) IN ALGERIA. Genet. Biodivers. J. 2017, 1, 43–65. [Google Scholar]
- Babelhadj, B. Camel Prion Disease: A Possible Emerging Disease in Dromedary Camel Populations? Available online: https://oiebulletin.com/wp-content/uploads/2019/12/OIE-News-December-2019-Camel-prion-disease.pdf (accessed on 21 January 2021).
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J. Virol. 2016, 90, 3003–3017. [Google Scholar] [CrossRef]
- Baker, C.A.; Manuelidis, L. Unique Inflammatory RNA Profiles of Microglia in Creutzfeldt-Jakob Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679. [Google Scholar] [CrossRef]
- Lewicki, H.; Tishon, A.; Homann, D.; Mazarguil, H.; Laval, F.; Asensio, V.C.; Campbell, I.L.; DeArmond, S.; Coon, B.; Teng, C.; et al. T Cells Infiltrate the Brain in Murine and Human Transmissible Spongiform Encephalopathies. J. Virol. 2003, 77, 3799–3808. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. Region-Specific Glial Homeostatic Signature in Prion Diseases Is Replaced by a Uniform Neuroinflammation Signature, Common for Brain Regions and Prion Strains with Different Cell Tropism. Neurobiol. Dis. 2020, 137, 104783. [Google Scholar] [CrossRef] [PubMed]
- Ano, Y.; Sakudo, A.; Onodera, T. Effect of Microglial Inflammation in Prion Disease. Curr. Issues Mol. Biol. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The Immunobiology of Prion Diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Ragagnin, A.; Ezpeleta, J.; Guillemain, A.; Boudet-Devaud, F.; Haeberlé, A.-M.; Demais, V.; Vidal, C.; Demuth, S.; Béringue, V.; Kellermann, O.; et al. Cerebellar Compartmentation of Prion Pathogenesis: Prions in Cerebellum. Brain Pathol. 2018, 28, 240–263. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef]
- Kranich, J.; Krautler, N.J.; Falsig, J.; Ballmer, B.; Li, S.; Hutter, G.; Schwarz, P.; Moos, R.; Julius, C.; Miele, G.; et al. Engulfment of Cerebral Apoptotic Bodies Controls the Course of Prion Disease in a Mouse Strain–Dependent Manner. J. Exp. Med. 2010, 207, 2271–2281. [Google Scholar] [CrossRef]
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the Degenerating Brain Are Capable of Phagocytosis of Beads and of Apoptotic Cells, but Do Not Efficiently Remove PrPSc, Even upon LPS Stimulation. Glia 2010, 58, 2017–2030. [Google Scholar] [CrossRef]
