
SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors





Abstract
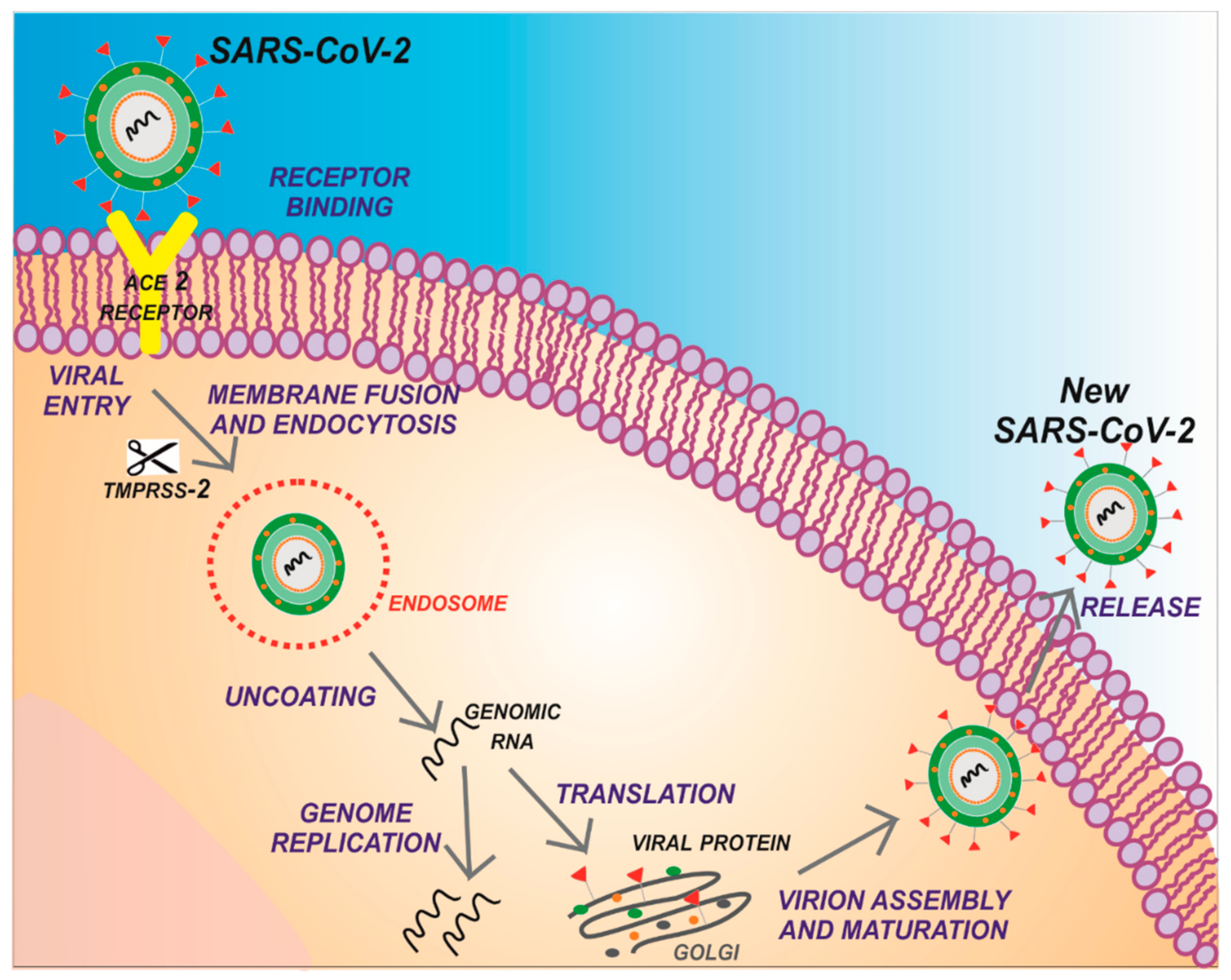
1. Introduction
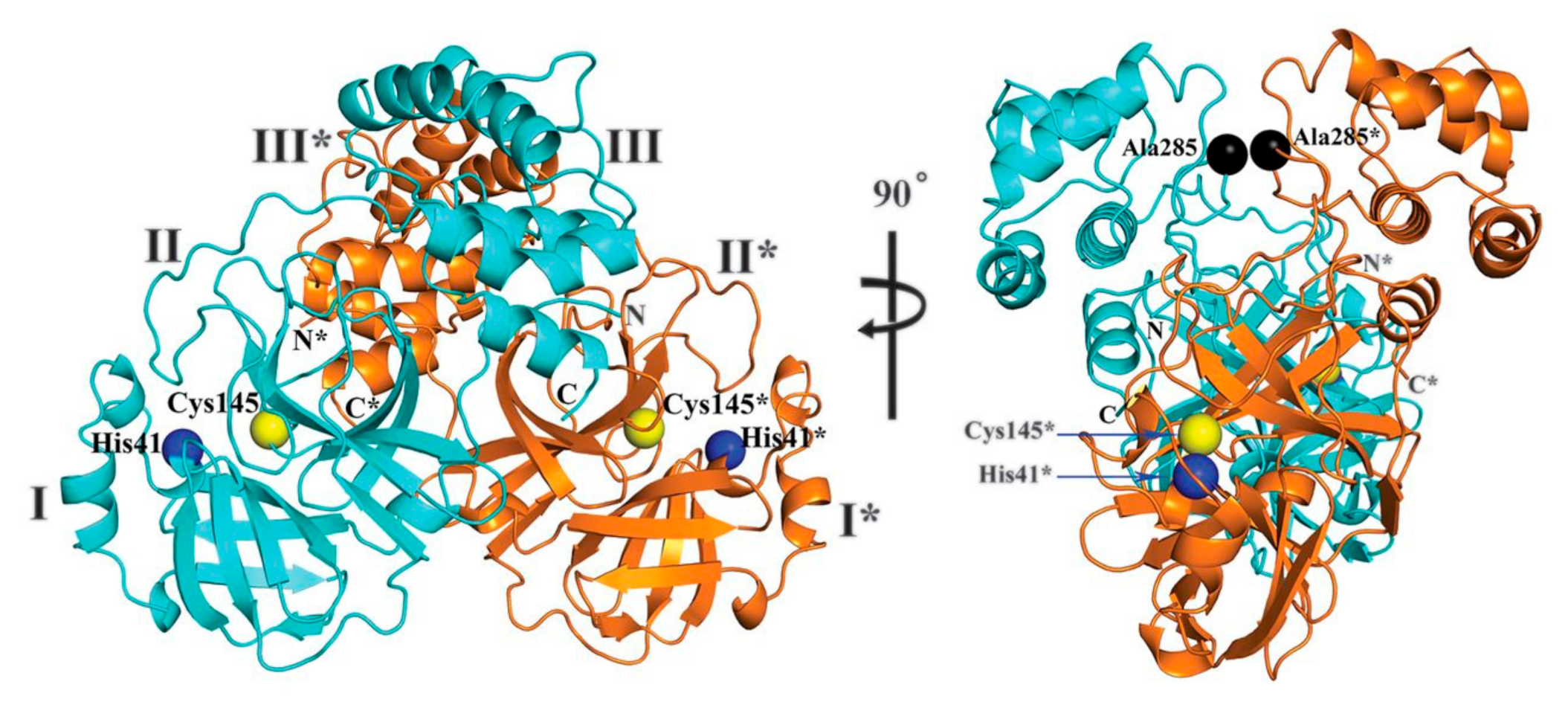
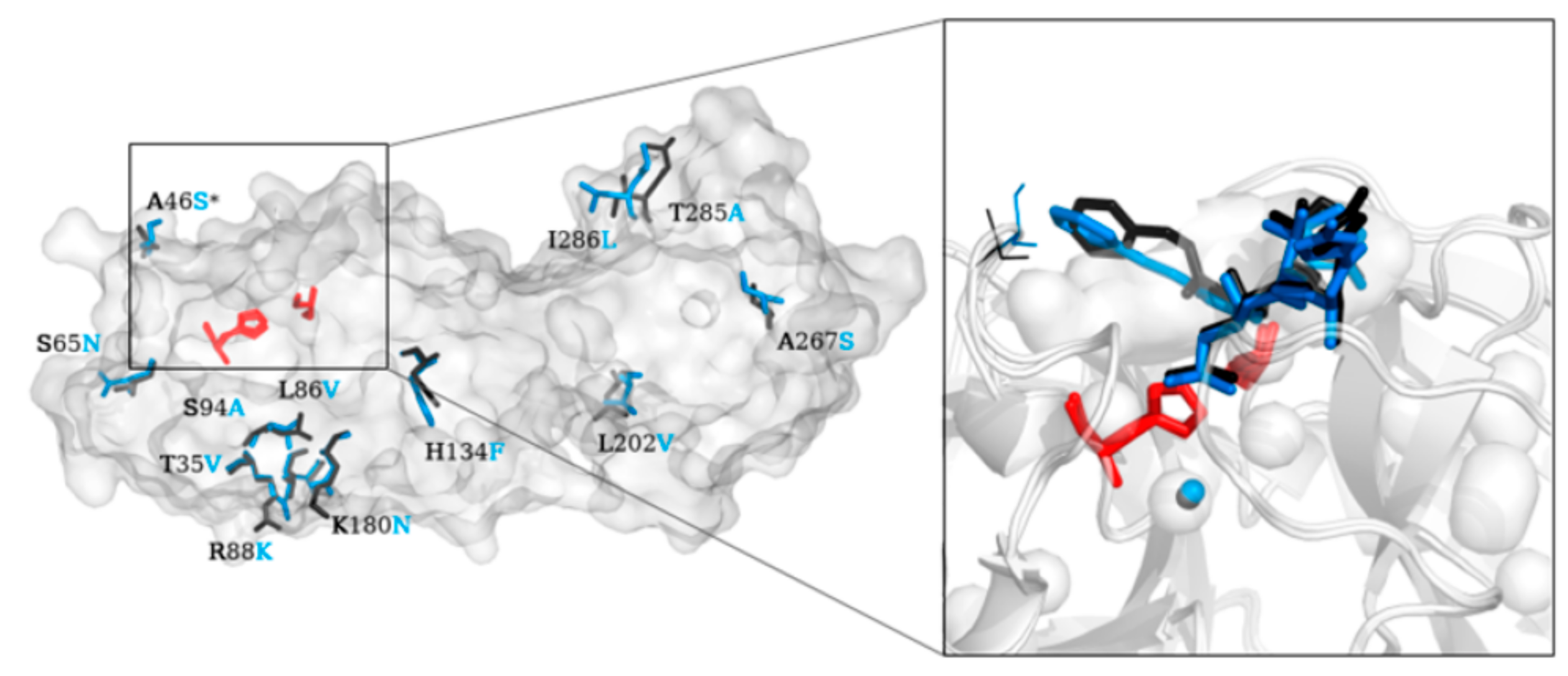
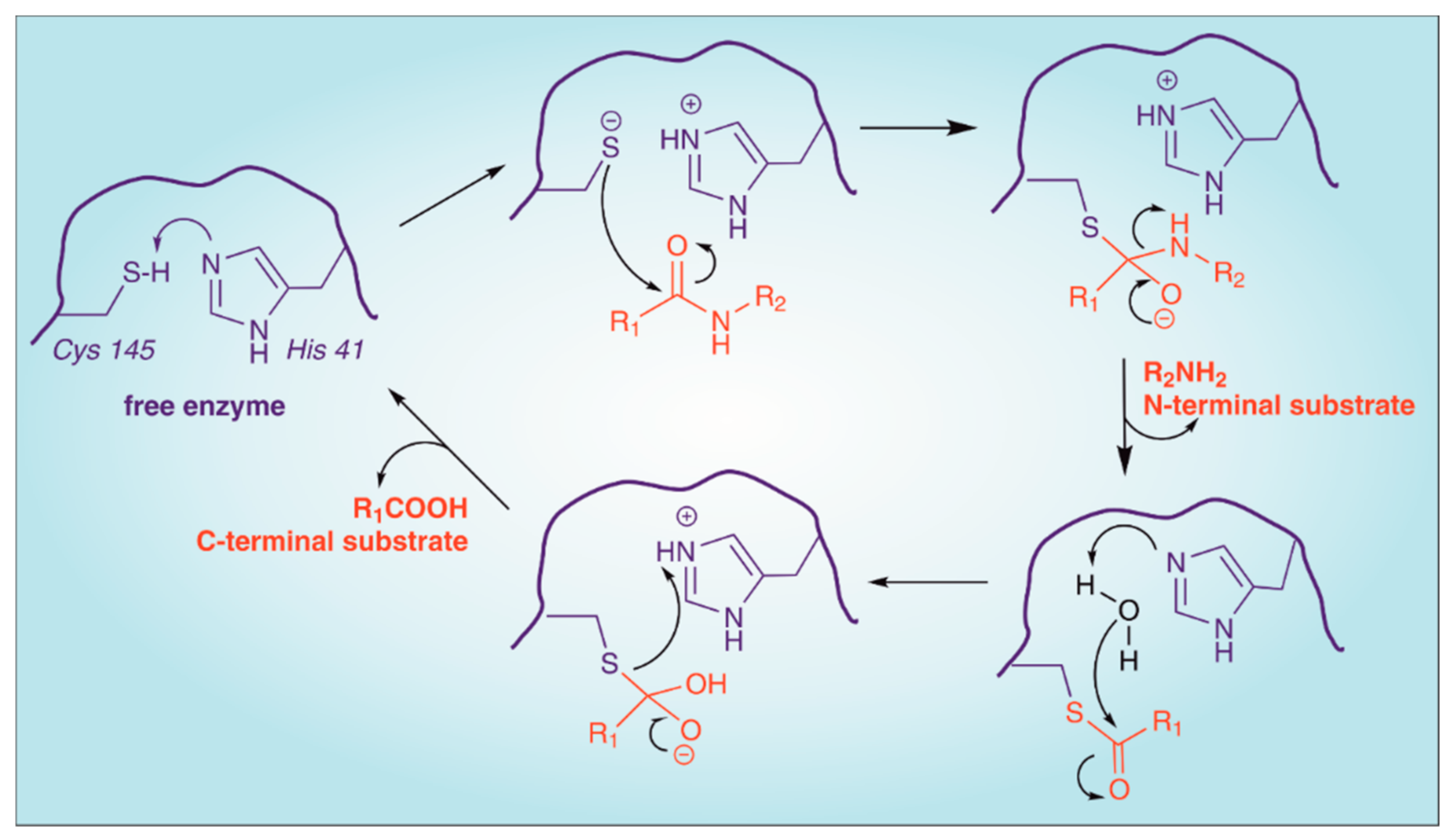
2. Structure of SARS-CoV-2 Main Protease (Mpro)
3. SARS-CoV-2 Mpro Inhibitors
- FDA-approved drugs, proposed for the treatment of COVID-19 (Drug Repurposing).
- Peptidomimetic and non-peptidic compounds (small-molecules), derived from studies on SARS-CoV Mpro and other viral/retroviral targets [41].
3.1. Drug Repurposing
3.2. Peptidomimetic Inhibitors
- Aldehydes;
- α-Ketoamides;
- Vinyl esters;
- Fluoromethyl ketones (FMKs);
- Hydroxymethyl ketones;
- Acyloxymethyl ketones.
