
The Role of Exosomes in Lysosomal Storage Disorders



Abstract
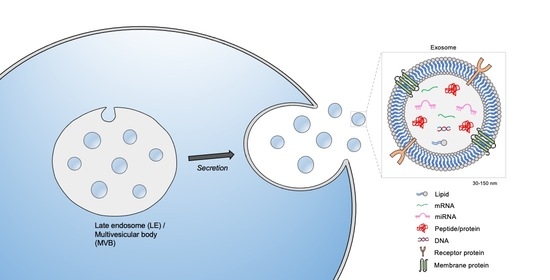
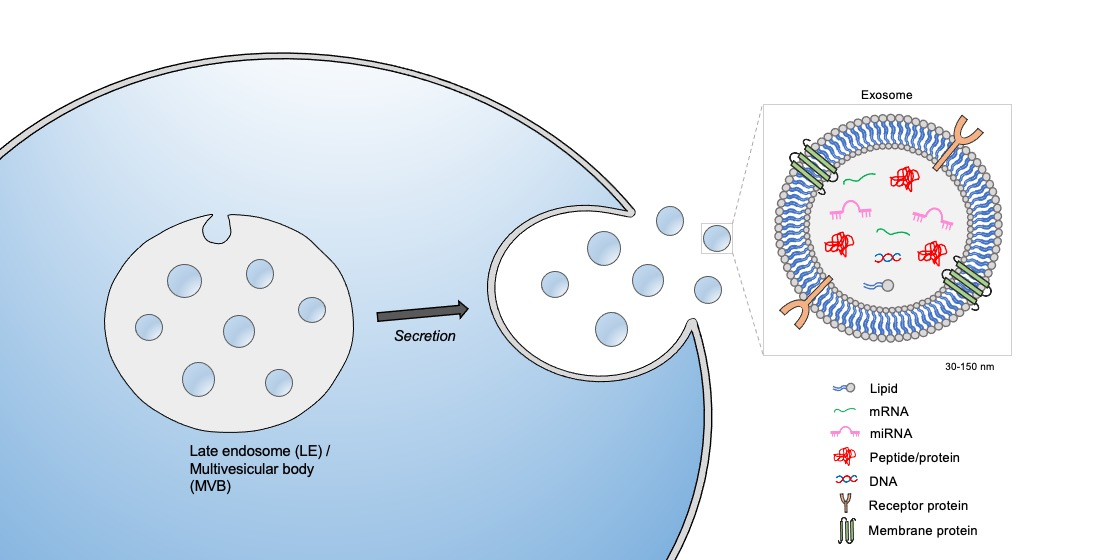
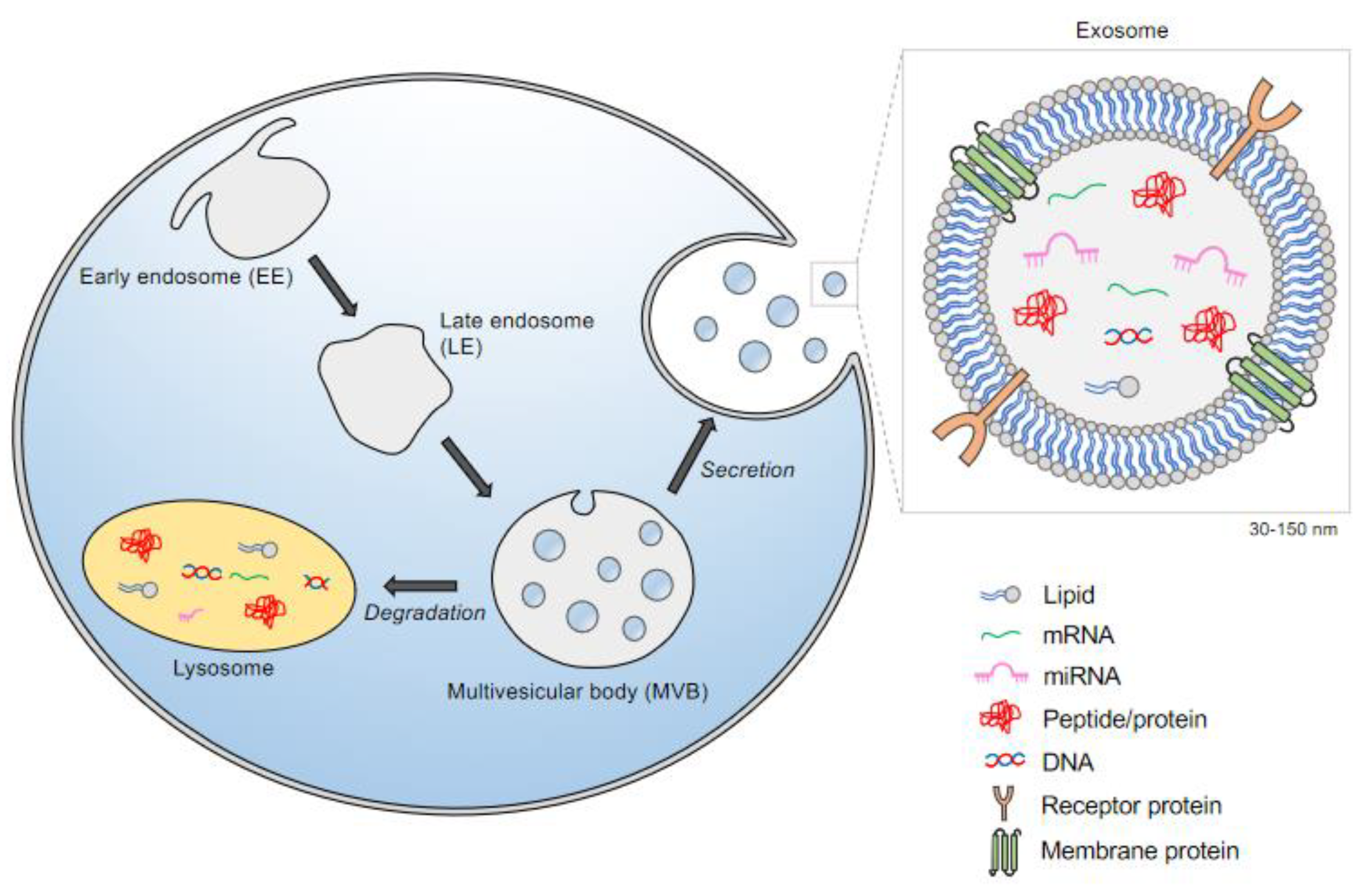
1. Introduction
2. The Biogenesis of Exosomes: A Subclass of Extracellular Vesicles (EVs)
3. Exosome Cargo and Transport:
4. Exosomal Signatures: Lessons from the CNS
4.1. Exosomes from Astrocytes
4.2. Exosomes from Cortical Neurons
4.3. Exosomes from Oligodendrocytes
5. Studies of Exosomes in Specific Lysosome Storage Disorders (LSDs)
5.1. Metachromatic Leukodystrophy
5.2. Mucolipidosis Type IV
5.3. Sialidosis
5.4. Cystinosis
5.5. Niemann-Pick Type C
5.6. GaucherDdisease
5.7. Lysosomal Storage Disorders in General
6. Exosomes as Therapeutic Agents for Treating Lysosomal Storage Disorders
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and exosomal microRNA: Trafficking, sorting, and function. Genom. Proteom. Bioinform. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell Vesicles 2014, 3. [Google Scholar] [CrossRef]
- Gruenberg, J.; Stenmark, H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 317–323. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Schey, K.L.; Luther, J.M.; Rose, K.L. Proteomics characterization of exosome cargo. Methods 2015, 87, 75–82. [Google Scholar] [CrossRef]
- Chen, S.; Datta-Chaudhuri, A.; Deme, P.; Dickens, A.; Dastgheyb, R.; Bhargava, P.; Bi, H.; Haughey, N.J. Lipidomic characterization of extracellular vesicles in human serum. J. Circ. Biomark. 2019, 8, 1849454419879848. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, Y.; Lu, M.; Ding, Q.; Wang, P.; Zhang, H.; Tian, X.; Lu, P.; Meng, D.; Sun, N.; et al. ALIX increases protein content and protective function of iPSC-derived exosomes. J. Mol. Med. (Berl.) 2019, 97, 829–844. [Google Scholar] [CrossRef]
- Yang, F.; Ning, Z.; Ma, L.; Liu, W.; Shao, C.; Shu, Y.; Shen, H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol. Cancer 2017, 16, 148. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J. Lipid Res. 2019, 60, 9–18. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Johnstone, R. Selective externalization of the transferrin receptor by sheep reticulocytes in vitro. Response to ligands and inhibitors of endocytosis. J. Biol. Chem. 1984, 259, 9776–9782. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, L.; Giurato, G.; Cicchini, C.; Montaldo, C.; Mancone, C.; Tarallo, R.; Battistelli, C.; Alonzi, T.; Weisz, A.; Tripodi, M. The RNA-Binding Protein SYNCRIP Is a Component of the Hepatocyte Exosomal Machinery Controlling MicroRNA Sorting. Cell Rep. 2016, 17, 799–808. [Google Scholar] [CrossRef]
