
The Pseudo-Symmetric N-benzyl Hydroxyethylamine Core in a New Series of Heteroarylcarboxyamide HIV-1 Pr Inhibitors: Synthesis, Molecular Modeling and Biological Evaluation

 ,
,  ,
,  ,
,  and
and 
Abstract
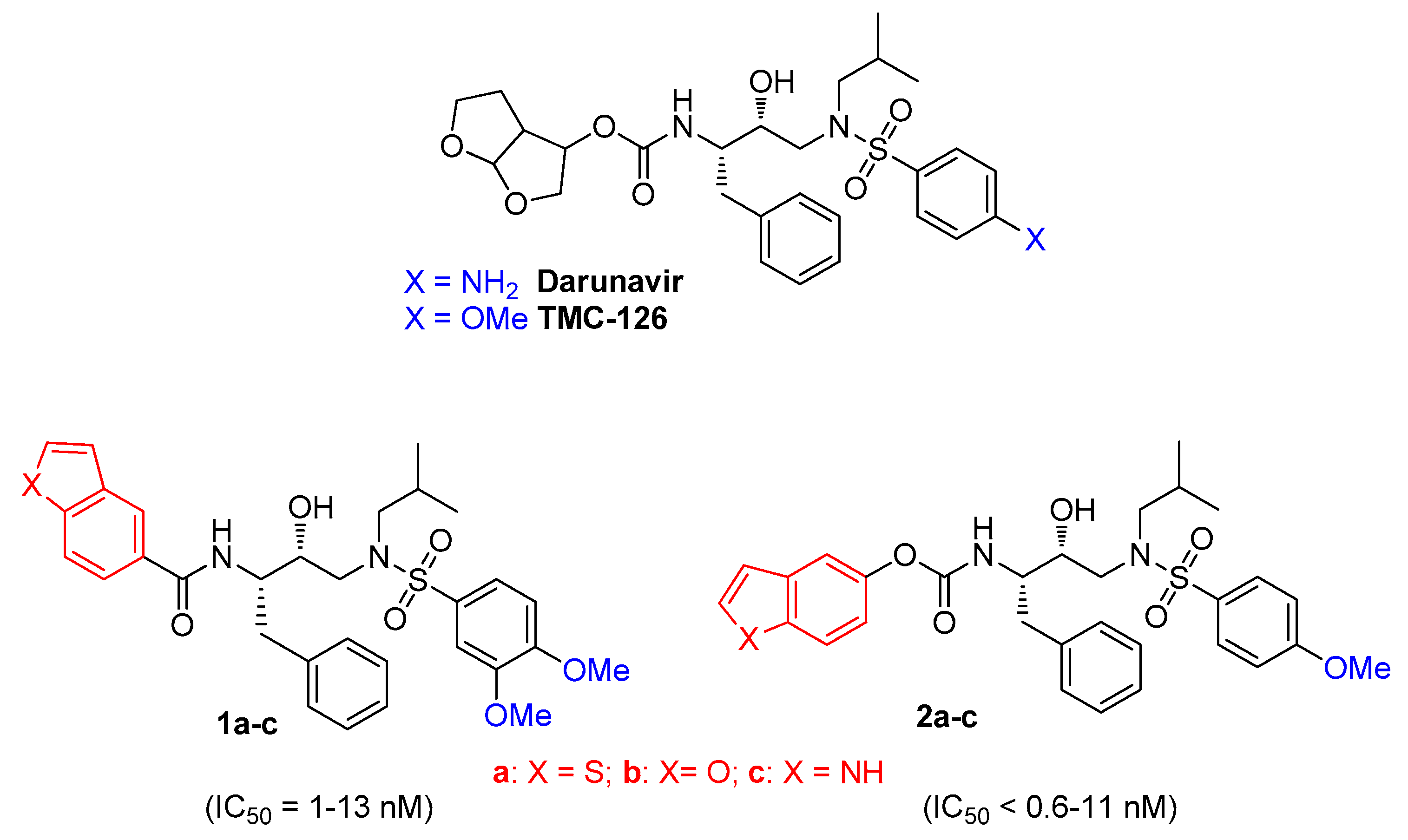
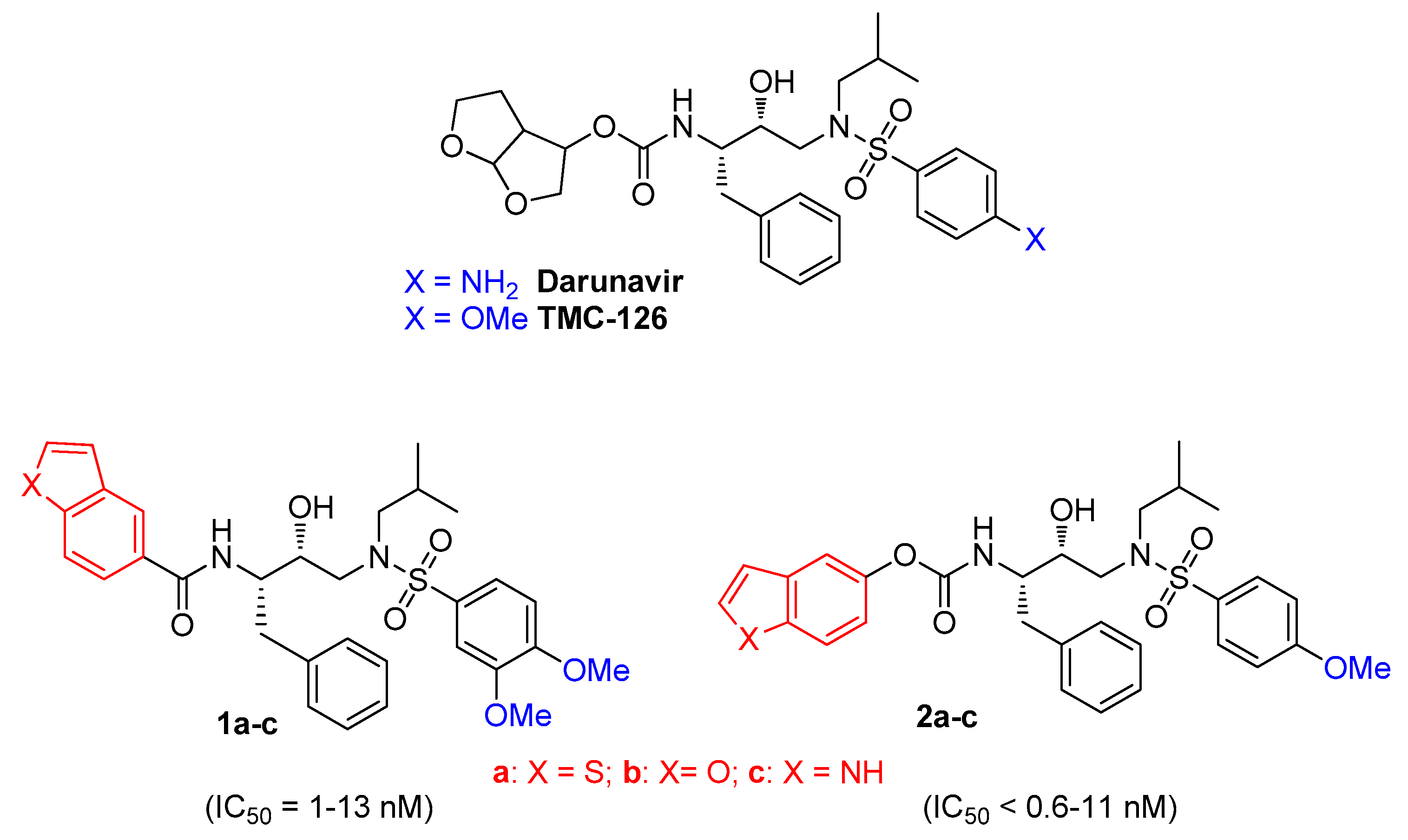
:1. Introduction
2. Materials and Methods
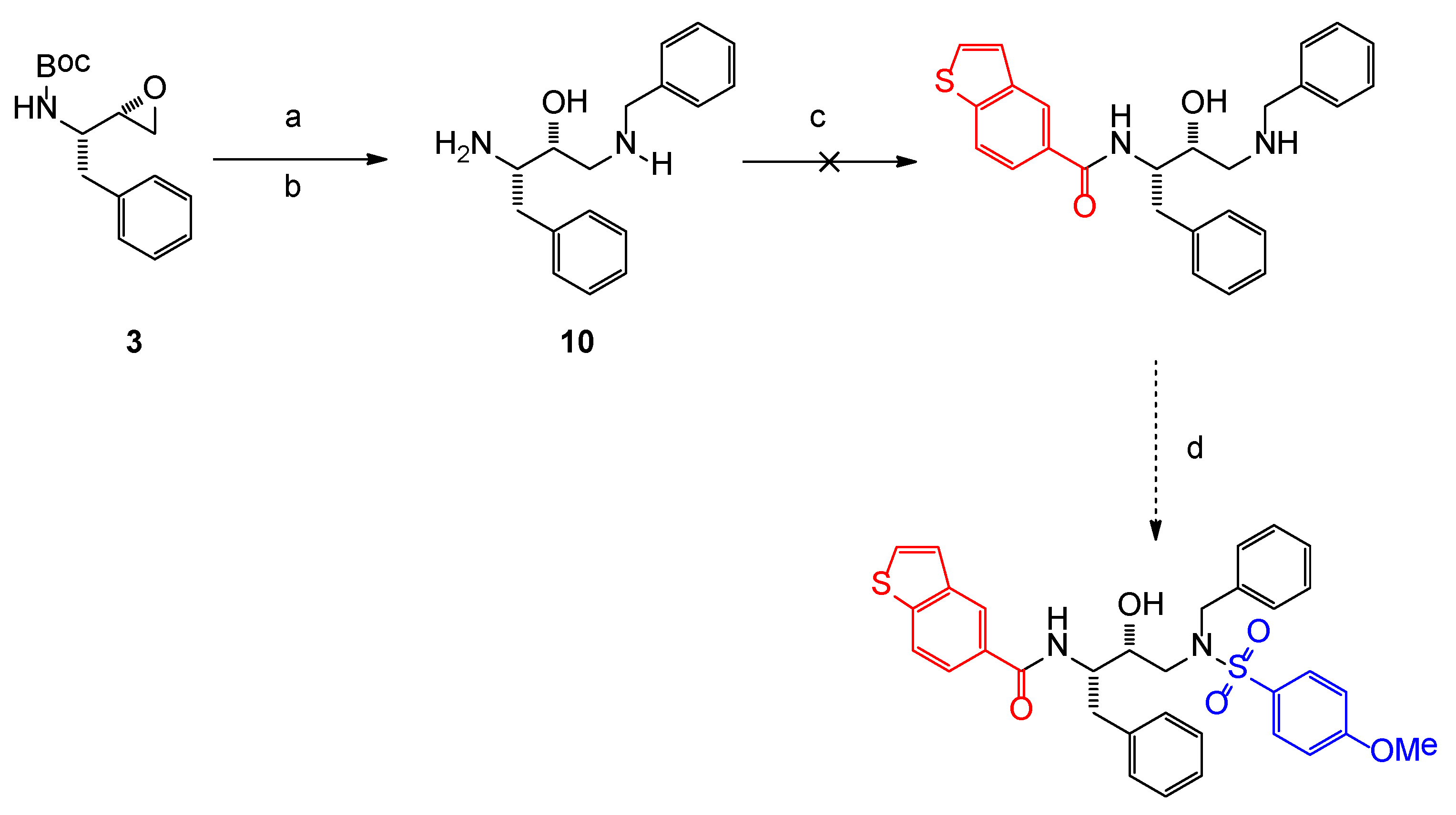
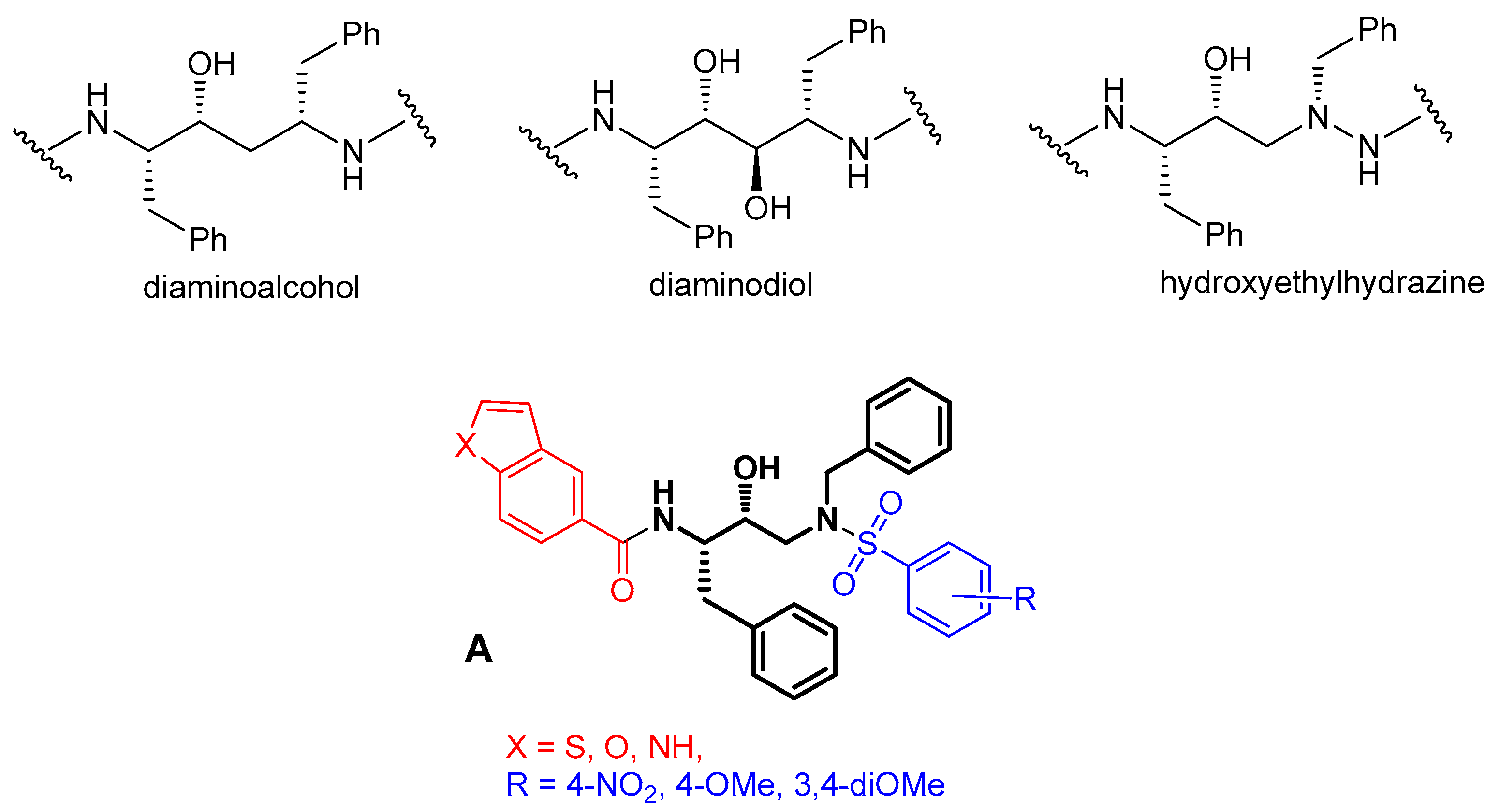