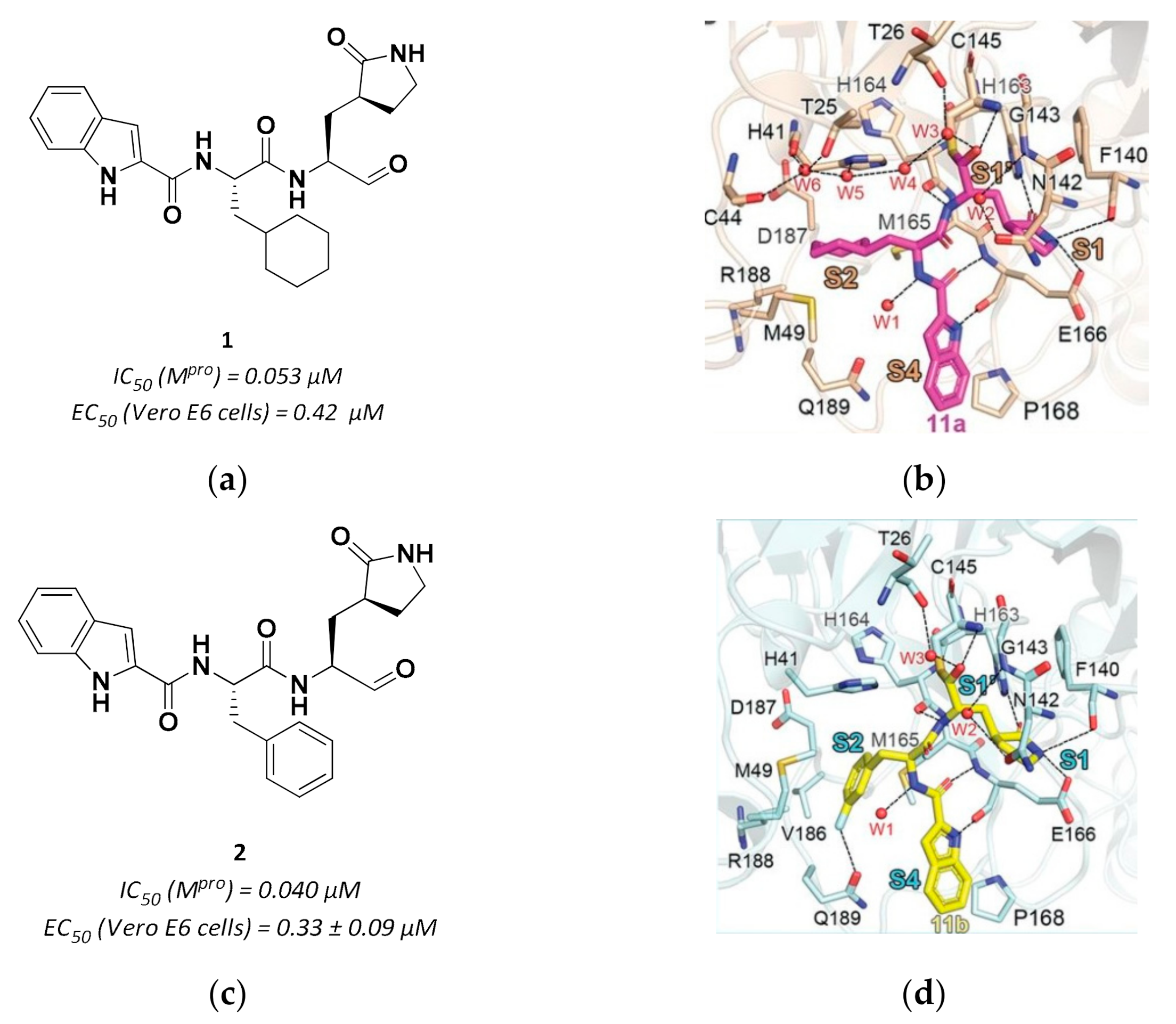
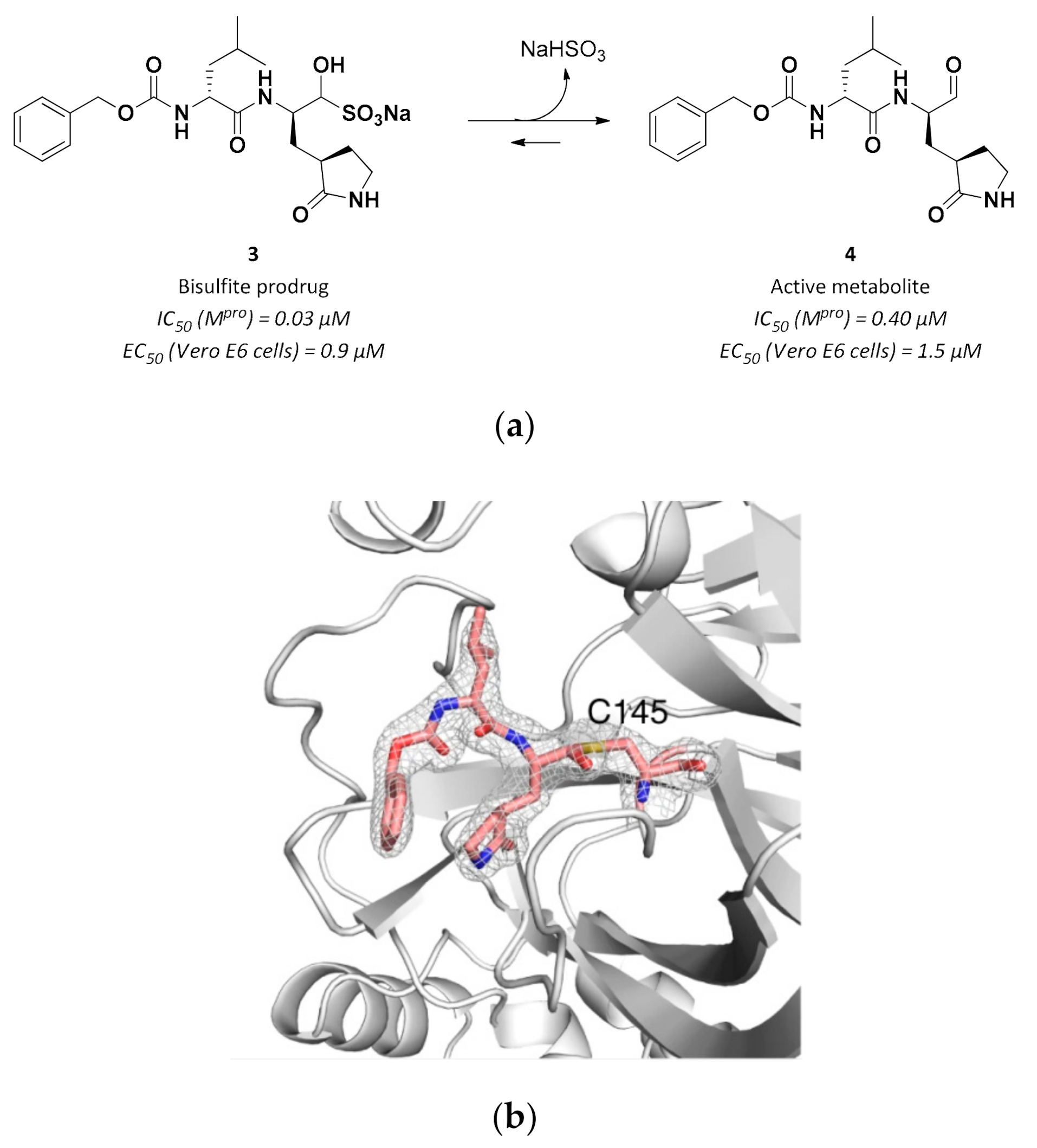
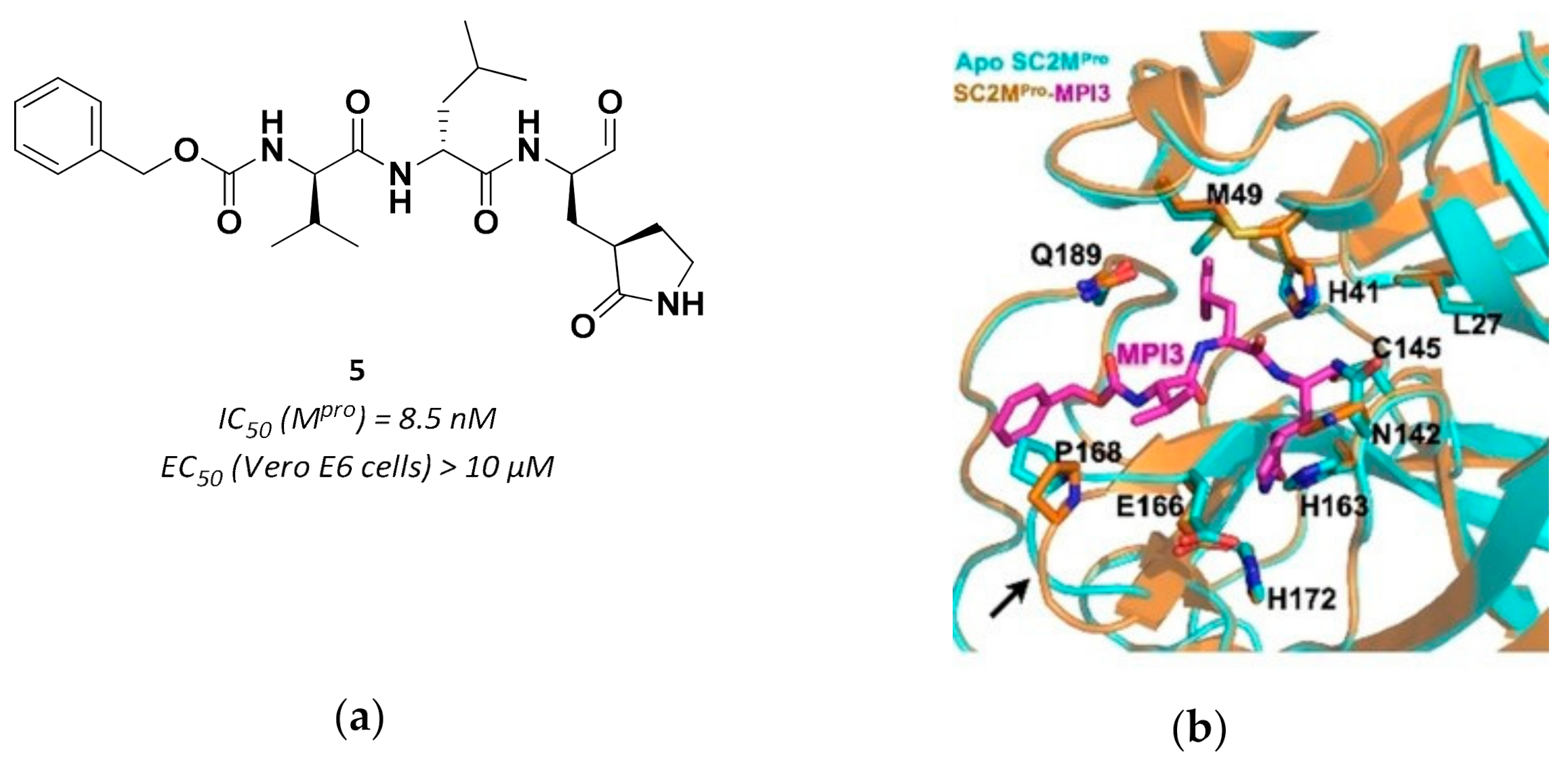
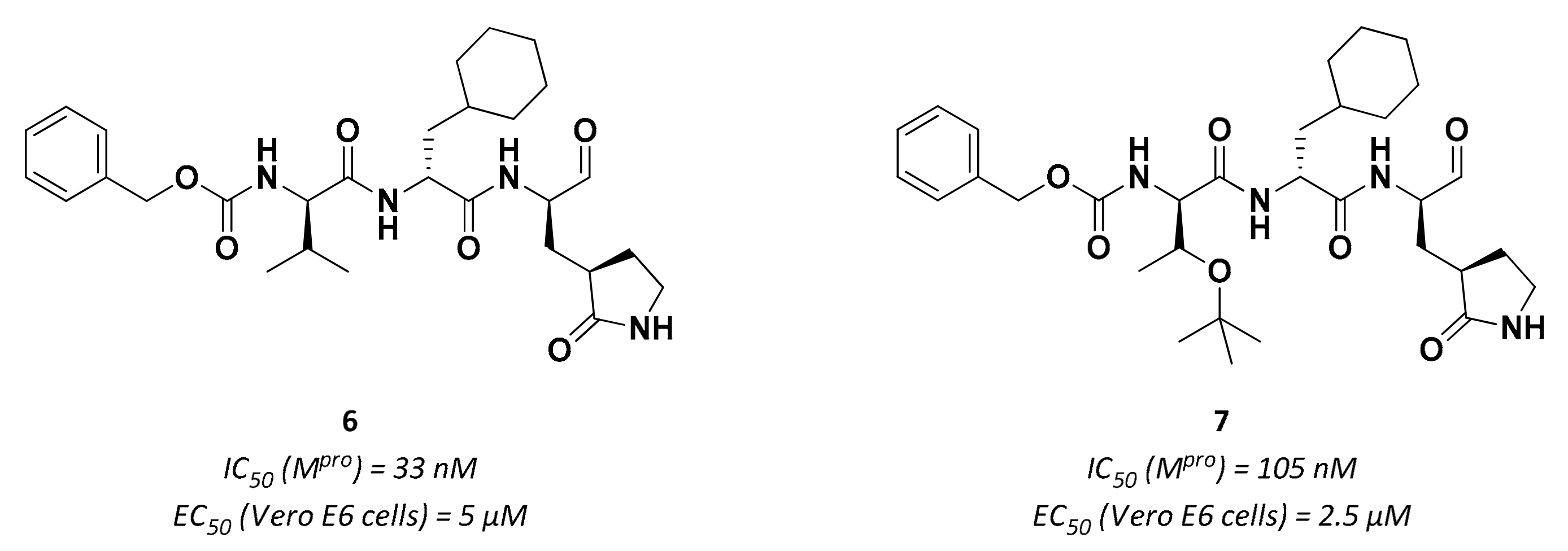
3.2.1. Peptidyl Aldehydes
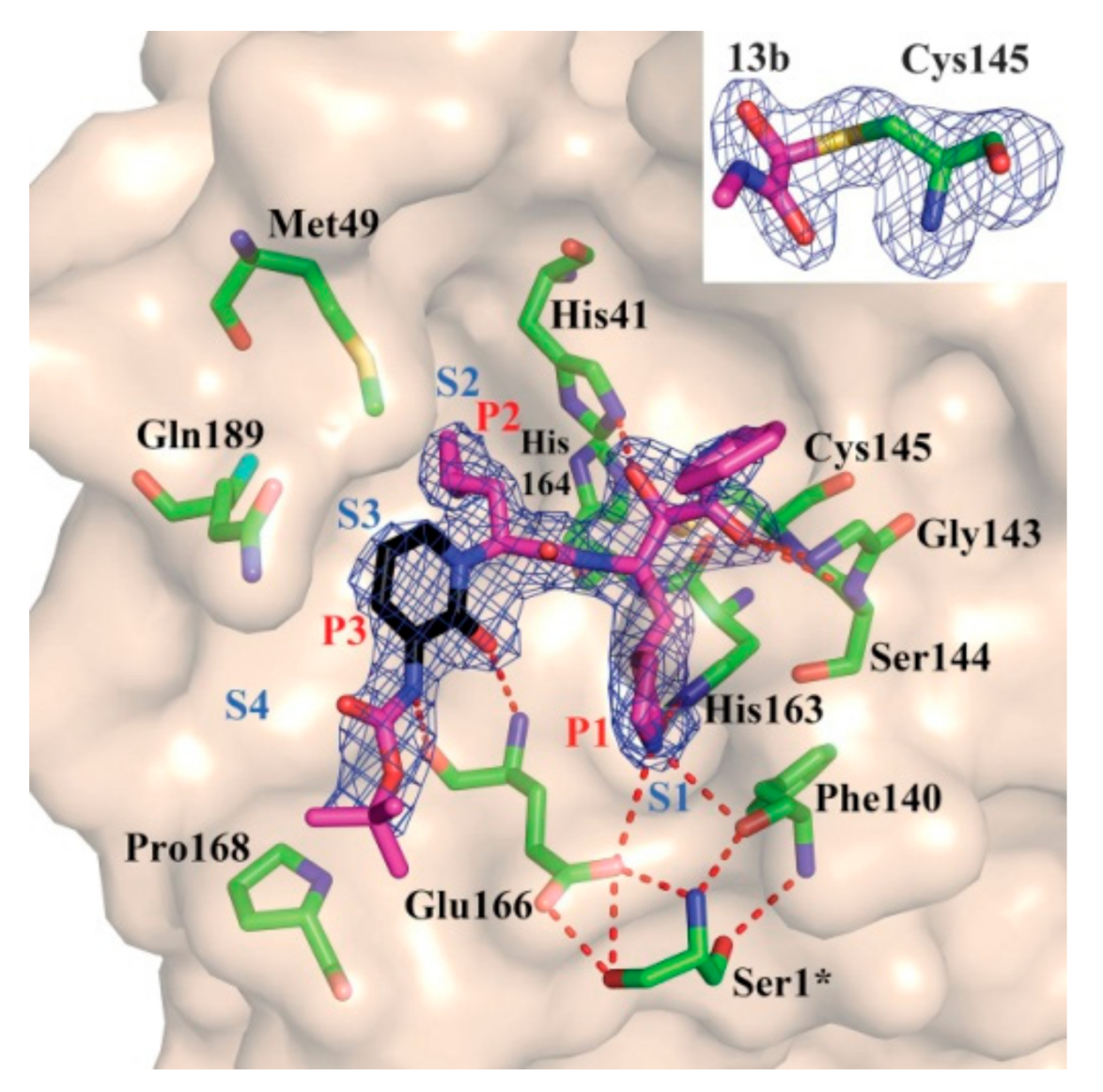
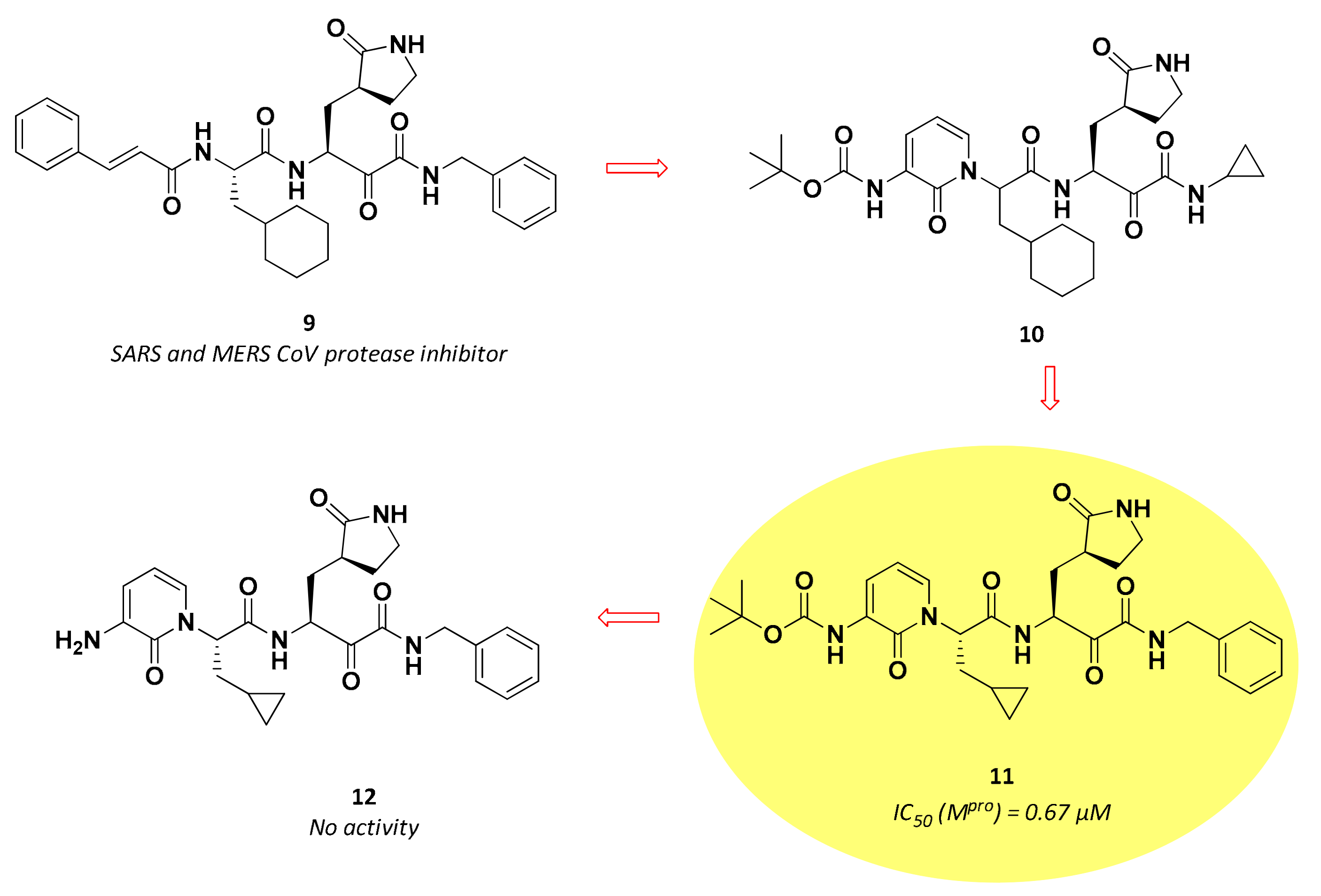
3.2.2. α-Ketoamides
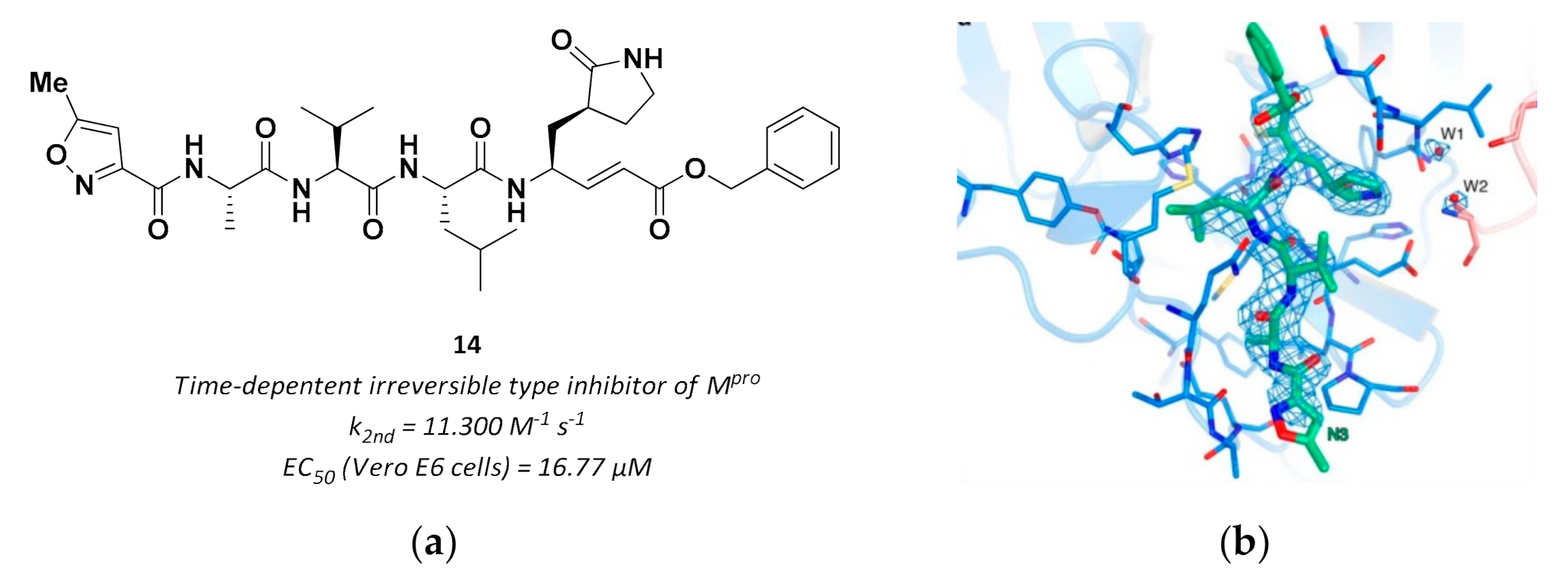
3.2.3. Vinyl Esters
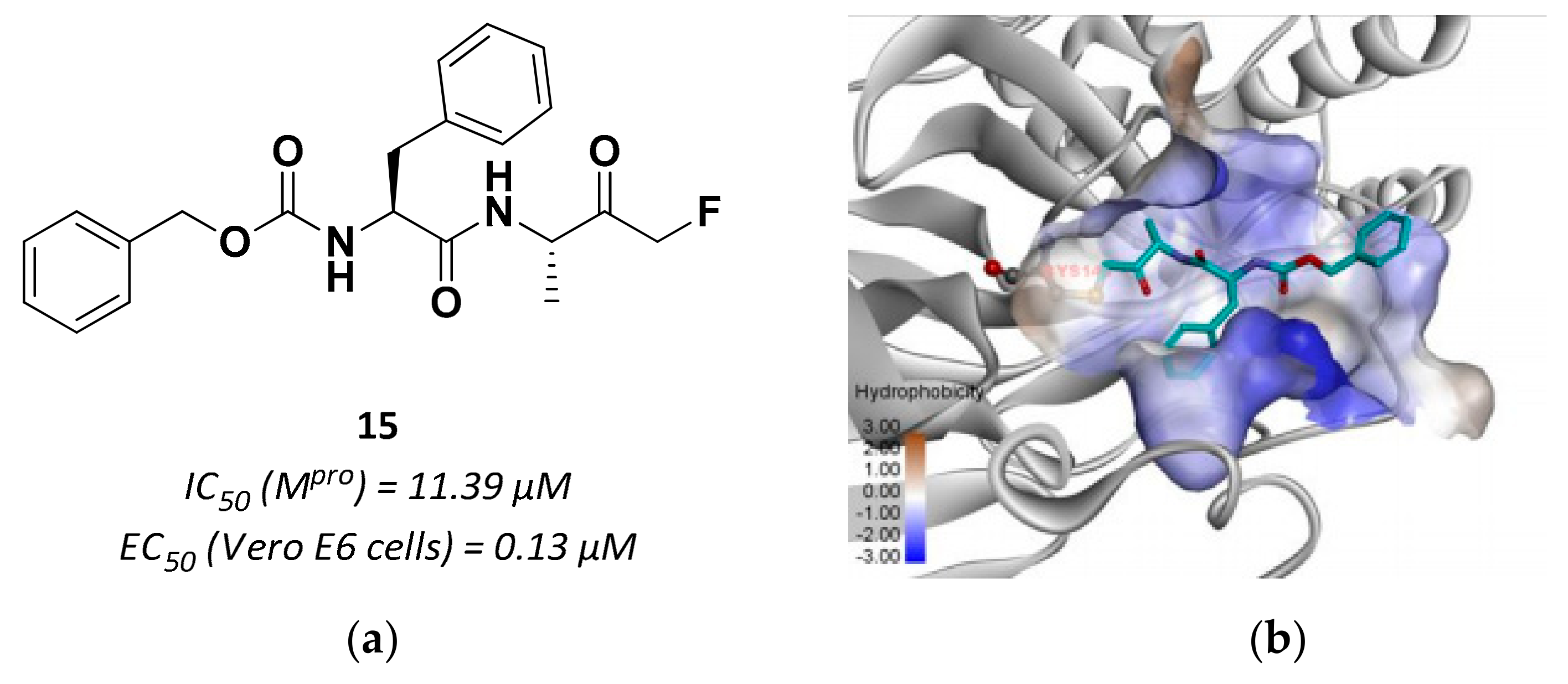
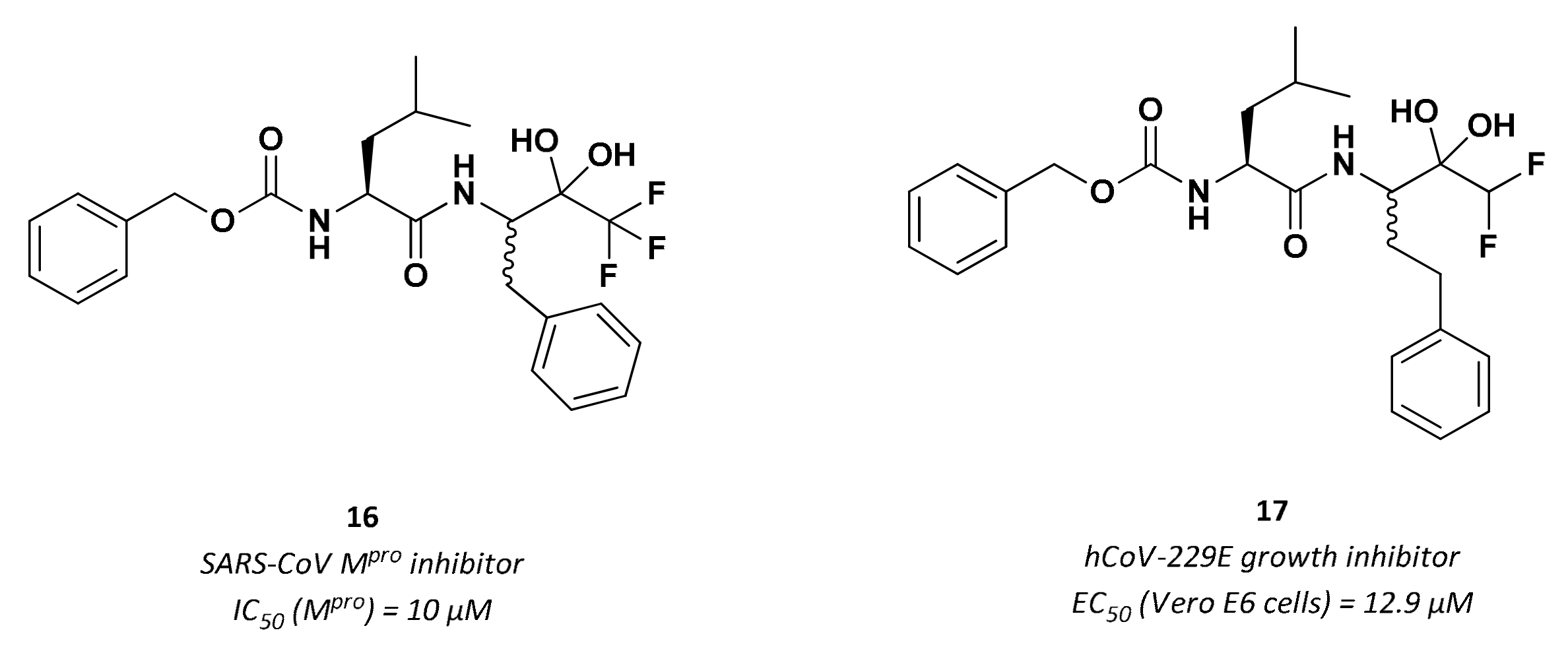
3.2.4. Peptidyl Fluoromethyl Ketones (pFMKs)
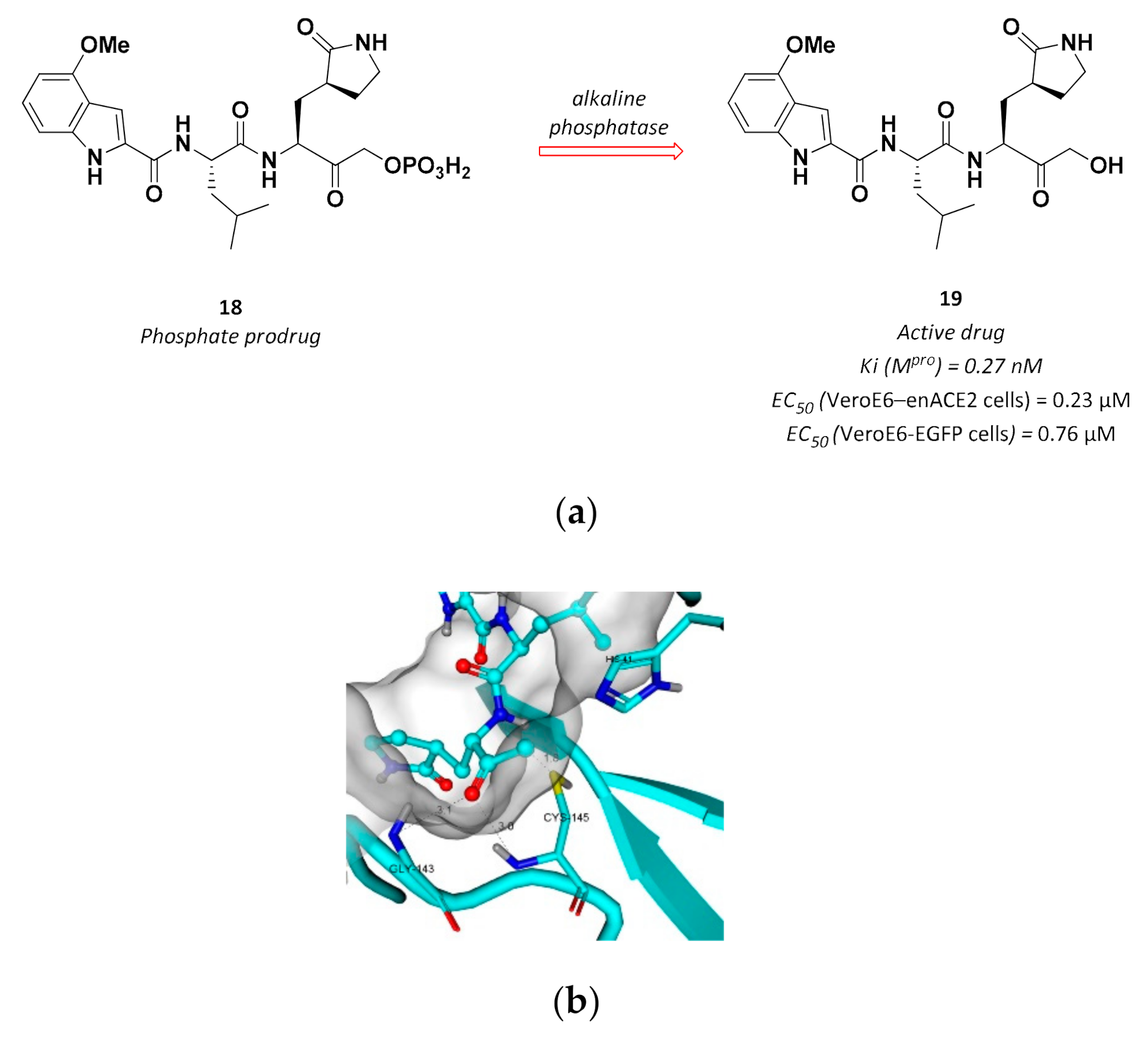
3.2.5. Hydroxymethyl Ketones
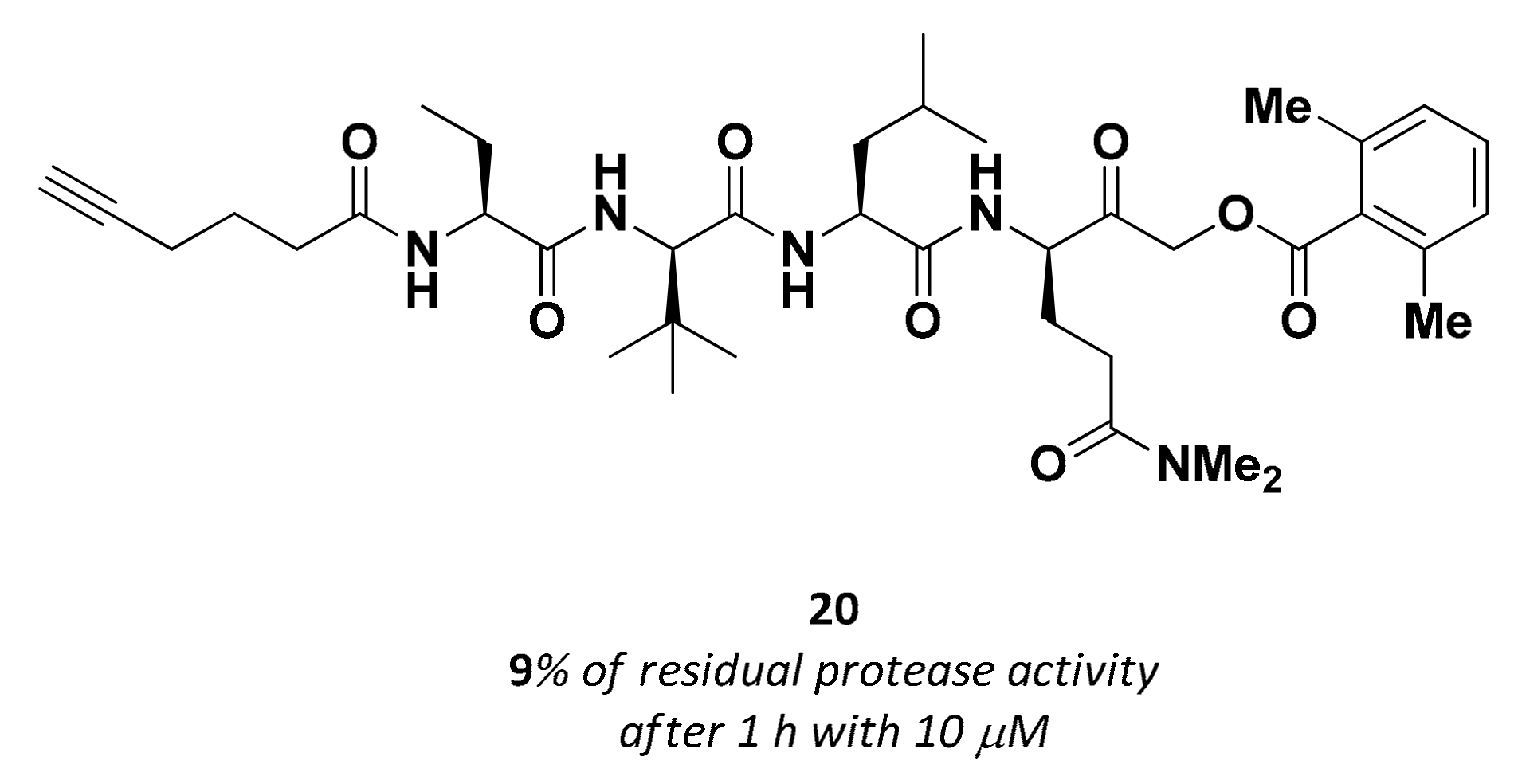
3.2.6. Acyloxymethyl Ketones
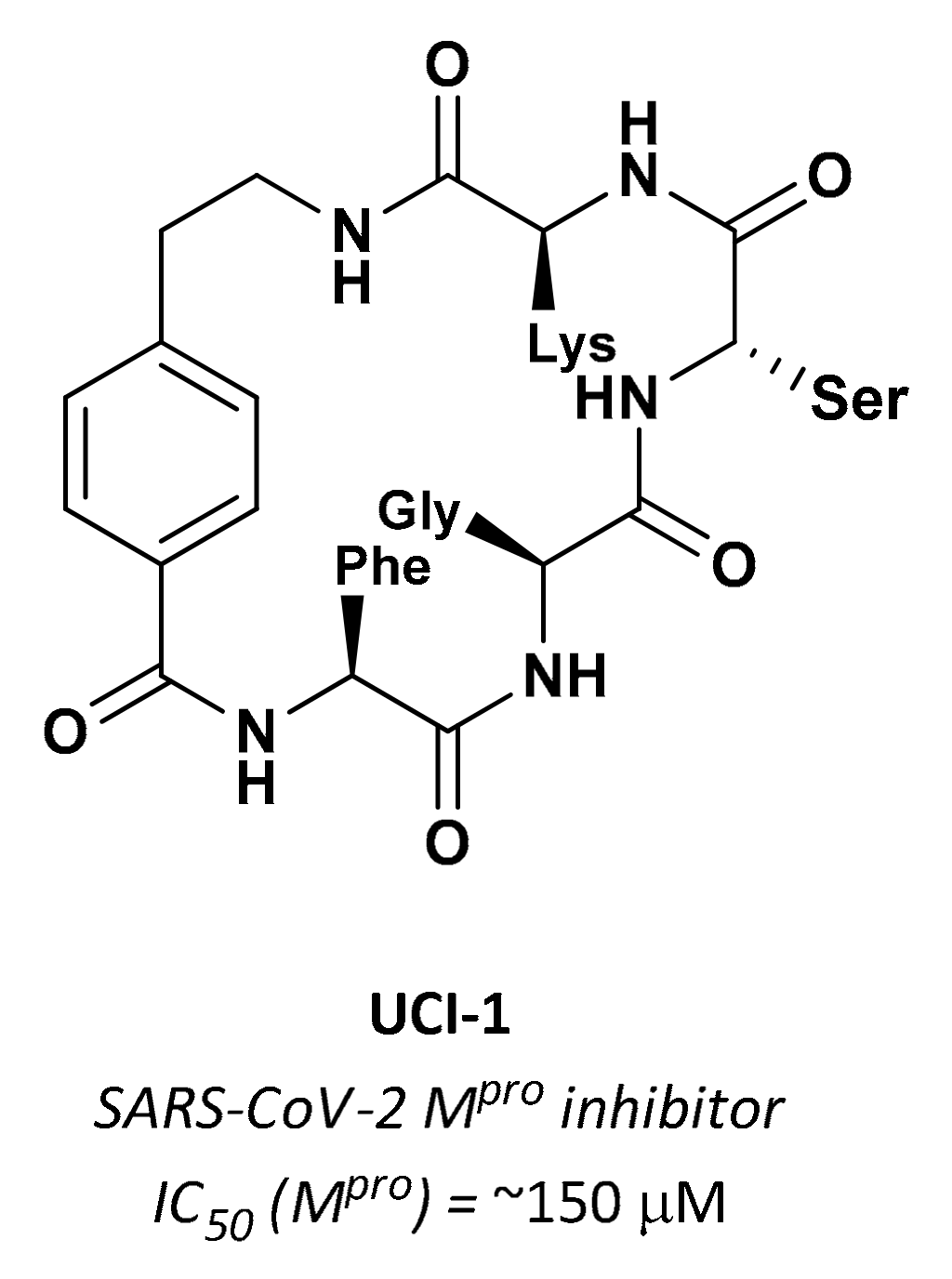
3.2.7. Cyclic Peptides
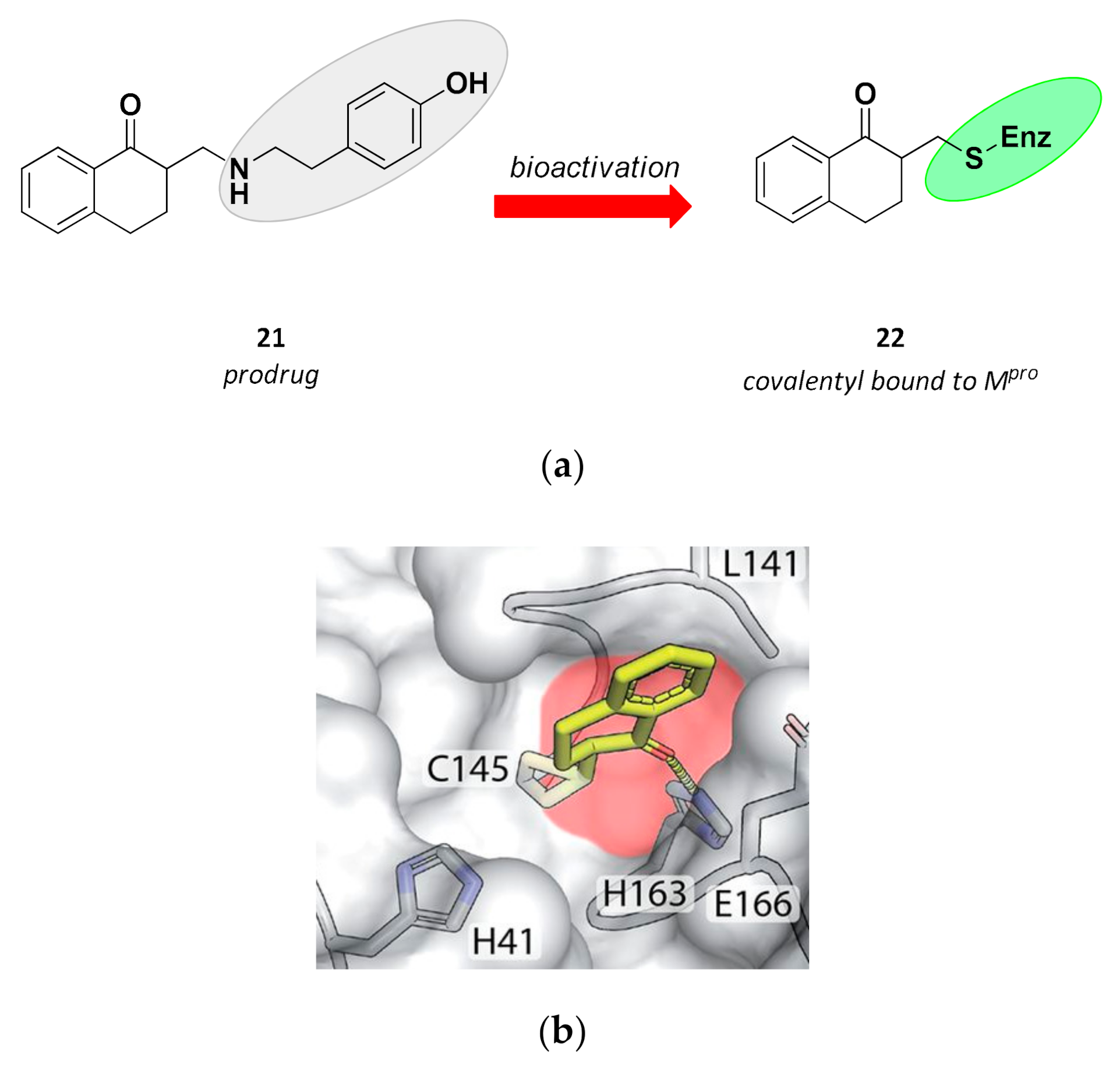
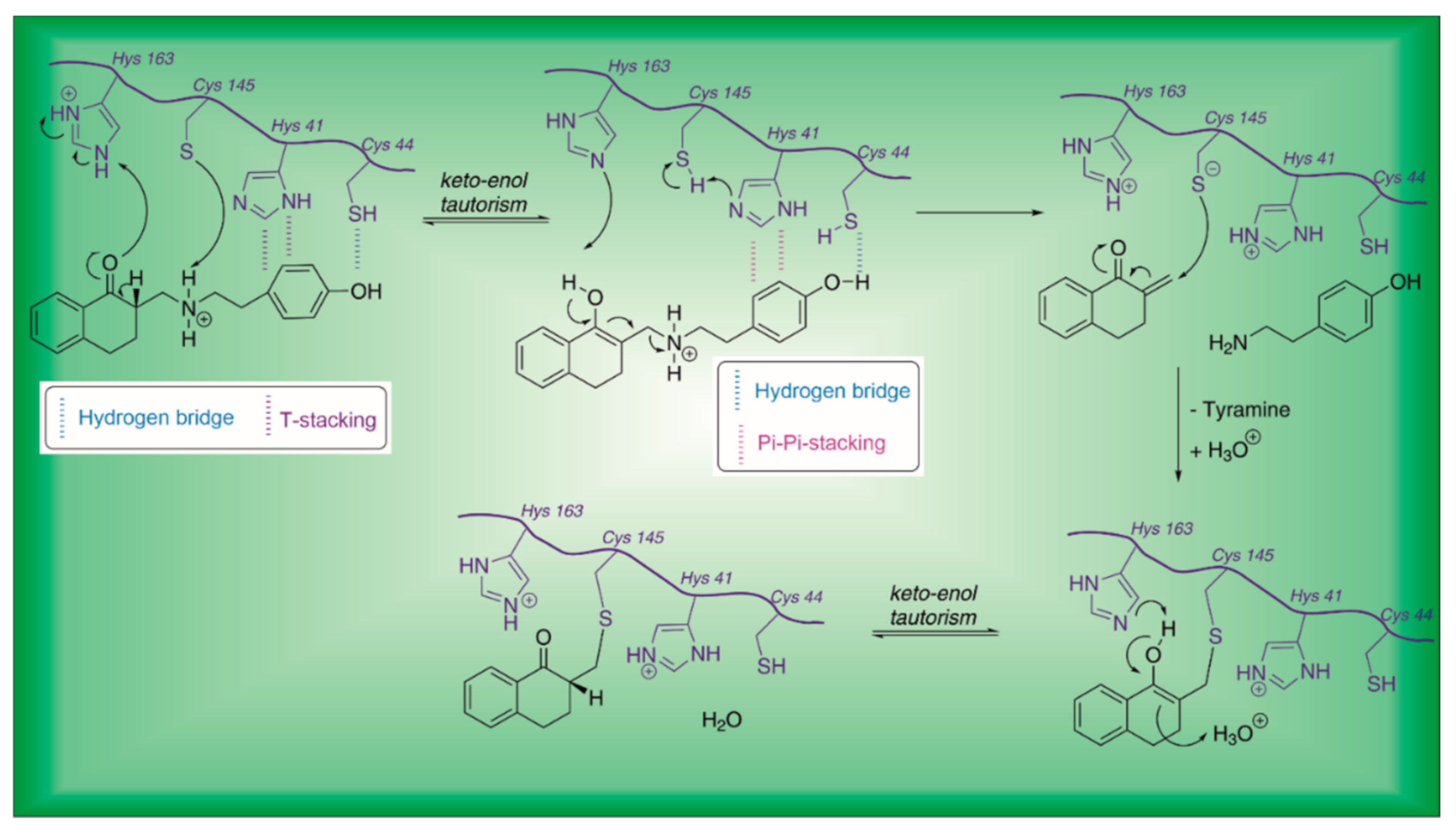
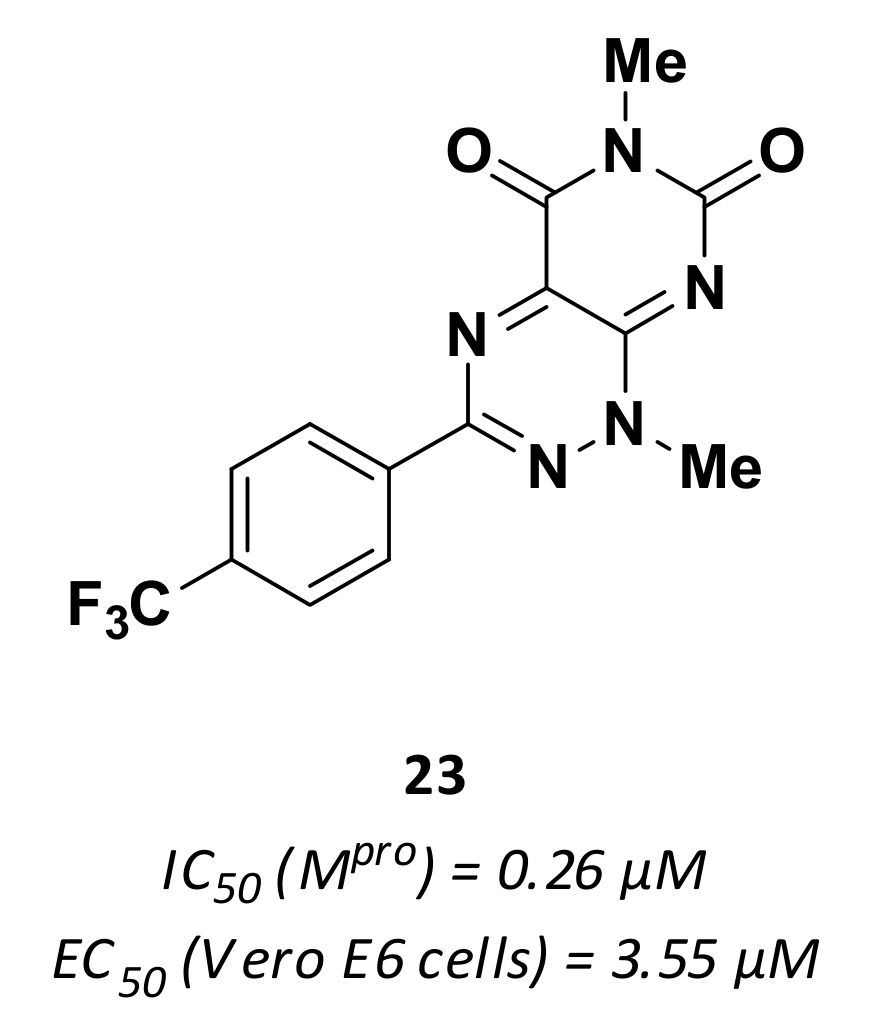
3.3. Non-Peptidic Inhibitors
4. Enzymatic Assays
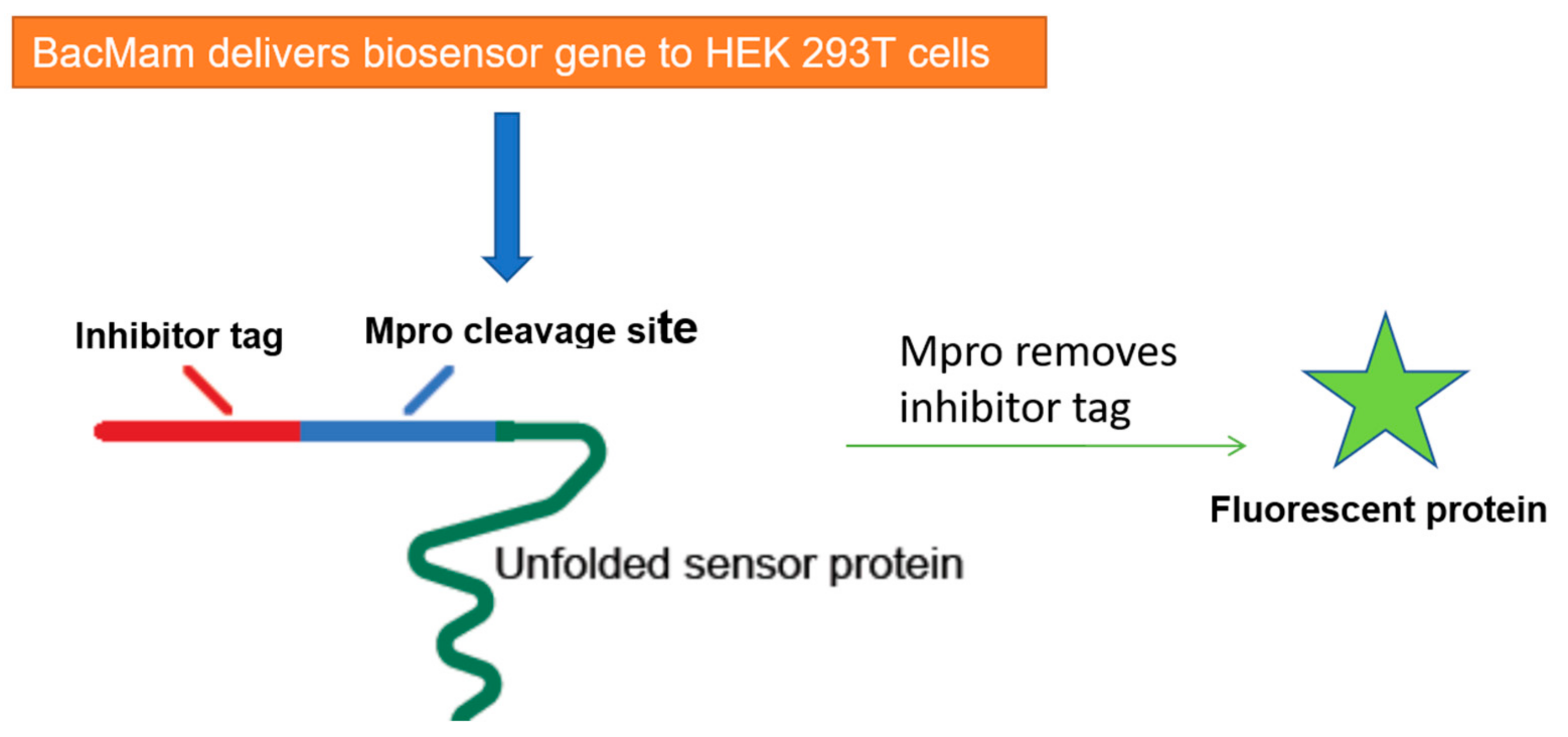
5. Cell-Based Assays to Screen Novel SARS-CoV-2 Antiviral Drugs
Author Contributions
Funding
Conflicts of Interest
References
- Adachi, S.; Koma, T.; Doi, N.; Nomaguchi, M.; Adachi, A. Commentary: Origin and Evolution of Pathogenic Coronaviruses. Front. Immunol. 2020, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günter, S.; Preiser, W.; Van Der Weft, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Li, K.S.M.; To, K.K.W.; Cheng, V.C.C.; Chen, H.; Yuen, K.-Y. Is the Discovery of the Novel Human Betacoronavirus 2c EMC/2012 (Hcov-EMC) the Beginning of Another SARS-Like Pandemic? J. Infect. 2012, 65, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 26 March 2021).
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic Characterization of a Novel SARS-Cov-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.S.; Zamzami, M.A.; Choudhry, H.; Murtaza, B.N.; Kazmi, I.; Ahmad, H.; Shakoori, A.R. Origin, Potential Therapeutic Targets and Treatment for Coronavirus Disease (COVID-19). Pathogens 2020, 9, 307. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-Cov-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; Mclellan, J.S. Cryo-EM Structure of the 2019-Ncov Spike in the Perfusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host Cell Entry of Middle East Respiratory Syndrome Coronavirus After Two-Step, Furin-Mediated Activation of the Spike Protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, D.; Yan, H.; Chong, H.; He, Y. Design of Potent Membrane Fusion Inhibitors Against SARS-Cov-2, an Emerging Coronavirus with High Fusogenic Activity. J. Virol. 2020, 94, e00635–20. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-Cov-2 Main Protease Provides a Basis for Design of Improved A-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides As Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef]
- Thiel, V.; Ivanov, K.A.; Putics, Á.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanism and Enzymes Involved in SARS Coronavirus Genome Expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Hegyi, A.; Ziebuhr, J. Conservation of Substrate Specificities among Coronavirus Main Proteases. J. Gen. Virol. 2002, 83, 595–599. [Google Scholar] [CrossRef]
- Ziebuhr, J. Molecular Biology of Severe Acute Respiratory Syndrome Coronavirus. Curr. Opin. Microbiol. 2004, 7, 412–419. [Google Scholar] [CrossRef]
- Du, Q.-S.; Wang, S.-Q.; Zhu, Y.; Wei, D.-Q.; Guo, H.; Sirois, S.; Chou, K.-C. Polyprotein Cleavage Mechanism of SARS Cov Mpro and Chemical Modification of the Octapeptide. Peptides 2004, 25, 1857–1864. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-Cov-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-Ncov. Chembiochem 2020, 21, 730–738. [Google Scholar] [CrossRef]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and Evolutionary Analysis Indicate That the SARS-Cov-2 Mpro is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-Cov-2 Main Protease from a Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Zhang, S.; Zou, P.; Chen, J.; Kang, X.; Li, Z.; Liang, C.; Jin, C.; Xia, B. Without its N-Finger, the Main Protease of Severe Acute Respiratory Syndrome Coronavirus Can form a Novel Dimer Through its C-Terminal Domain. J. Virol. 2008, 82, 4227–4234. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII inhibit SARS-Cov-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The Crystal Structures of Severe Acute Respiratory Syndrome Virus Main Protease and its Complex with an Inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef]