- You, Y.; Borgmann, K.; Edara, V.V.; Stacy, S.; Ghorpade, A.; Ikezu, T. Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. J. Extracell Vesicles 2020, 9, 1706801. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; Mayer-Proschel, M.; Bieberich, E. Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): Potential mechanism of apoptosis induction in Alzheimer disease (AD). J. Biol. Chem. 2012, 287, 21384–21395. [Google Scholar] [CrossRef] [PubMed]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Chivet, M.; Hemming, F.; Pernet-Gallay, K.; Fraboulet, S.; Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 2012, 3, 145. [Google Scholar] [CrossRef]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Albers, E.M.; Bretz, N.; Tenzer, S.; Winterstein, C.; Mobius, W.; Berger, H.; Nave, K.A.; Schild, H.; Trotter, J. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Proteom. Clin. Appl. 2007, 1, 1446–1461. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeis, C.; Frohlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Mobius, W.; Goebbels, S.; Nave, K.A.; et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef]
- Frohlich, D.; Kuo, W.P.; Fruhbeis, C.; Sun, J.J.; Zehendner, C.M.; Luhmann, H.J.; Pinto, S.; Toedling, J.; Trotter, J.; Kramer-Albers, E.M. Multifaceted effects of oligodendroglial exosomes on neurons: Impact on neuronal firing rate, signal transduction and gene regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef]
- Kaye, E.M. Lysosomal Storage Diseases. Curr. Treat. Options Neurol. 2001, 3, 249–256. [Google Scholar] [CrossRef]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Polten, A.; Fluharty, A.L.; Fluharty, C.B.; Kappler, J.; von Figura, K.; Gieselmann, V. Molecular basis of different forms of metachromatic leukodystrophy. N. Engl. J. Med. 1991, 324, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Gieselmann, V.; Krageloh-Mann, I. Metachromatic leukodystrophy—An update. Neuropediatrics 2010, 41, 1–6. [Google Scholar] [CrossRef]
- Klein, D.; Bussow, H.; Fewou, S.N.; Gieselmann, V. Exocytosis of storage material in a lysosomal disorder. Biochem. Biophys. Res. Commun. 2005, 327, 663–667. [Google Scholar] [CrossRef]
- Bach, G. Mucolipidosis type IV. Mol. Genet. Metab. 2001, 73, 197–203. [Google Scholar] [CrossRef]