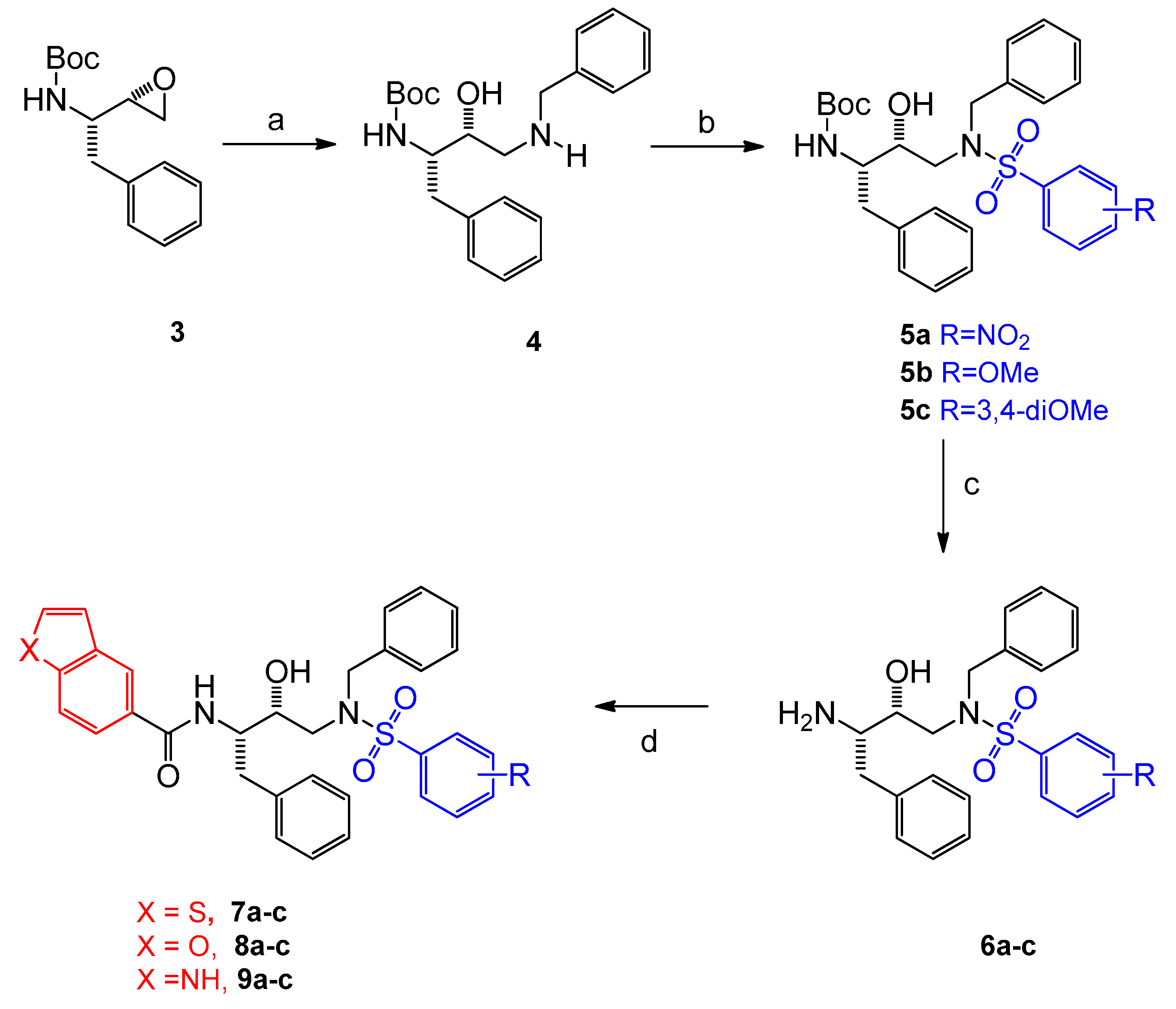
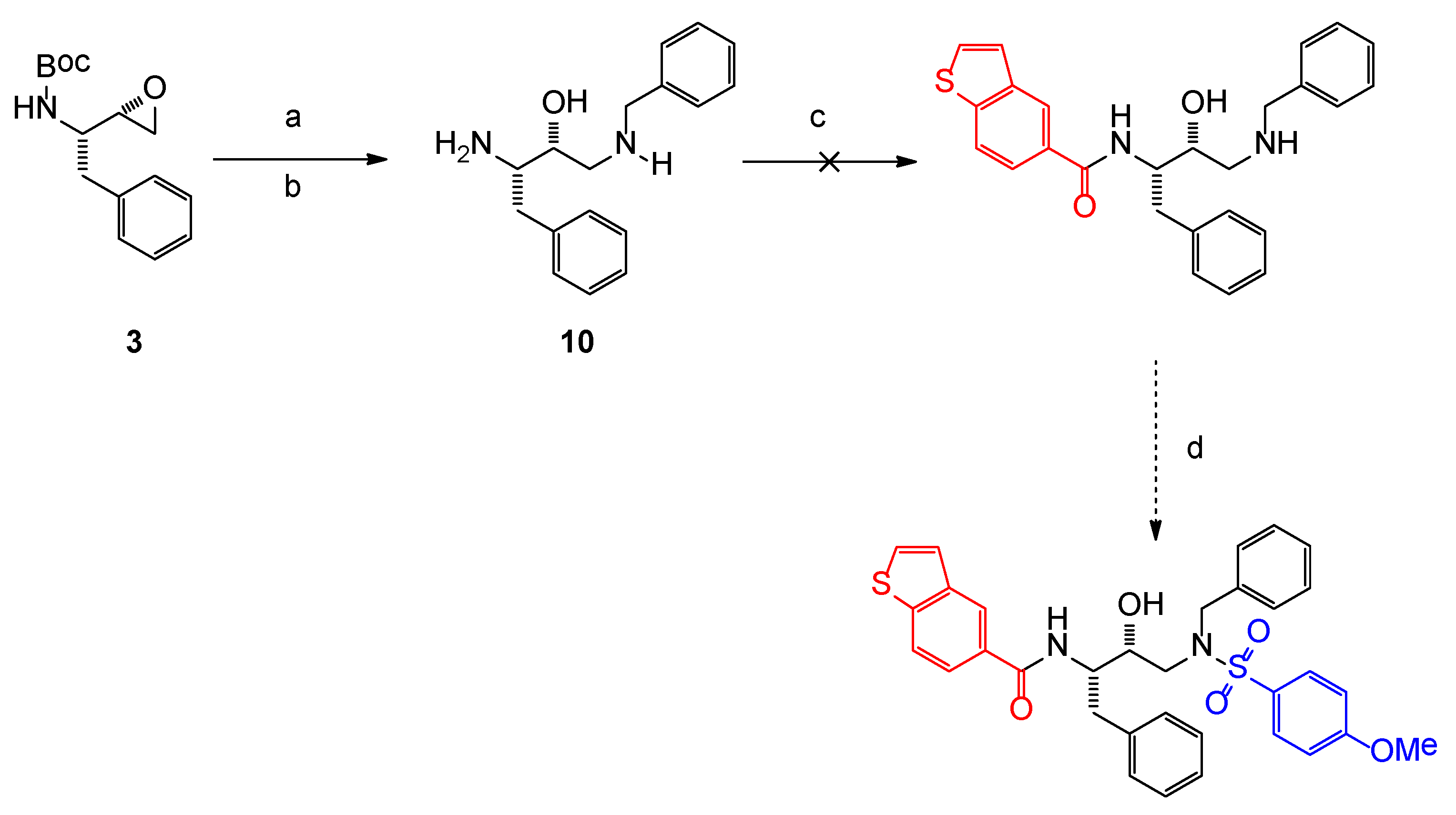
2.1. Chemistry
2.2. In Vitro Activity Test
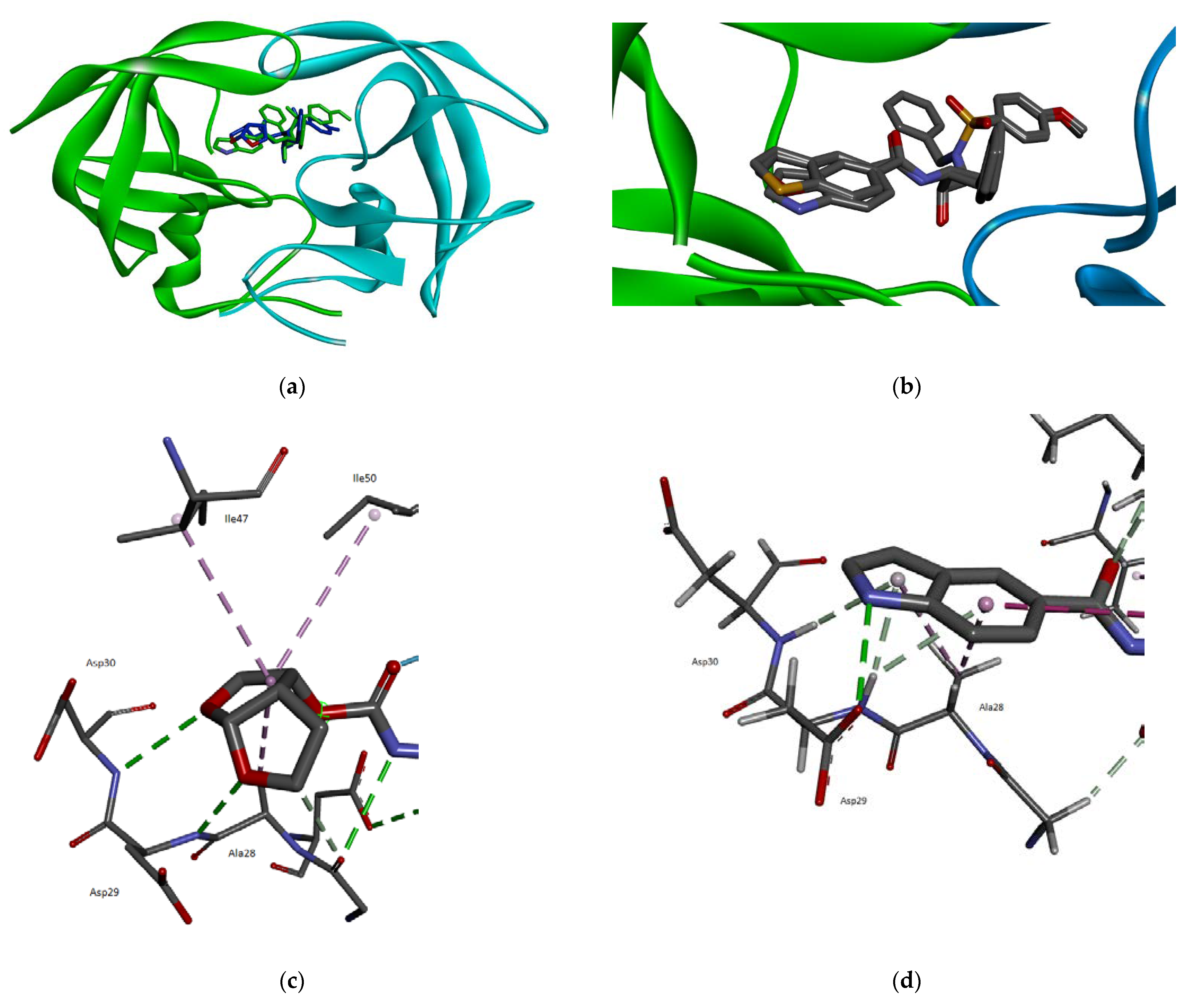
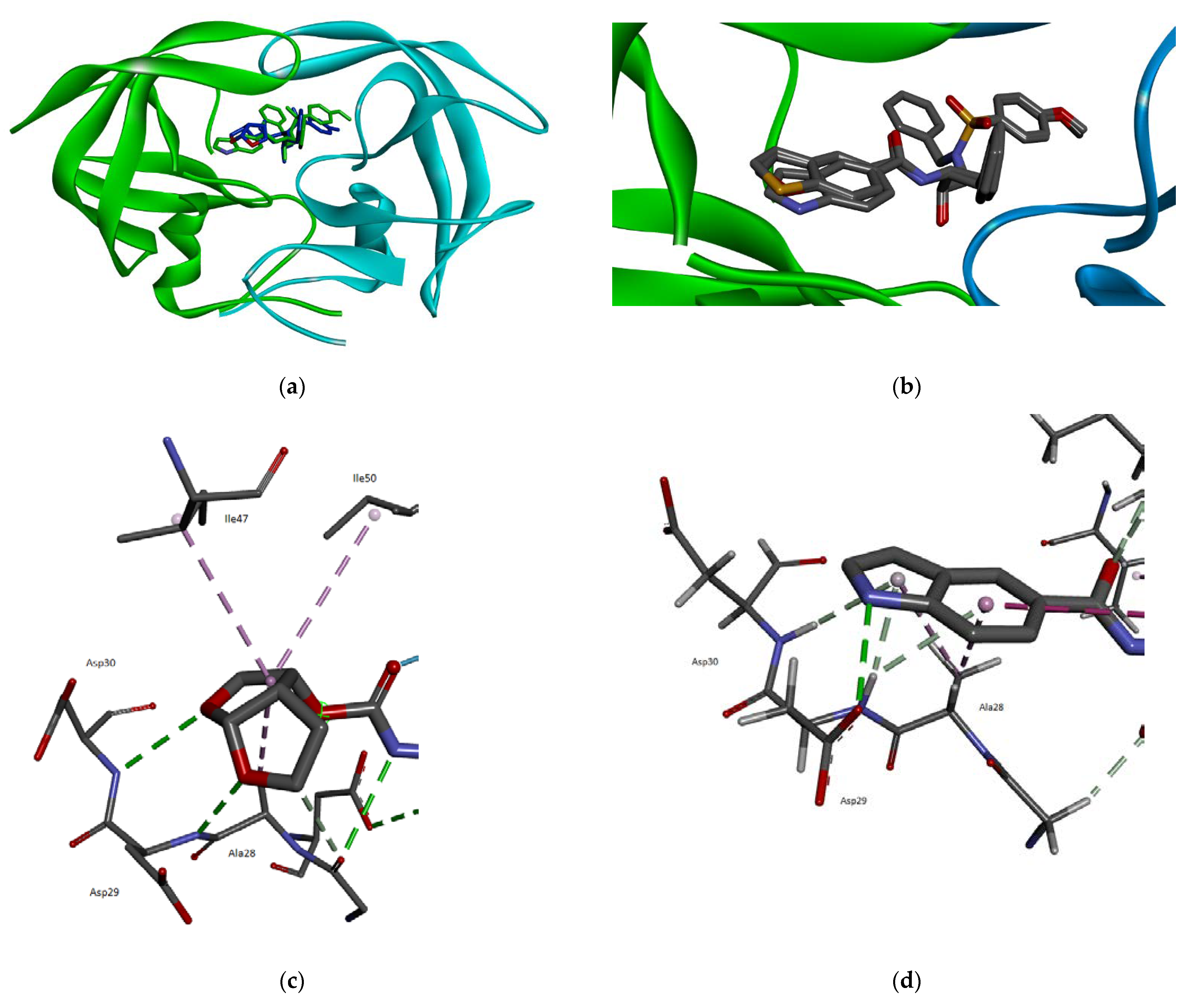
2.3. Molecular Modeling
3. Results and Discussion
3.1. Chemistry
3.2. In Vitro Activity
3.3. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Global Report: UNAIDS Report on the Global AIDS Epidemic 2021. Available online: https://www.unaids.org/sites/default/files/media_asset/2021-global-aids-update_en.pdf. (accessed on 17 September 2021).
- Wensing, A.M.J.; Van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antiviral Res. 2010, 85, 59–74. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization, Geneva, Switzerland. Available online: https://www.who.int/hiv/pub/arv/chapter4.pdf. (accessed on 17 September 2021).