- Hsu, M.-F.; Kuo, C.-J.; Chang, K.-T.; Chang, H.-C.; Chou, C.-C.; Ko, T.-P.; Shr, H.-L.; Chang, G.-G.; Wang, A.H.-J.; Liang, P.-H. Mechanism of Maturation Process of SARS-Cov 3CL Protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef]
- Chou, C.-Y.; Chang, H.-C.; Hsu, W.-C.; Lin, T.-Z.; Lin, C.-H.; Chang, G.-G. Quaternary Structure of the Severe Acute Respiratory Syndrome (SARS) Coronavirus Main Protease. Biochemistry 2004, 43, 14958–14970. [Google Scholar] [CrossRef]
- Tahir Ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural Basis SARS-Cov-2 3clpro and Anti-COVID-19 Drug Discovery from Medicinal Plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, Y.; Li, L.; Wang, K.; Chen, S.; Chen, J.; Ding, J.; Jiang, H.; Shen, X. Two Adjacent Mutations of the Dimer Interface of SARS Coronavirus 3C-Like Protease Cause Different Conformational Changes in Crystal Structure. Virology 2009, 388, 324–334. [Google Scholar] [CrossRef]
- He, F.; Deng, Y.; Li, W. Coronavirus Disease 2019: What We Know? J. Med. Virol. 2020, 92, 719–725. [Google Scholar] [CrossRef]
- Muramatsu, T.; Takemoto, C.; Kim, Y.-T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-Cov 3CL Protease Cleaves Its C-Terminal Autoprocessing Site by Novel Subsite Cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Protease (3clpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Ho, B.-L.; Cheng, S.-C.; Shi, L.; Wang, T.-Y.; Ho, K.-I.; Chou, C.-Y. Critical Assessment of the Important Residues Involved in the Dimerization and Catalysis of MERS Coronavirus Main Protease. PLoS ONE 2015, 10, e0144865. [Google Scholar] [CrossRef]
- Zhou, X.; Zhong, F.; Lin, C.; Hu, X.; Zhang, Y.; Xiong, B.; Yin, X.; Fu, J.; He, W.; Duan, J.; et al. Structure of SARS-Cov-2 Main Protease in the Apo State Reveals the Inactive Conformation. Biorxiv 2020. [Google Scholar] [CrossRef]