- Bargal, R.; Avidan, N.; Olender, T.; Ben Asher, E.; Zeigler, M.; Raas-Rothschild, A.; Frumkin, A.; Ben-Yoseph, O.; Friedlender, Y.; Lancet, D.; et al. Mucolipidosis type IV: Novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. Hum. Mutat. 2001, 17, 397–402. [Google Scholar] [CrossRef] [PubMed]
- LaPlante, J.M.; Sun, M.; Falardeau, J.; Dai, D.; Brown, E.M.; Slaugenhaupt, S.A.; Vassilev, P.M. Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol. Genet. Metab. 2006, 89, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Muallem, S.; Kim, S.H.; Kwon, K.B.; Kim, M.S. Exosomal release through TRPML1-mediated lysosomal exocytosis is required for adipogenesis. Biochem. Biophys. Res. Commun. 2019, 510, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.; van der Spoel, A.; Fornerod, M.; Grosveld, G.; d’Azzo, A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996, 10, 3156–3169. [Google Scholar] [CrossRef]
- Rottier, R.J.; Bonten, E.; d’Azzo, A. A point mutation in the neu-1 locus causes the neuraminidase defect in the SM/J mouse. Hum. Mol. Genet. 1998, 7, 313–321. [Google Scholar] [CrossRef]
- Yogalingam, G.; Bonten, E.J.; van de Vlekkert, D.; Hu, H.; Moshiach, S.; Connell, S.A.; d’Azzo, A. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev. Cell 2008, 15, 74–86. [Google Scholar] [CrossRef]
- van de Vlekkert, D.; Demmers, J.; Nguyen, X.X.; Campos, Y.; Machado, E.; Annunziata, I.; Hu, H.; Gomero, E.; Qiu, X.; Bongiovanni, A.; et al. Excessive exosome release is the pathogenic pathway linking a lysosomal deficiency to generalized fibrosis. Sci. Adv. 2019, 5, eaav3270. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, G.; Gahl, W.A. Cystinosis: The evolution of a treatable disease. Pediatr. Nephrol. 2013, 28, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Naphade, S.; Sharma, J.; Gaide Chevronnay, H.P.; Shook, M.A.; Yeagy, B.A.; Rocca, C.J.; Ur, S.N.; Lau, A.J.; Courtoy, P.J.; Cherqui, S. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 2015, 33, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef]
- Chang, T.Y.; Reid, P.C.; Sugii, S.; Ohgami, N.; Cruz, J.C.; Chang, C.C. Niemann-Pick type C disease and intracellular cholesterol trafficking. J. Biol. Chem. 2005, 280, 20917–20920. [Google Scholar] [CrossRef]
- Strauss, K.; Goebel, C.; Runz, H.; Mobius, W.; Weiss, S.; Feussner, I.; Simons, M.; Schneider, A. Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J. Biol. Chem. 2010, 285, 26279–26288. [Google Scholar] [CrossRef]
