
Role of Cdkn2a in the Emery–Dreifuss Muscular Dystrophy Cardiac Phenotype

 , , , ,
, , , ,
Abstract
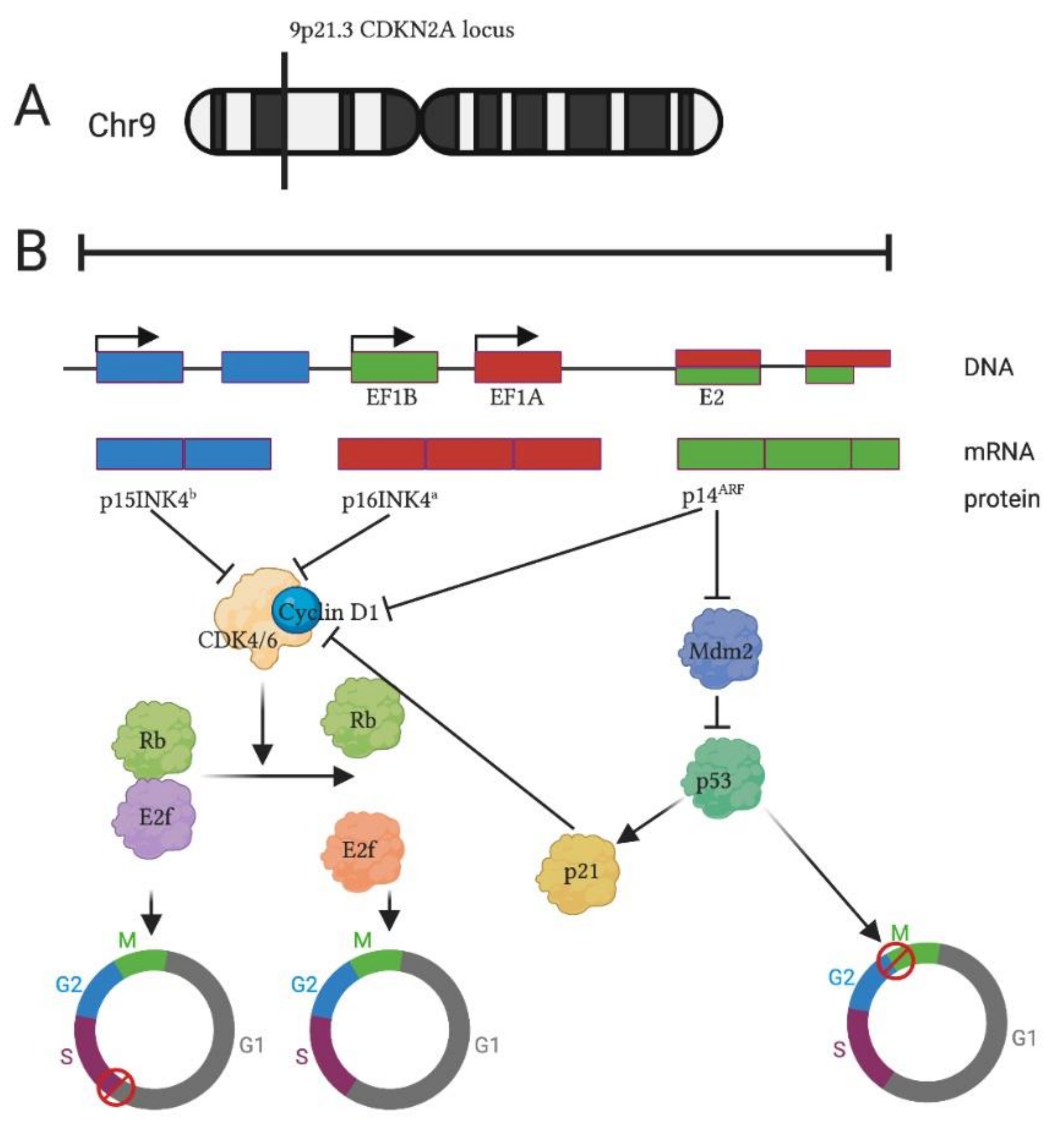
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Mice Genotyping