- Li, J.; Zhou, X.; Zhang, Y.; Zhong, F.; Lin, C.; Mccormick, P.J.; Jiang, F.; Luo, J.; Zhou, H.; Wang, Q.; et al. Crystal Structure of SARS-Cov-2 Main Protease in Complex with the Natural Product Inhibitor Shikonin Illuminates a Unique Binding Mode. Sci. Bull. 2021, 66, 661–663. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, X.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-Cov-2 Main Protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural Basis for the Inhibition of SARS-Cov-2 Main Protease by Antineoplastic Drug Carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef]
- Rut, W.; Groborz, K.; Zhang, L.; Sun, X.; Zmudzinski, M.; Pawlik, B.; Młynarski, W.; Hilgenfeld, R.; Drag, M. Substrate Specificity Profiling of SARS-Cov-2 Main Protease Enables Design of Activity-Based Probes for Patient-Sample Imaging. Biorxiv 2020. [Google Scholar] [CrossRef]
- Chuck, C.-P.; Chow, H.-F.; Wan, D.C.-C.; Wong, K.-B. Profiling and Substrate Specificities of 3C-Like Proteases from Group 1, 2a, 2b, and 3 Coronaviruses. PLoS ONE 2011, 6, e27228. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.-H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-Cov) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Huang, C.; Wei, P.; Fan, K.; Liu, Y.; Lai, L. 3C-Like Proteinase from SARS Coronavirus Catalyzes Substrate Hydrolysis by a General Base Mechanism. Biochemistry 2004, 43, 4568–4574. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbial. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Sultana, J.; Crisafulli, S.; Gabbay, F.; Lynn, E.; Shakir, S.; Trifirò, G. Challenges for Drug Repurposing in the COVID-19 Pandemic Era. Front. Pharmacol. 2020, 11, 588654. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Tinivella, A.; Caporuscio, F.; Rastelli, G. Drug Repurposing and Polypharmacology to Fight SARS-Cov-2 Through Inhibition of the Main Protease. Front. Pharmacol. 2021, 12, 636989. [Google Scholar] [CrossRef] [PubMed]
- WHO. Solidarity Clinical Trial for COVID-19 Treatments. 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov/solidarity-clinical-trial-for-covid-19-treatments (accessed on 26 March 2021).