- Tatiana, S.; Stanislav, N.; Darya, K.; Luiza, G.; Konstantin, S.; Sergey, L.; Elena, V.; Galina, S.; Nikolai, V.; Arthur, K.; et al. Altered level of plasma exosomes in patients with Gaucher disease. Eur. J. Med. Genet. 2020, 63, 104038. [Google Scholar] [CrossRef] [PubMed]
- Sarko, D.K.; McKinney, C.E. Exosomes: Origins and Therapeutic Potential for Neurodegenerative Disease. Front. Neurosci. 2017, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, M.J.; Maegawa, G.H.B. CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges. Front. Mol. Biosci. 2020, 7, 559804. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Choi, H.; Yim, N.; Ryu, S.-W.; Choi, C. Exosome-based delivery of glucocerebrosidase lysosomal enzyme for treatment of Gaucher disease. Mol. Genet. Metab. 2018, 123, S31–S32. [Google Scholar] [CrossRef]
- Do, M.A.; Levy, D.; Brown, A.; Marriott, G.; Lu, B. Targeted delivery of lysosomal enzymes to the endocytic compartment in human cells using engineered extracellular vesicles. Sci. Rep. 2019, 9, 17274. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Harrison, E.B.; Zhao, Y.; Kabanov, A.V.; Batrakova, E.V. TPP1 Delivery to Lysosomes with Extracellular Vesicles and their Enhanced Brain Distribution in the Animal Model of Batten Disease. Adv. Healthc. Mater. 2019, 8, e1801271. [Google Scholar] [CrossRef] [PubMed]
- Zipkin, M. Exosome redux. Nat. Biotechnol. 2019, 37, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Carnino, J.M.; Lee, H.; Jin, Y. Isolation and characterization of extracellular vesicles from Broncho-alveolar lavage fluid: A review and comparison of different methods. Respir. Res. 2019, 20, 240. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Kenney, D.L.; Benarroch, E.E. The autophagy-lysosomal pathway: General concepts and clinical implications. Neurology 2015, 85, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef]
- Machado, E.; White-Gilbertson, S.; van de Vlekkert, D.; Janke, L.; Moshiach, S.; Campos, Y.; Finkelstein, D.; Gomero, E.; Mosca, R.; Qiu, X.; et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 2015, 1, e1500603. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Chatrchyan, S.; Khachatryan, V.; Sirunyan, A.M.; Tumasyan, A.; Adam, W.; Bergauer, T.; Dragicevic, M.; Ero, J.; Fabjan, C.; Friedl, M.; et al. Search for new physics with a monojet and missing transverse energy in pp collisions at radicals = 7 TeV. Phys. Rev. Lett. 2011, 107, 201804. [Google Scholar] [CrossRef] [PubMed]