2.3. Survival and Weight Control
2.4. Immunohistochemistry
2.5. Real-Time PCR
2.6. Ecocardiography Measurements
2.7. Statistics
2.8. Graphics
3. Results
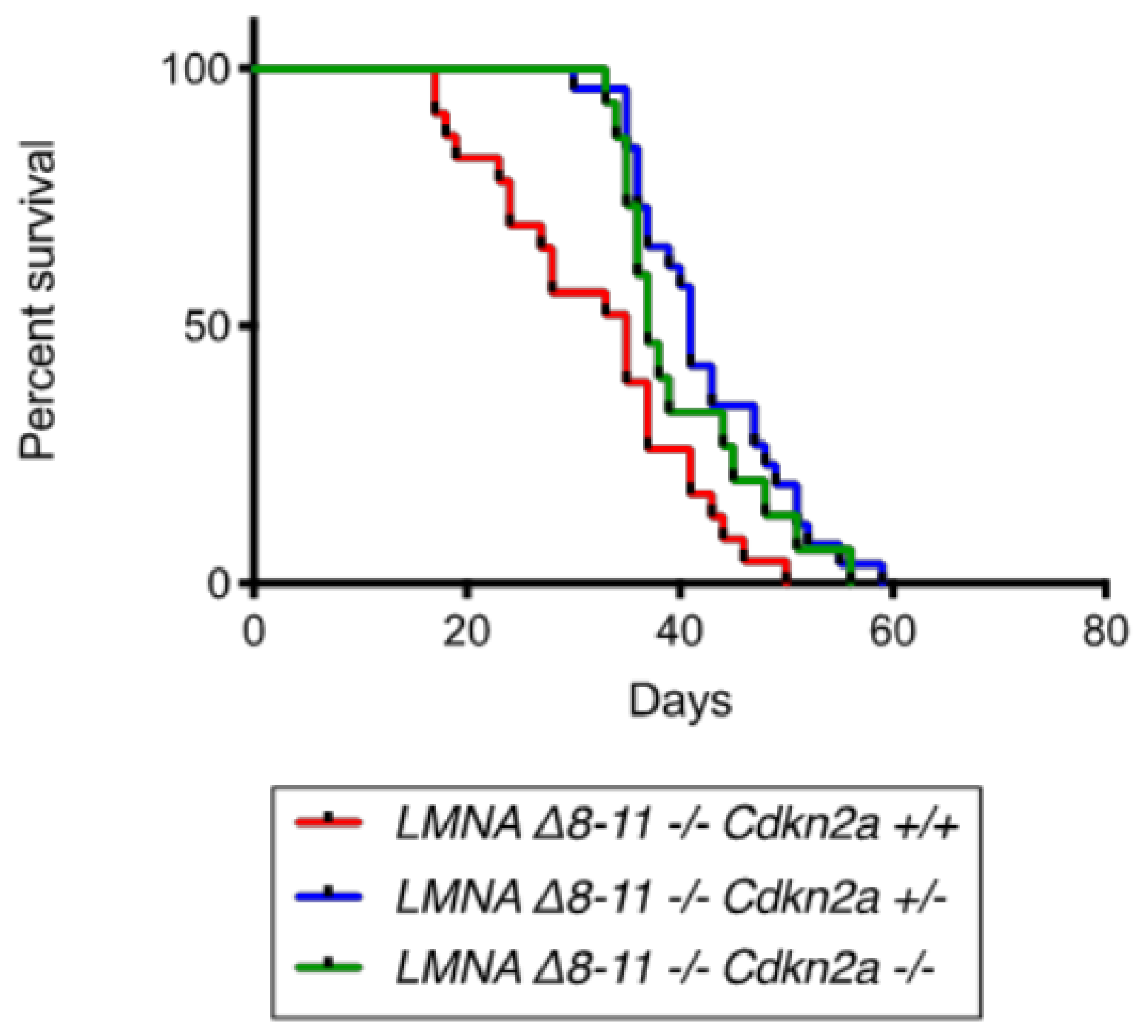