- Ghosh, A.K.; Chapsal, B.D. Aspartic acid proteases as therapeutic targets. In Methods and Principles in Medicinal Chemistry; Ghosh, A.K., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Volume 45, pp. 169–204. [Google Scholar]
- Tomasselli, A.G.; Heinrikson, R.L. Targeting the HIV-protease in AIDS therapy: A current clinical perspective. Biochim. et Biophys. Acta 2000, 1477, 189–214. [Google Scholar] [CrossRef]
- Brik, A.; Wong, C.H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Rao, K.V.; Nyalapatla, P.R.; Kovela, S.; Brindisi, M.; Osswald, H.L.; Reddy, B.S.; Agniswamy, J.; Wang, Y.-F.; Aoki, M.; et al. Design of Highly Potent, Dual-Acting and Central-Nervous-System-Penetrating HIV-1 Protease Inhibitors with Excellent Potency against Multidrug-Resistant HIV-1 Variants. Chem. Med. Chem. 2018, 13, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Williams, J.N.; Ho, R.Y.; Simpson, H.M.; Hatton, S.-I.; Hayashi, H.; Agniswamy, J.; Wang, Y.-F.; Weber, I.T. Design and Synthesis of Potent HIV-1 Protease Inhibitors Containing Bicyclic Oxazolidinone Scaffold as the P2 Ligands: Structure-Activity Studies and Biological and X-ray Structural Studies. J. Med. Chem. 2018, 61, 9722–9737. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jia, Q.; Zhang, Z. Applications of amide isosteres in medicinal chemistry. Bioorg. Med. Chem. Lett. 2019, 29, 2535–2550. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Chiummiento, L.; De Bonis, M.; Funicello, M.; Lupattelli, P.; Pandolfo, R. Application of Sharpless asymmetric dihydroxylation to thienyl- and benzothienyl acrylates and crotonates. Tetrahedron Asymmetry 2006, 17, 2919–2924. [Google Scholar] [CrossRef]
- Bochicchio, A.; Cefola, R.; Choppin, S.; Colobert, F.; Di Noia, M.A.; Funicello, M.; Hanquet, G.; Pisano, I.; Todisco, S. Chiummiento, L. Selective Claisen rearrangement and iodination for the synthesis of polyoxygenated allyl phenol derivatives. Tetrahedron Lett. 2016, 57, 4053–4055. [Google Scholar] [CrossRef]
- Cerminara, I.; Chiummiento, L.; Funicello, M.; Guarnaccio, A.; Lupattelli, P. Heterocycles in Peptidomimetics and Pseudopeptides: Designand Synthesis. Pharmaceuticals 2012, 5, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Chiummiento, L.; De Bonis, M.; Di Blasio, N.; Funicello, M.; Lupattelli, P.; Pandolfo, R.; Tramutola, F.; Berti, F. Synthesis of New Thienyl Ring Containing HIV-1 Protease Inhibitors: Promising Preliminary Pharmacological Evaluation against Recombinant HIV-1 Proteases. J. Med. Chem. 2010, 53, 1451–1457. [Google Scholar] [CrossRef]
- Bonini, C.; Chiummiento, L.; De Bonis, M.; Funicello, M.; Lupattelli, P.; Suanno, G.; Berti, F.; Campaner, P. Synthesis, biological activity and modelling studies of two novel anti HIV PR inhibitors with a thiophene containing hydroxyethylamino core. Tetrahedron 2005, 61, 6580–6589. [Google Scholar] [CrossRef]
- Bonini, C.; Chiummiento, L.; De Bonis, M.; Funicello, M.; Lupattelli, P. Synthesis of a First Thiophene Containing Analog of the HIV Protease Inhibitor Nelfinavir. Tetrahedron Lett. 2004, 45, 2797–2799. [Google Scholar] [CrossRef]
- Chiummiento, L.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Campaner, P. New indolic non-peptidic HIV protease inhibitors from (S)-glycidol: Synthesis and preliminary biological activity. Tetrahedron 2009, 65, 5984–5989. [Google Scholar] [CrossRef]
- Chiummiento, L.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Berti, F.; Marino-Merlo, F. Synthesis and biological evaluation of novel small non-peptidic HIV-1 PIs: The benzothiophene ring as an effective moiety. Bioorg. Med. Chem. Lett. 2012, 22, 2948–2950. [Google Scholar] [CrossRef]
- Bonini, C.; Chiummiento, L.; Di Blasio, N.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Berti, F.; Ostric, A.; Miertus, S.; Frecer, V.; et al. Synthesis and biological evaluation of new simple indolic non peptidic HIV Protease inhibitors: The effect of different substitution patterns. Bioorg. Med. Chem. 2014, 22, 4792–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Xu, C.X.; Rao, K.V.; Baldridge, A.; Agniswamy, J.; Wang, Y.F.; Weber, I.T.; Aoki, M.; Miguel, S.G.P.; Amano, M.; et al. Probing multidrug-resistance/protein-ligand interaction with oxatricyclic designed ligands in HIV-1 Protease inhibitors. ChemMedChem 2010, 5, 1850–1854. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Anderson, D.D.; Weber, I.T.; Mitsuya, H. Enhancing Protein Backbone Binding-A Fruitful Concept for Combating Drug-Resistant HIV. Angew. Chem. Int. Ed. Engl. 2012, 51, 1778–1802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.F.; Shen, C.H.; Agniswamy, J.; Rao, K.V.; Xu, X.; Ghosh, A.K.; Harrison, R.W.; Weber, I.T. Novel P2 Tris-tetrahydrofuran Group in Antiviral Compound 1 (GRL-0519) Fills the S2 Binding Pocket of Selected Mutants of HIV-1 Protease. J. Med. Chem. 2013, 56, 1074–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funicello, M.; Chiummiento, L.; Tramutola, F.; Armentano, M.F.; Bisaccia, F.; Miglionico, R.; Milella, L.; Benedetti, F.; Berti, F.; Lupattelli, P. Synthesis and biological evaluation in vitro and in mammalian cells of new heteroaryl carboxyamides as HIV-protease inhibitors. Bioorg. Med. Chem. 2017, 25, 4715–4722. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, F.; Armentano, M.F.; Berti, F.; Chiummiento, L.; Lupattelli, P.; D’Orsi, R.; Miglionico, R.; Milella, L.; Bisaccia, F.; Funicello, M. New heteroaryl carbamates: Synthesis and biological screening in vitro and in mammalian cells of wild-type and mutant HIV-protease inhibitors. Bioorg. Med. Chem. 2019, 27, 1863–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidaka, K.; Kimura, T.; Hayashi, Y.; McDaniel, K.F.; Dekhtyar, T.; Colletti, L.; Kiso, Y. Design and Synthesis of Pseudo-Symmetric HIV Protease Inhibitors Containing a Novel Hydroxymethylcarbonyl (HMC)-Hydrazide Isostere. Bioorg. Med. Chem. Lett. 2003, 13, 93–96. [Google Scholar] [CrossRef]
- Facchinetti, V.; Moreth, M.; Gomes, C.R.B.; do Ó Pessoa, C.; Rodrigues, F.A.R.; Cavalcanti, B.C.; Oliveira, A.C.A.; Carneiro, T.R.; Gama, I.L.; de Souza, M.V.N. Evaluation of (2S,3R)-2-(amino)-[4-(N-benzylarenesulfonamido)-3-hydroxy-1-phenylbutane derivatives: A promising class of anticancer agents. Med. Chem. Res. 2015, 24, 533–542. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-3: MacroModel. Schrödinger, LLC.: New York, NY, USA, 2021; The inhibitors were then docked inside the catalytic site of the enzyme with the Schrödinger Glide protocol.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Trucks, G.W.; Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (2009) Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bai, X.; Yang, Z.; Zhu, M.; Dong, B.; Zhu, L.; Zhang, G.; Wang, J.; Wang, Y. Design and synthesis of potent HIV-1 protease inhibitors with (S)-tetrahydrofuran-tertiary amine-acetamide as P2ligand: Structure activity studies and biological evaluation. Eur. J. Med. Chem. 2017, 137, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-H.; Bai, X.-G.; Zhou, L.; Wang, J.-X.; Liu, H.-T.; Wang, Y.-C. Synthesis and biological evaluation of novel HIV-1 protease inhibitors using tertiary amine as P2-ligands. Bioorg. Med. Chem. Lett. 2015, 25, 1880–1883. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.A.; Jiang, C.; Guan, M. Anti-HIV drugs for cancer therapeutics: Back to the future? Lancet Oncol. 2009, 10, 61–71. [Google Scholar] [CrossRef]
- Toupkanloo, H.A.; Rahmani, Z. An in-depth study on non-covalent stacking interactions between DNA bases and aromatic drug fragments using DFT method and AIM analysis: Conformers, binding energies, and charge transfer. App. Biol. Chem. 2018, 61, 209–226. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 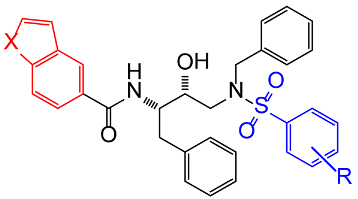 Compound Compound | X | R | IC50 (nM) | Std. Error (nM) |
|---|---|---|---|---|---|
| 1 | 7a | S | 4-NO2 | 4.2 | 0.6 |
| 2 | 7b | S | 4-OMe | 2.3 | 0.4 |
| 3 | 7c | S | 3,4-diOMe | 6.2 | 2.1 |
| 4 | 8a | O | 4-NO2 | 47.0 | 6.0 |
| 5 | 8b | O | 4-OMe | 9.6 | 0.1 |
| 6 | 8c | O | 3,4-diOMe | 82.0 | 7.0 |
| 7 | 9a | NH | 4-NO2 | 27.7% * | 3.5% |
| 8 | 9b | NH | 4-OMe | 36.3% * | 5.1% |
| 9 | 9c | NH | 3,4-diOMe | 10.2% * | 2.4% |
| 10 | darunavir | 1.8 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Orsi, R.; Funicello, M.; Laurita, T.; Lupattelli, P.; Berti, F.; Chiummiento, L. The Pseudo-Symmetric N-benzyl Hydroxyethylamine Core in a New Series of Heteroarylcarboxyamide HIV-1 Pr Inhibitors: Synthesis, Molecular Modeling and Biological Evaluation. Biomolecules 2021, 11, 1584. https://doi.org/10.3390/biom11111584
D’Orsi R, Funicello M, Laurita T, Lupattelli P, Berti F, Chiummiento L. The Pseudo-Symmetric N-benzyl Hydroxyethylamine Core in a New Series of Heteroarylcarboxyamide HIV-1 Pr Inhibitors: Synthesis, Molecular Modeling and Biological Evaluation. Biomolecules. 2021; 11(11):1584. https://doi.org/10.3390/biom11111584
Chicago/Turabian StyleD’Orsi, Rosarita, Maria Funicello, Teresa Laurita, Paolo Lupattelli, Federico Berti, and Lucia Chiummiento. 2021. "The Pseudo-Symmetric N-benzyl Hydroxyethylamine Core in a New Series of Heteroarylcarboxyamide HIV-1 Pr Inhibitors: Synthesis, Molecular Modeling and Biological Evaluation" Biomolecules 11, no. 11: 1584. https://doi.org/10.3390/biom11111584
APA StyleD’Orsi, R., Funicello, M., Laurita, T., Lupattelli, P., Berti, F., & Chiummiento, L. (2021). The Pseudo-Symmetric N-benzyl Hydroxyethylamine Core in a New Series of Heteroarylcarboxyamide HIV-1 Pr Inhibitors: Synthesis, Molecular Modeling and Biological Evaluation. Biomolecules, 11(11), 1584. https://doi.org/10.3390/biom11111584