- Slagsvold, T.; Pattni, K.; Malerod, L.; Stenmark, H. Endosomal and non-endosomal functions of ESCRT proteins. Trends Cell Biol. 2006, 16, 317–326. [Google Scholar] [CrossRef]
- Kolba, M.D.; Dudka, W.; Zareba-Koziol, M.; Kominek, A.; Ronchi, P.; Turos, L.; Chroscicki, P.; Wlodarczyk, J.; Schwab, Y.; Klejman, A.; et al. Tunneling nanotube-mediated intercellular vesicle and protein transfer in the stroma-provided imatinib resistance in chronic myeloid leukemia cells. Cell Death Dis. 2019, 10, 817. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and Functions in Brain Pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K. Cortical neuron specification: It has its time and place. Neuron 2005, 46, 373–376. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, E.; Coumans, F.; Varga, Z.; Krumrey, M.; Nieuwland, R. Innovation in detection of microparticles and exosomes. J. Thromb Haemost 2013, 11 (Suppl. 1), 36–45. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| LSD | Metabolite Accumulation | Consequence |
|---|---|---|
| Metachromatic leukodystrophy | Sulfatide (spingolipid-3-O-sulfogalactosylceramide) | Increased lysosomal exocytosis |
| Mucolipidosis type IV | Phospholipids, gangliosides | Increased lysosomal exocytosis |
| Sialidosis | Sialylated oligosaccharides and glycopeptides | Increased lysosomal exocytosis Identified NEU1 as a negative regulator in lysosomal exocytosis |
| Cystinosis | Cystine | Microvesicles/exosomes containing wildtype CTNS protein can correct |
| Niemann-Pick type C | Cholesterol | Increased exosomal-cholesterol secretion in vitro |
| Gaucher disease |
Glucosylceramide, glucosylsphingosine | Increased number of exosomes, aberrant morphology |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gleason, A.M.; Woo, E.G.; McKinney, C.; Sidransky, E. The Role of Exosomes in Lysosomal Storage Disorders. Biomolecules 2021, 11, 576. https://doi.org/10.3390/biom11040576
Gleason AM, Woo EG, McKinney C, Sidransky E. The Role of Exosomes in Lysosomal Storage Disorders. Biomolecules. 2021; 11(4):576. https://doi.org/10.3390/biom11040576
Chicago/Turabian StyleGleason, Adenrele M., Elizabeth G. Woo, Cindy McKinney, and Ellen Sidransky. 2021. "The Role of Exosomes in Lysosomal Storage Disorders" Biomolecules 11, no. 4: 576. https://doi.org/10.3390/biom11040576
APA StyleGleason, A. M., Woo, E. G., McKinney, C., & Sidransky, E. (2021). The Role of Exosomes in Lysosomal Storage Disorders. Biomolecules, 11(4), 576. https://doi.org/10.3390/biom11040576