- RECOVERY. Randomised Evaluation of COVID-19 Therapy. Available online: https://www.recoverytrial.net (accessed on 26 March 2021).
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, J.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Eng. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Zeldin, R.K.; Petruschke, R.A. Pharmacological and Therapeutic Properties of Rotonavir-Boosted Protease Inhibitor Therapy in HIV-Infected Patients. J. Antimicrob. Chemother. 2004, 53, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef]
- Bolcato, G.; Bissaro, M.; Pavan, M.; Sturlese, M.; Moro, S. Targeting the Coronavirus SARS-Cov-2: Computational Insights into the Mechanism of Action of the Protease Inhibitors Lopinavir, Rotonavir and Nelfinavir. Sci. Rep. 2020, 10, 20927. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsuyama, S.; Hoshino, T.; Yamamoto, N. Nelfinavir Inhibits Replication of Severe Acute Respiratory Syndrome Coronavirus 2 in vitro. Biorxiv 2020. [Google Scholar] [CrossRef]
- Vatansever, E.C.; Yang, K.; Kratch, K.C.; Drelich, A.; Cho, C.-C.; Mellott, D.M.; Xu, S.; Tseng, C.-T.K.; Liu, W.R. Bepridil is Potent Against SARS-Cov-2 in vitro. Biorxiv 2020. [Google Scholar] [CrossRef]
- Mahdi, M.; Mótyán, J.A.; Szojka, Z.I.; Golda, M.; Miczi, M.; Tőzsér, J. Analysis of the Efficacy of HIV Protease Inhibitors Against SARS-Cov-2′S Main Protease. Virol. J. 2020, 17, 190. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z.; Wang, L.; Huang, Z.; Gong, F.; Li, X.; Chen, Y.; Wu, J.J. First Clinical Study Using HCV Protease Inhibitor Danoprevir to Treat Naïve and Experienced COVID-19 Patients. Medrxiv 2020. [Google Scholar] [CrossRef]
- Fintelman-Rodrigues, N.; Sacramento, C.Q.; Ribeiro Lima, C.; Souza Da Silva, F.; Ferreira, A.C.; Mattos, M.; De Freitas, C.S.; Cardoso Soares, V.; Da Silva Gomes Dias, S.; Temerozo, J.R.; et al. Atazanavir, Alone or in Combination with Ritonavir, Inhibits SARS-Cov-2 Replication and Proinflammatory Cytokine Production. Antimicrob. Agents Chemother. 2020, 64, e00825-20. [Google Scholar] [CrossRef]
- Xu, Z.; Yao, H.; Shen, J.; Wu, N.; Xu, Y.; Lu, X.; Zhu, W.; Li, L. Nelfinavir is Active Against SARS-Cov-2 in Vero E6 Cells. Chemrxiv 2020. [Google Scholar] [CrossRef]
- Xu, Z.; Peng, C.; Shi, Y.; Zhu, Z.; Mu, K.; Wang, X.; Zhu, W. Nelfinavir Was Predicted to Be a Potential Inhibitor of 2019-Ncov Main Protease by an Integrative Approach Combining Homology Modeling, Molecular Docking and Binding Free Energy Calculation. Biorxiv 2020. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, X.; Mu, K.; Peng, C.; Zhu, Z.; Wang, X.; Yang, Y.; Xu, Z.; Zhu, W. D3Targets-2019-Ncov: A Web Server to Identify Potential Targets for Antivirals Against 2019-Ncov. Chemrxiv 2020. [Google Scholar] [CrossRef]
- Degertekin, B.; Lok, A.S.F. Update on Viral Hepatitis: 2007. Curr. Opin. Gastroenterol. 2008, 24, 306–311. [Google Scholar] [CrossRef]
- Njoroge, F.G.; Chen, K.X.; Shih, N.-Y.; Piwinski, J.J. Challenges in Modern Drug Discovery: A Case Study of Boceprevir, an HCV Protease Inhibitor for the Treatment of Hepatitis C Virus Infection. Acc. Chem. Res. 2008, 41, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Agbowuro, A.A.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Proteases and Protease Inhibitors in Infectious Diseases. Med. Res. Rev. 2018, 38, 1295–1331. [Google Scholar] [CrossRef] [PubMed]
- Lynch, E.; Kil, J. Development of Ebselen, a Glutathione Peroxidase Mimic, for the Prevention and Treatment of Noise-Induced Hearing Loss. Semin. Hear 2009, 30, 47–55. [Google Scholar] [CrossRef]
- Kil, J.; Lobarinas, E.; Spankovich, C.; Griffiths, S.K.; Antonelli, P.J.; Lynch, E.D.; Le Prell, C.G. Safety and Efficacy of Ebselen for the Prevention of Noise-Induced Hearing Loss: A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet 2017, 390, 969–979. [Google Scholar] [CrossRef]
- Masaki, C.; Sharpley, A.L.; Cooper, C.M.; Godlewska, B.R.; Singh, N.; Vasudevan, S.R.; Harmer, C.J.; Churchill, G.C.; Sharp, T.; Rogers, R.D.; et al. Effects of Potential Lithium-Mimetic, Ebselen, on Impulsivity and Emotional Processing. Psychopharmacology 2016, 233, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xiao, D.; Wang, Y.; Liu, L.; Zhou, X.; Wu, Z. Research Progress on Repositioning Drugs and Specific Therapeutic Drugs for SARS-Cov-2. Future Med. Chem. 2020, 12, 1565–1578. [Google Scholar] [CrossRef] [PubMed]
- Holshue, M.L.; Debolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. Washington State 2019-Ncov Case Investigation Team. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.-X.; et al. Compassionate use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef]
- Garibaldi, B.T.; Wang, K.; Robinson, M.L.; Zeger, S.L.; Bandeen-Roche, K.; Wang, M.-C.; Alexander, G.C.; Gupta, A.; Bollinger, R.; Xu, Y. Comparison of Time to Clinical Improvement with vs without Remdesivir Treatment in Hospitalized Patients with COVID-19. JAMA Netw. Open 2021, 4, e213071. [Google Scholar] [CrossRef]
- Gilead, U.S. Food and Drug Administration Approves Gilead’s Antiviral Veklury (remdesivir) for Treatment of COVID-19. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2020/10/us-food-and-drug-administration-approves-gileads-antiviral-veklury-remdesivir-for-treatment-of-covid19 (accessed on 26 March 2021).
- Gilead Veklury Global Marketing Authorization Status. Available online: https://www.gilead.com/purpose/advancing-global-health/covid-19/veklury-global-marketing-authorization (accessed on 26 March 2021).
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-Ncov) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Huynh, T.; Wang, H.; Luan, B. In silico Exploration of the Molecular Mechanism of Clinically Oriented Drugs for Possibly Inhibiting SARS-Cov-2′S Main Protease. J. Phys. Chem. Lett. 2020, 11, 4413–4420. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Liu, S.; Sun, J.; Chen, Z.; Jiang, M.; Zhang, Q.; Wei, Y.; Wang, X.; Huang, Y.-Y.; et al. Potential Therapeutic Effects of Dipyridamole in the Severely Ill Patients with COVID-19. Acta Pharmaceutica Sinica B 2020, 10, 1205–1215. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a Less Toxic Derivative of Chloroquine, is Effective in Inhibiting SARS-Cov-2 Infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-Cov-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef]
- Touret, F.; Gilles, M.; Barral, K.; Nougairède, A.; Van Helden, J.; Decroly, E.; De Lamballerie, X.; Coutard, B. In vitro Screening of a FDA Approved Chemical Library Reveals Potential Inhibitors of SARS-Cov-2 Replication. Sci. Rep. 2020, 10, 13093. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a Potent Inhibitor of SARS Coronavirus Infection and Spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Huang, Y.-Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Indentify Potent SARS-Cov-2 Main Protease Inhibitors via Accelerated Free Energy Perturbation-Based Virtual Screening of Existing Drugs. Proc. Natl Acad. Sci. USA 2020, 117, 27381–27387. [Google Scholar] [CrossRef]
- Gentile, D.; Fuochi, V.; Rescifina, A.; Furneri, P.M. New Anti-SARS-Cov-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine. Int. J. Mol. Sci. 2020, 21, 5856. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a Synthetic Tool of Drug Discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef]
- Vanpatten, S.; He, M.; Altiti, A.; Cheng, K.F.; Ghanem, M.H.; Al-Abed, Y. Evidence Supporting the use of Peptides and Peptidomimetics as Potential SARS-Cov-2 (COVID-19) Therapeutics. Future Med. Chem. 2020, 12, 1647–1656. [Google Scholar] [CrossRef]
- Alagumuthu, M.; Rajpoot, S.; Baig, M.S. Structure-Based Design of Novel Peptidomimetics Targeting the SARS-Cov-2 Spike Protein. Cell. Mol. Bioeng. 2020. [Google Scholar] [CrossRef]
- Zhu, L.; George, S.; Schmidt, M.F.; Al.Gharabli, S.I.; Rademann, J.; Hilgenfeld, R. Peptide Aldehyde Inhibitors Challenge the Substrate Specificity of the SARS-Coronavirus Main Protease. Antiviral Res. 2011, 92, 204–212. [Google Scholar] [CrossRef]
- Al-Gharabli, S.I.; Ali Shah, S.T.; Weik, S.; Schmidt, M.F.; Mesters, J.R.; Kuhn, D.; Klebe, G.; Hilgenfeld, R.; Rademann, J. An Efficient Method for the Synthesis of Peptide Aldehyde Libraries Employed in the Discovery of Reversible SARS Coronavirus Main Protease (SARS-Cov Mpro) Inhibitors. Chembiochem. 2006, 7, 1048–1055. [Google Scholar] [CrossRef]
- Malcolm, B.A.; Lowe, C.; Shechosky, S.; Mckay, R.T.; Yang, C.C.; Shah, V.J.; Simon, R.J.; Vederas, J.C.; Santi, D.V. Peptide Aldehyde Inhibitors of Hepatitis A Virus 3C Proteinase. Biochemistry 1995, 34, 8172–8179. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Shivanna, V.; Narayanan, S.; Prior, A.M.; Weerasekara, S.; Hua, D.H.; Galasiti Kankanamalage, A.C.; Groutas, W.C.; Chang, K.O. Broad-Spectrum Inhibitors Against 3C-Like Proteases of Feline Coronaviruses and Feline Caliciviruses. J. Virol. 2015, 89, 4942–4950. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.-O. Efficacy of a 3C-Like Prorease Inhibitor in Treating Various forms of Acquired Feline Infectious Peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. [Google Scholar] [CrossRef]
- Vuong, W.; Bashir Khan, M.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; Mckay, R.T.; Van Belkum, M.J.; Joyce, M.A.; et al. Feline Coronavirus Drug Inhibits the Main Protease of SARS-Cov-2 and Blocks Virus Replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.W.; Hakmi, M.; Essabbar, A.; Laamarti, M.; Hlali, H.; Allam, L.; El Hafidi, N.; et al. Large Scale Genomic Analysis of 3067 SARS-Cov-2 Genomes Reveals a Clonal Geo-Distribution and a Rich Genetic Variations of Hotspots Mutations. PLoS ONE 2020, 15, e0240345. [Google Scholar] [CrossRef]
- Yang, K.S.; Ma, X.R.; Ma, Y.; Alugubelli, Y.R.; Scott, D.A.; Vatansever, E.C.; Drelich, A.K.; Sankaran, B.; Geng, Z.Z.; Blankenship, L.R.; et al. A Quick Route to Multiple Potent SARS-Cov-2 Main Protease Inhibitors. ChemMedChem 2020. [Google Scholar] [CrossRef]
- Sasaki, T.; Kishi, M.; Saito, M.; Tanaka, T.; Higuchi, N.; Kominami, E.; Katunuma, N.; Murachi, T. Inhibitory Effect of Di- And Tripeptidyl Aldehydes on Calpains And Cathepsins. J. Enzyme Inhib. 1990, 3, 195–201. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of Cathepsin L Prevent Severe Acute Respiratory Syndrome Coronavirus Entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Santos, M.M.M.; Moreira, R. Michael Acceptors as Cysteine Proteases Inhibitors. Mini Rev. Med. Chem. 2007, 7, 1040–1050. [Google Scholar] [CrossRef]
- Citarella, A.; Micale, N. Fluoromethyl Ketones and Their Applications in Medicinal Chemistry. Molecules 2020, 25, 4431. [Google Scholar] [CrossRef]
- Citarella, A.; Gentile, D.; Rescifina, A.; Piperno, A.; Mognetti, B.; Gribaudo, G.; Sciortino, M.T.; Holzer, W.; Pace, V.; Micale, N. Pseudo-Dipeptide Bearing A,A-Difluoromethyl Ketone Moiety as Electrophilic Warhead with Activity Against Coronaviruses. Int. J. Mol. Sci. 2021, 22, 1398. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-Cov-2- 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef]
- Shao, Y.-M.; Yang, W.-B.; Kuo, T.-H.; Tsai, K.-C.; Lin, C.-H.; Yang, A.-S.; Liang, P.-H.; Wong, C.-H. Design, Synthesis, And Evaluation of Trifluoromethyl Ketones as Inhibitors of SARS-Cov 3CL Protease. Bioorg. Med. Chem. 2008, 16, 4652–4660. [Google Scholar] [CrossRef]
- Miele, M.; Citarella, A.; Langer, T.; Urban, E.; Zehl, M.; Holzer, W.; Ielo, L.; Pace, V. Chemoselective Homologation-Deoxygenation Strategy Enabling the Direct Conversion of Carbonyls Into (N + 1)-Halomethyl-Alkanes. Org. Lett. 2020, 22, 7629–7634. [Google Scholar] [CrossRef]
- Miele, M.; Citarella, A.; Micale, N.; Holzer, W.; Pace, V. Direct and Chemoselective Synthesis of Tertiary Difluorketones Via Weinreb Amide Homologation with a CHF2-Carbene Equivalent. Org. Lett. 2019, 21, 8261–8265. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Protease for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2021, 63, 12725–12747. [Google Scholar] [CrossRef]
- Single Ascending Dose Study of Intravenous Infusion of PF 07304814 in Healthy Adults Participants. Available online: https://clinicaltrials.gov/ct2/show/nct04627532 (accessed on 26 March 2021).