- Xiang, W.; Windl, O.; Wünsch, G.; Dugas, M.; Kohlmann, A.; Dierkes, N.; Westner, I.M.; Kretzschmar, H.A. Identification of Differentially Expressed Genes in Scrapie-Infected Mouse Brains by Using Global Gene Expression Technology. J. Virol. 2004, 78, 11051–11060. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Roomp, K.; Jurkowski, W.; Kitano, H.; del Sol, A. Gene Regulatory Network Analysis Supports Inflammation as a Key Neurodegeneration Process in Prion Disease. BMC Syst. Biol. 2012, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Striebel, J.F.; Race, B.; Phillips, K.; Chesebro, B. Prion Infection of Mouse Brain Reveals Multiple New Upregulated Genes Involved in Neuroinflammation or Signal Transduction. J. Virol. 2015, 89, 2388–2404. [Google Scholar] [CrossRef]
- Uskokovic, A.; Dinic, S.; Mihailovic, M.; Grigorov, I.; Ivanovic-Matic, S.; Bogojevic, D.; Grdovic, N.; Arambasic, J.; Vidakovic, M.; Martinovic, V.; et al. STAT3/NFκB Interplay in the Regulation of Α2-Macroglobulin Gene Expression During Rat Liver Development and the Acute Phase Response. IUBMB Life 2017, 59, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Albrecht, U.; Häussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic Acute Phase Proteins—Regulation by IL-6- and IL-1-Type Cytokines Involving STAT3 and Its Crosstalk with NF-ΚB-Dependent Signaling. Eur. J. Cell Biol. 2012, 91, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Castelli, J.C.; Hassel, B.A.; Maran, A.; Paranjape, J.; Hewitt, J.A.; Li, X.; Hsu, Y.-T.; Silverman, R.H.; Youle, R.J. The Role of 2′-5′ Oligoadenylate-Activated Ribonuclease L in Apoptosis. Cell Death Differ. 1998, 5, 313–320. [Google Scholar] [CrossRef]
- Valerio, A.; Ferrario, M.; Martinez, F.O.; Locati, M.; Ghisi, V.; Bresciani, L.G.; Mantovani, A.; Spano, P. Gene Expression Profile Activated by the Chemokine CCL5/RANTES in Human Neuronal Cells. J. Neurosci. Res. 2004, 78, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Potu, H.; Sgorbissa, A.; Brancolini, C. Identification of USP18 as an Important Regulator of the Susceptibility to IFN-α and Drug-Induced Apoptosis. Cancer Res. 2010, 70, 655–665. [Google Scholar] [CrossRef]
- Van Coillie, E.; Van Damme, J.; Opdenakker, G. The MCP/Eotaxin Subfamily of CC Chemokines. Cytokine Growth Factor Rev. 1999, 10, 61–86. [Google Scholar] [CrossRef]
- Fabrizi, C.; Businaro, R.; Lauro, G.M.; Starace, G.; Fumagalli, L. Activated Α2Macroglobulin Increases β-Amyloid (25–35)-Induced Toxicity in LAN5 Human Neuroblastoma Cells. Exp. Neurol. 1999, 155, 252–259. [Google Scholar] [CrossRef]
- Kovacs, D. Α2-Macroglobulin in Late-Onset Alzheimer’s Disease. Exp. Gerontol. 2000, 35, 473–479. [Google Scholar] [CrossRef]