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Zhang, L.; Kasperkiewicz, P.; Poreba, M.; Hilgenfeld, R.; Drąg, M. Extended Substrate Specificity and First Potent Irreversible Inhibitor/Activity-Based Probe Design for Zika Virus NS2B-NS3 Protease. Antiviral Res. 2017, 139, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Groborz, K.; Zhang, L.; Modrzycka, S.; Poreba, M.; Hilgenfeld, R.; Drag, M. Profiling of Flaviviral NS2B-NS3 Protease Specificity Provides a Structural Basis for the Development of Selective Chemical Tools That Differentiate Dengue from Zika and West Nile Viruses. Antiviral Res. 2020, 175, 104731. [Google Scholar] [CrossRef] [PubMed]
- Van De Plassche, M.A.T.; Barniol-Xicota, M.; Verhelst, S.H.L. Peptidyl Acyloxymethyl Ketones as Activity-Based Probes for the Main Protease of SARS-Cov-2. Chembiochem 2020, 21, 3383–3388. [Google Scholar] [CrossRef]
- Kreutzer, A.G.; Krumberger, M.; Parrocha, C.M.T.; Morris, M.A.; Guaglianone, G.; Nowick, J.S. Structure-Based Design of a Cyclic Peptide Inhibitor of the SARS-Cov-2 Main Protease. Biorxiv 2020. [Google Scholar] [CrossRef]
- Ellinger, B.; Bojkova, D.; Zaliani, A.; Cinatl, J.; Claussen, C.; Westhaus, S.; Reinshagen, J.; Kuzikov, M.; Wolf, M.; Geisslinger, G.; et al. Identification of Inhibitors of SARS-Cov-2 In-Vitro Cellular Toxicity in Human (Caco-2) Cells Using a Large Scale Drug Repurposing Collection. Res. Square 2020. [Google Scholar] [CrossRef]
- Günter, S.; Reinke, P.Y.A.; Oberthuer, D.; Yefanov, O.; Ginn, H.; Meier, S.; Lane, T.J.; Lorenzen, K.; Gelisio, L.; Brehm, W.; et al. X-Ray Screening Identifies Active Site and Allosteric Inhibitors of SARS-Cov-2 Main Protease. Science 2021. [Google Scholar] [CrossRef]
- Hattori, S.-I.; Higshi-Kuwata, N.; Raghavaiah, J.; Das, D.; Bulut, H.; Davis, D.A.; Takamatsu, Y.; Matsuda, K.; Takamune, N.; Kishimoto, N.; et al. GRL-0020, an Indole Chloropyridinyl Ester, Completely Blocks SARS-Cov-2 Infection. Mbio 2020, 11, e01833. [Google Scholar] [CrossRef]
- Luo, H.; Tang, Q.-L.; Shang, Y.-X.; Liang, S.-B.; Yang, M.; Robinson, N.; Liu, J.-P. Can Chinese Medicine Be Used for Prevention of Corona Virus Disease 2019 (COVID-19)? A Review of Historical Classics, Research Evidence and Current Prevention Programs. Chin. J. Integr. Med. 2020, 26, 243–250. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Guo, L.; Li, J.; Zhong, D.; Zhang, Y.; Clarke, M.; Jin, R. Traditional Chinese Herbal Medicine for Treting Novel Coronavirus (COVID-19) Pneumonia: Protocol for a Systematic Review and Meta-Analysis. Syst. Rev. 2020, 9, 75. [Google Scholar] [CrossRef]
- Staniforth, V.; Wang, S.-Y.; Shyur, L.-F.; Yang, N.-S. Shikonins, Phytocompounds from Lithospermum Erythrorhizon, Inhibit the Transcriptional Activation of Human Tumor Necrosis Factor Alpha Promoter in vivo. J. Biol. Chem. 2004, 279, 5877–5885. [Google Scholar] [CrossRef]
- Gurard-Levin, Z.A.; Liu, C.; Jekle, A.; Jaisinghani, R.; Ren, S.; Vandyck, K.; Jochmans, D.; Leyssen, P.; Neyts, J.; Blatt, L.M.; et al. Evaluation of SARS-Cov-2 3C-Like Protease Inhibitors Using Self-Assembled Monolayer Desorption Ionization Mass Spectrometry. Antiviral Res. 2020, 182, 104924. [Google Scholar] [CrossRef]
- Fang, P.; Yu, M.; Shi, M.; Bo, P.; Gu, X.; Zhang, Z. Baicalin and its Aglycone: A Novel Approach for Treatment of Metabolic Disorders. Pharmacol. Rep. 2020, 72, 13–23. [Google Scholar] [CrossRef]
- Liang, S.; Deng, X.; Lei, L.; Zheng, Y.; Ai, J.; Chen, L.; Xiong, H.; Mei, Z.; Cheng, Y.-C.; Ren, Y. The Comparative Study of the Therapeutic Effects and Mechanism of Baicalin, Baicalein, and Their Combination on Ulcerative Colitis Rat. Front. Pharmacol. 2019, 10, 1466. [Google Scholar] [CrossRef] [PubMed]
- Dinda, B.; Dinda, S.; Dassharma, S.; Banik, R.; Chakraborty, A.; Dinda, M. Therapeutic Potentials of Baicalin and Its Aglycone, Baicalein Against Inflammatory Disorders. Eur. J. Med. Chem. 2017, 131, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria Baicalensis Extract and Baicalein Inhibit Replication of SARS-Cov-2 and Its 3C-Like Protease in vitro. J. Enzyme Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.; Xie, H.; Ke, C.; Hu, H.; Gao, M.; et al. Anti-SARS-Cov-2 Activities in Vitro of Shuanghuanglian Preparations and Bioactive Ingredients. Acta Pharmacologica Sinica 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
- Khare, P.; Sahu, U.; Pandey, S.C.; Samant, M. Current Approaches for Target-Specific Drug Discovery Using Natural Compounds Against SARS-Cov-2 Infection. Virus Res. 2020, 290, 198169. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of SARS-Cov 3CL Protease by Flavonoids. J. Enzyme Inhib. Med. Chem. 2020, 35, 145–151. [Google Scholar] [CrossRef]
- Pitsillou, E.; Liang, J.; Karagiannis, C.; Ververis, K.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.G. Interaction of Small Molecules with SARS-Cov-2 Main Protease in silico and in vitro Validation of Potential Lead Compounds Using an Enzyme-Linked Immunosorbent Assay. Comput. Biol. Chem. 2020, 89, 107408. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease for the Potential Treatment of COVID-19. Biorxiv 2020. [Google Scholar] [CrossRef]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-Cov-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef]