- Sui, Y.; Stehno-Bittel, L.; Li, S.; Loganathan, R.; Dhillon, N.K.; Pinson, D.; Nath, A.; Kolson, D.; Narayan, O.; Buch, S. CXCL10-Induced Cell Death in Neurons: Role of Calcium Dysregulation. Eur. J. Neurosci. 2006, 23, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Severini, C.; Passeri, P.P.; Ciotti, M.; Florenzano, F.; Possenti, R.; Zona, C.; Di Matteo, A.; Guglielmotti, A.; Calissano, P.; Pachter, J.; et al. Bindarit, Inhibitor of CCL2 Synthesis, Protects Neurons Against Amyloid-β-Induced Toxicity. J. Alzheimers Dis. 2013, 38, 281–293. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
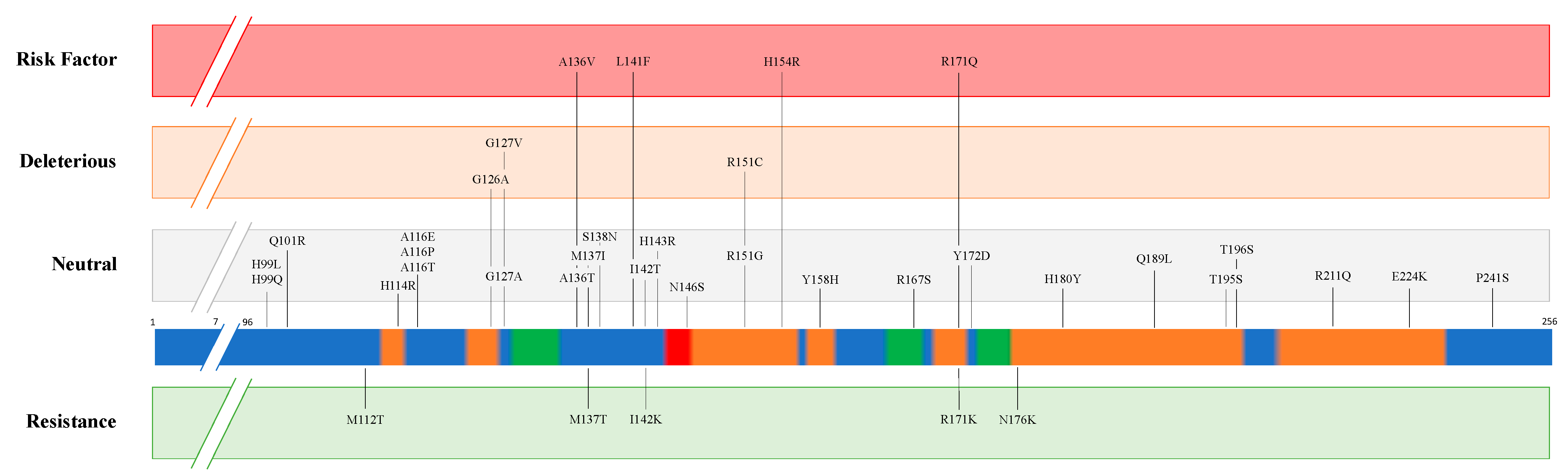
| Disease | Species | PRNP Predisposition to Resistance | PRNP Predisposition to Susceptibility | ||
|---|---|---|---|---|---|
| Position | Amino acid | Position | Amino acid | ||
| Classical Scrapie | Sheep | 112 136 137 141 142 154 171 171 176 | T [12] A [13] T [11] F [19] K [11] H [13] K [20] R [13] K [11] | 136 154 171 | V [13] R [13] Q [13] |
| Goat | 143 146 146 154 211 222 | R [21] D [22] S [22] H [13] Q [13] K [13] | |||
| Atypical Scrapie | Sheep | 141 154 | F [13] H [13] | ||
| Goat | 154 | H [23] | |||
| Chronic Wasting Disease | Elk | 132 | L [24] | ||
| White-tail deer | 95 96 | H [25] S [25] | |||
| Mule deer | 225 | F [26] | |||
| Classical Bovine Spongiform Encephalopathy | Cattle | 23-bp deletion (Promoter region) [27] 12-bp deletion (First intron) [28] | |||
| Atypical Bovine Spongiform Encephalopathy | 211 | K [29] | |||
| Indel 23-bp (Promoter region) [30] Indel 12-bp (First intron) [30] | |||||
| Camel Prion Disease | Dromedary camel | 134E to be determined [31] | To be determined [32] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orge, L.; Lima, C.; Machado, C.; Tavares, P.; Mendonça, P.; Carvalho, P.; Silva, J.; Pinto, M.d.L.; Bastos, E.; Pereira, J.C.; et al. Neuropathology of Animal Prion Diseases. Biomolecules 2021, 11, 466. https://doi.org/10.3390/biom11030466
Orge L, Lima C, Machado C, Tavares P, Mendonça P, Carvalho P, Silva J, Pinto MdL, Bastos E, Pereira JC, et al. Neuropathology of Animal Prion Diseases. Biomolecules. 2021; 11(3):466. https://doi.org/10.3390/biom11030466
Chicago/Turabian StyleOrge, Leonor, Carla Lima, Carla Machado, Paula Tavares, Paula Mendonça, Paulo Carvalho, João Silva, Maria de Lurdes Pinto, Estela Bastos, Jorge Cláudio Pereira, and et al. 2021. "Neuropathology of Animal Prion Diseases" Biomolecules 11, no. 3: 466. https://doi.org/10.3390/biom11030466
APA StyleOrge, L., Lima, C., Machado, C., Tavares, P., Mendonça, P., Carvalho, P., Silva, J., Pinto, M. d. L., Bastos, E., Pereira, J. C., Gonçalves-Anjo, N., Gama, A., Esteves, A., Alves, A., Matos, A. C., Seixas, F., Silva, F., Pires, I., Figueira, L., ... Pires, M. d. A. (2021). Neuropathology of Animal Prion Diseases. Biomolecules, 11(3), 466. https://doi.org/10.3390/biom11030466