- Musharrafieh, R.; Ma, C.; Zhang, J.; Hu, Y.; Diesing, J.M.; Marty, M.T.; Wang, J. Validating Enterovirus D68-2Apro As an Antiviral Drug Target and the Discovery of Telaprevir As a Potent D68-2Apro Inhibitor. J. Virol. 2019, 93, e02221-18. [Google Scholar] [CrossRef]
- Resnick, S.J.; Iketani, S.; Hong, S.J.; Zask, A.; Liu, H.; Kim, S.; Melore, S.; Nair, M.S.; Huang, Y.; Tay, N.E.S.; et al. A Simplified Cell-Based Assay to Identify Coronavirus 3CL Protease Inhibitors. Biorxiv 2020. [Google Scholar] [CrossRef]
- Froggatt, H.M.; Heaton, B.E.; Heaton, N.S. Development of a Fluorescence-Based, High-Throughput SARS-Cov-2 3clpro Reporter Assay. J. Virol. 2020, 94, e01265-20. [Google Scholar]
- Montana Molecular Fluorescent Biosensors for Live Cell Discovery. Available online: https://montanamolecular.com/wp-content/uploads/2021/03/3CLglow-Assay_PRINT.pdf (accessed on 26 March 2021).
- Ruocco, K.M.; Goncharova, E.I.; Young, M.R.; Colburn, N.H.; Mcmahon, J.B.; Henrich, C.J. A High-Throughput Cell-Based Assay to Identify Specific Inhibitors of Transcriptional Factor AP-1. J. Biomol. Sreen. 2007, 12, 133–139. [Google Scholar] [CrossRef]
- Good, S.S.; Westover, J.; Jung, K.H.; La Colla, P.; Collu, G.; Moussa, A.; Canard, B.; Sommadossi, J.-P. AT-527 Is a Potent in Vitro Replication Inhibitor of SARS-Cov-2, the Virus Responsible for the COVID-19 Pandemic. Biorxiv 2020. [Google Scholar] [CrossRef]
- Zhu, N.; Wang, W.; Liu, Z.; Liang, C.; Wang, W.; Ye, F.; Huang, B.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and Cytopathic Effect of SARS-Cov-2 Infection in Human Airway Epithelial Cells. Nat. Commun. 2020, 11, 3910. [Google Scholar] [CrossRef]
- Wurtz, N.; Penant, G.; Jardot, P.; Duclos, N.; La Scola, B. Culture of SARS-Cov-2 in a Panel of Laboratory Cell Lines, Permissivity, and Differences in Growth Profile. Eur. J. Clin. Microb. Infect. Dis. 2021, 40, 477–484. [Google Scholar] [CrossRef]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.W.A.L.; Van Der Meer, Y.; Caly, L.; Druce, J.; De Vries, J.J.C.; Kikkert, M.; Bárcena, M.; et al. SARS-Coronavirus-2 Replication in Vero E6 Cells: Replication Kinetics, Rapid Adaptation and Cytopathology. J. Gen. Virol. 2020, 101, 925–940. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinski, L.E.; Dinnon, K.H.; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Potently Inhibits SARS-Cov-2 in Human Lung Cells and Chimeric SARS-Cov Expressing the SARS-Cov-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107904. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Therapeutic Indication | IC50 (μM) Mpro | EC50 (μM) Vero E6 cells 1 |
|---|---|---|---|
| Lopinavir | HIV | 486 | 5.73 |
| Ritonavir | HIV | 13.7 | 8.63 |
| Atazanavir | HIV | 10 2 | 2.0 |
| Nelfinavir | HIV | 234 | 1.13 |
| Boceprevir | HCV | 4.13 | 1.90 |
| Carmofur | Antineoplastic drug | 1.82 | 24.87 |
| Ebselen | Anti-inflammatory | 0.67 | 4.67 |
| Remdesivir | Ebola virus | n.d. | 0.77 |
| Dipyridamole | Vasodilatator | n.d. | 0.1 |
| Chloroquine | Malaria | 0.56 3 | 5.4 |
| Compound | Chemical Class | Structure | IC50 (μM) 1 | Mechanism | EC50 (μM) 2 | Development Status |
|---|---|---|---|---|---|---|
| 1 | Aldheydes |  | 0.053 | Reversible | 0.53 | Preclinical 3 |
| 2 | Aldehydes |  | 0.040 | Reversible | 0.72 | Preclinical 4 |
| 3 | Aldehydes |  | 0.030 | Reversible | 3.37 | Preclinical 5 |
| 4 | Aldehydes |  | 0.40 | Reversible | 1.5 | Research |
| 5 | Aldehydes |  | 0.0085 | Reversible | >10 | Research |
| 6 | Aldehydes |  | 0.03 | Reversible | 5 | Research |
| 7 | Aldehydes |  | 0.1 | Reversible | 2.5 | Research |
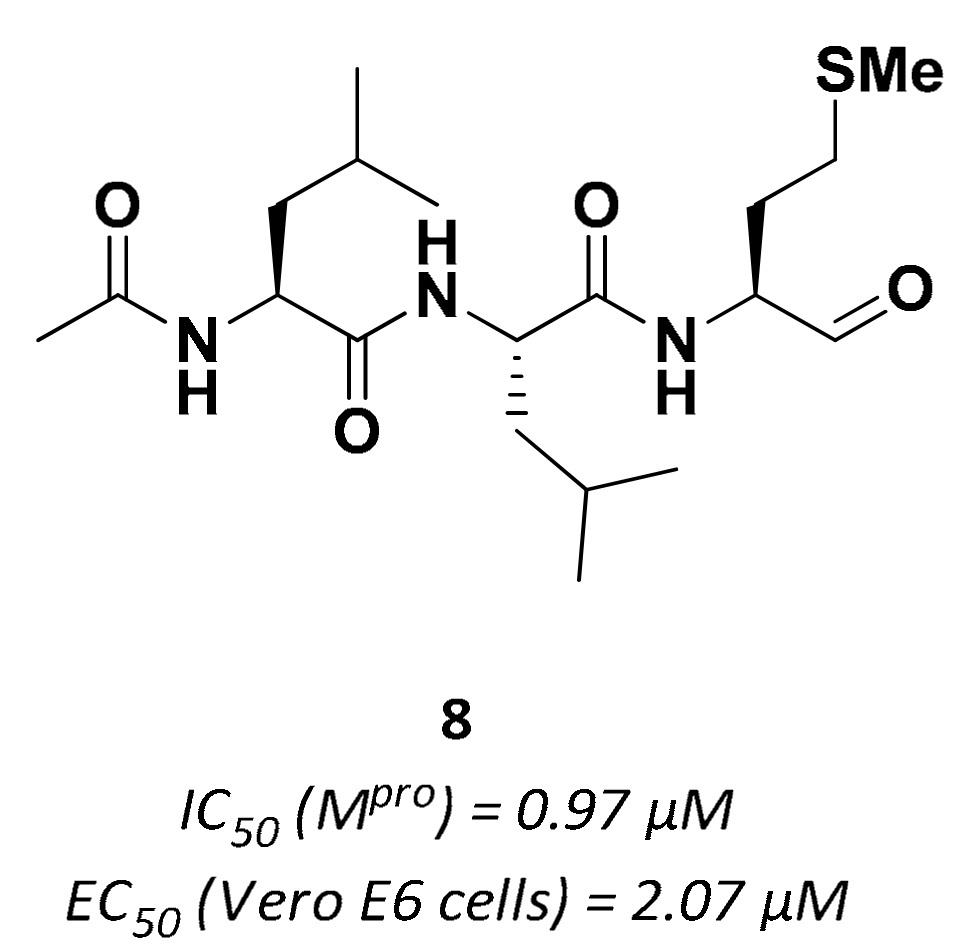
| 8 | Aldehydes |  | 0.97 | Reversible | 2.07 | Preclinical |
| Boceprevir | α-ketoamides |  | 4.13 | Irreversible | 1.90 | FDA approved drug |
| 11 | α-ketoamides |  | 0.67 | Irreversible | 4–5 6 | Preclinical |
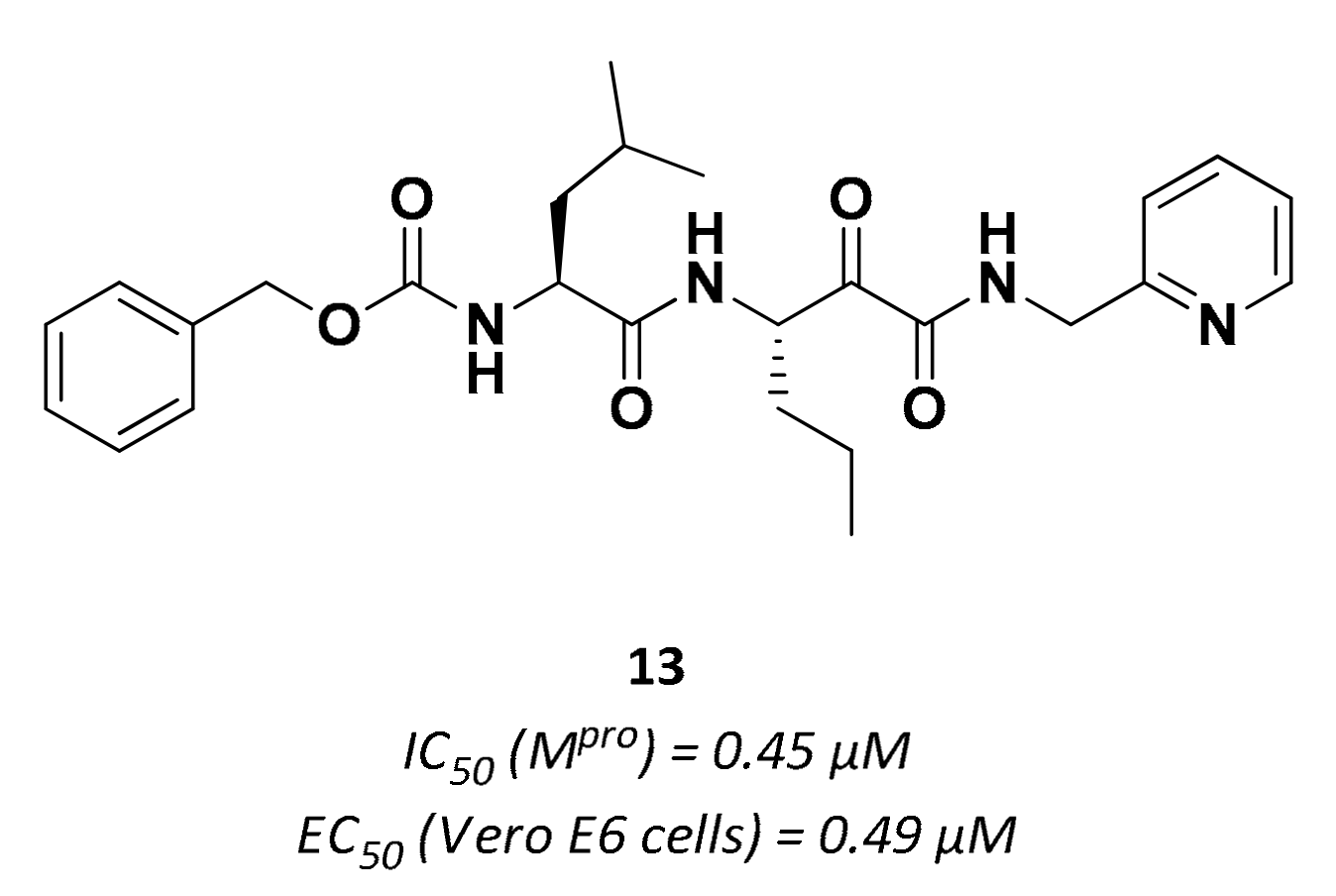
| 13 | α-ketoamides |  | 0.45 | Irreversible | 0.49 | Preclinical |
| 14 | Vinyl esters |  | - | Irreversible | 16.77 | Preclinical |
| 15 | Fluoromethyl ketones |  | 11.39 | Irreversible | 0.13 | Research |
| 19 | Hydroxymethyl ketones |  | 0.27 Ki | Reversible | 0.23 7 0.76 8 | Phase 1 clinical |
| 20 | Acyloxy methyl ketones |  | >200 nM 9 | Irreversbile | - | Research |
| UCI-1 | Cyclic peptides |  | ~150 µM | - | - | Research |
| 21 | Small molecules (tetralone) |  | - | Irreversible | - | Research |
| 23 | Small molecules |  | 0.26 | - | 3.55 | Research |
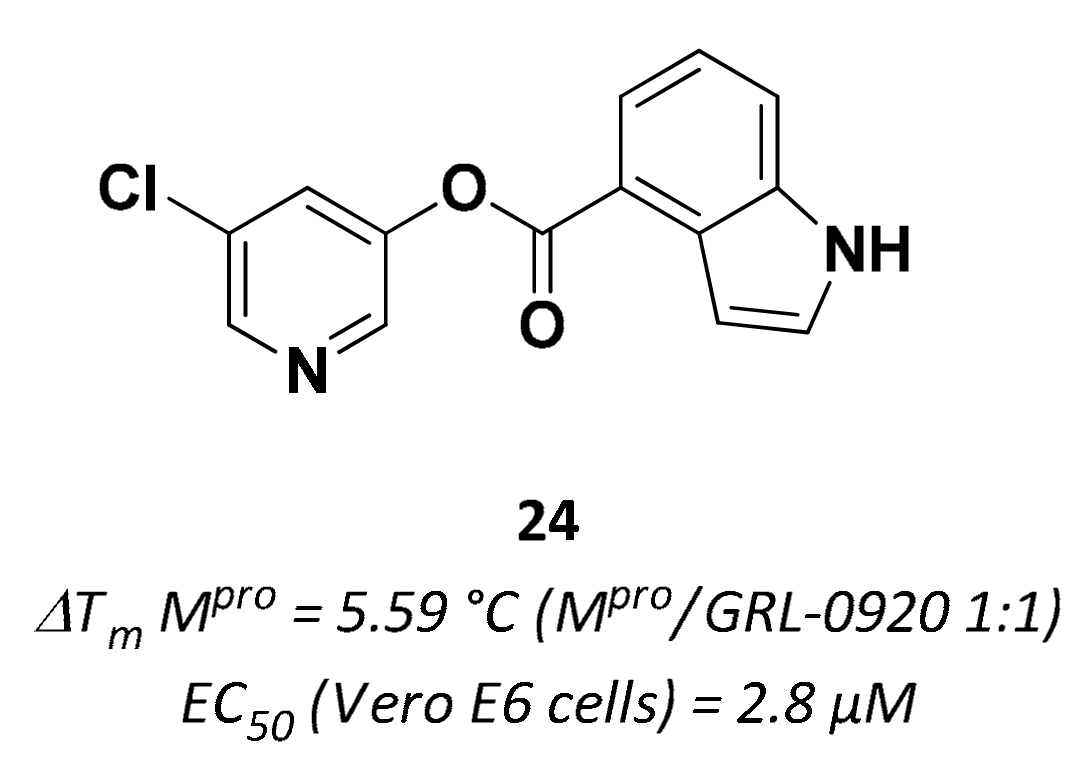
| 24 | Small molecules |  | 5.59 °C 10 | Irreversible | 2.8 | Research |
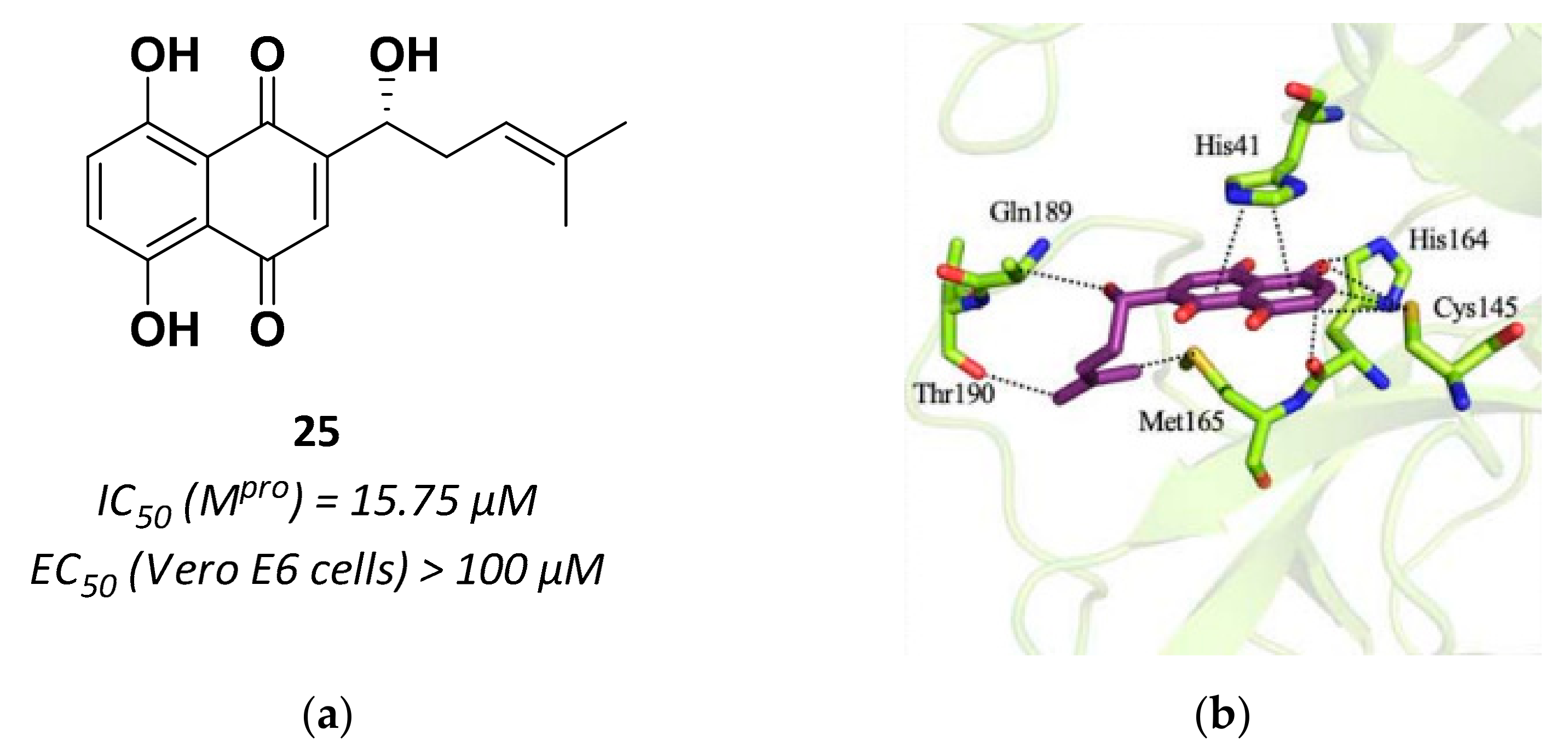
| 25 | Small molecules (Quinone) |  | 15.75 | Reversible | >100 | Research |
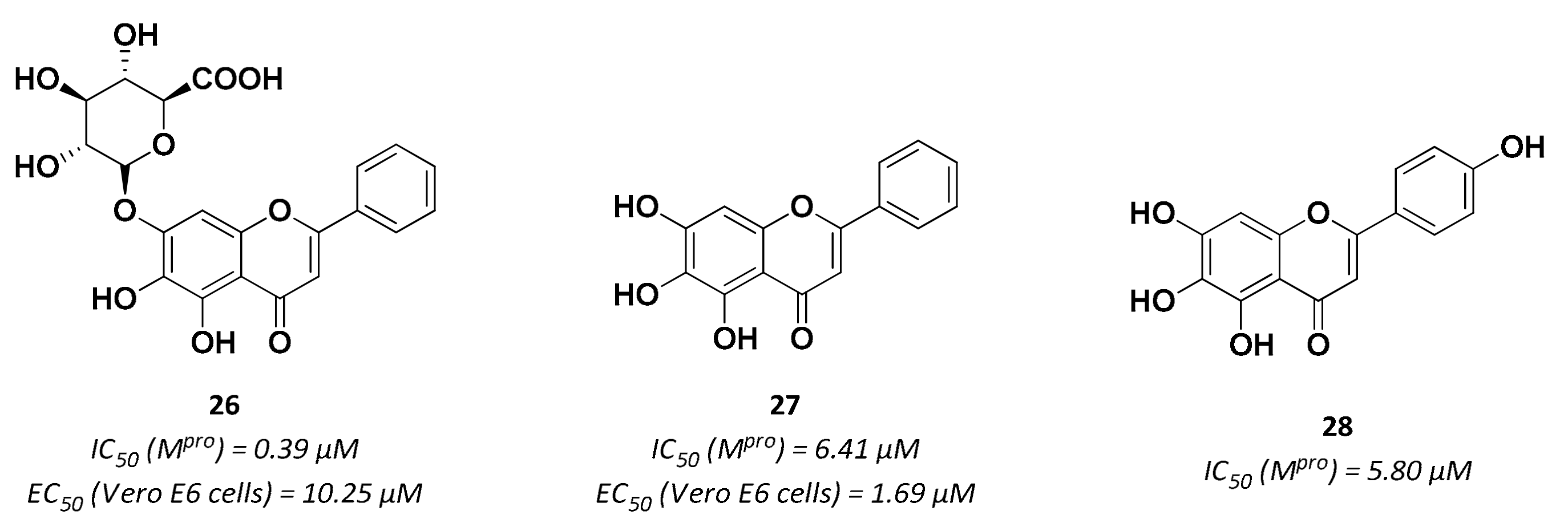
| 27 | Small molecules (Flavonoids) |  | 6.41 | Reversible | 1.69 | Research |
| 28 | Small molecules (Flavonoids) |  | 5.8 | Reversible | - | Research |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. https://doi.org/10.3390/biom11040607
Citarella A, Scala A, Piperno A, Micale N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules. 2021; 11(4):607. https://doi.org/10.3390/biom11040607
Chicago/Turabian StyleCitarella, Andrea, Angela Scala, Anna Piperno, and Nicola Micale. 2021. "SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors" Biomolecules 11, no. 4: 607. https://doi.org/10.3390/biom11040607
APA StyleCitarella, A., Scala, A., Piperno, A., & Micale, N. (2021). SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules, 11(4), 607. https://doi.org/10.3390/biom11040607