3.1. Cdkn2a KO Ameliorates Life Span of Dystrophic LMNA Δ8–11 −/− Mice
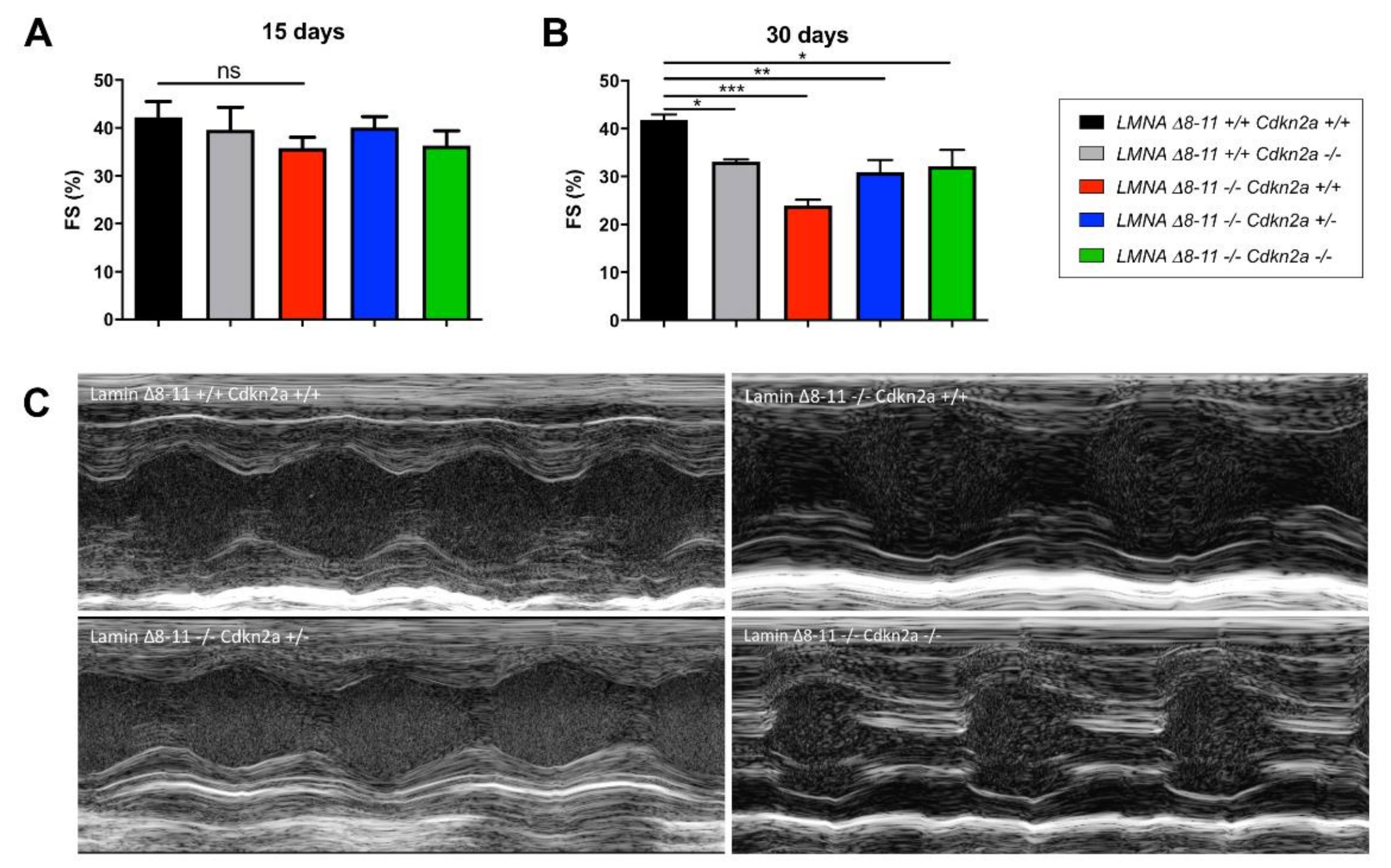
3.2. Cdkn2a KO Improves the Cardiac Function of Dystrophic LMNA Δ8–11 −/− Mice
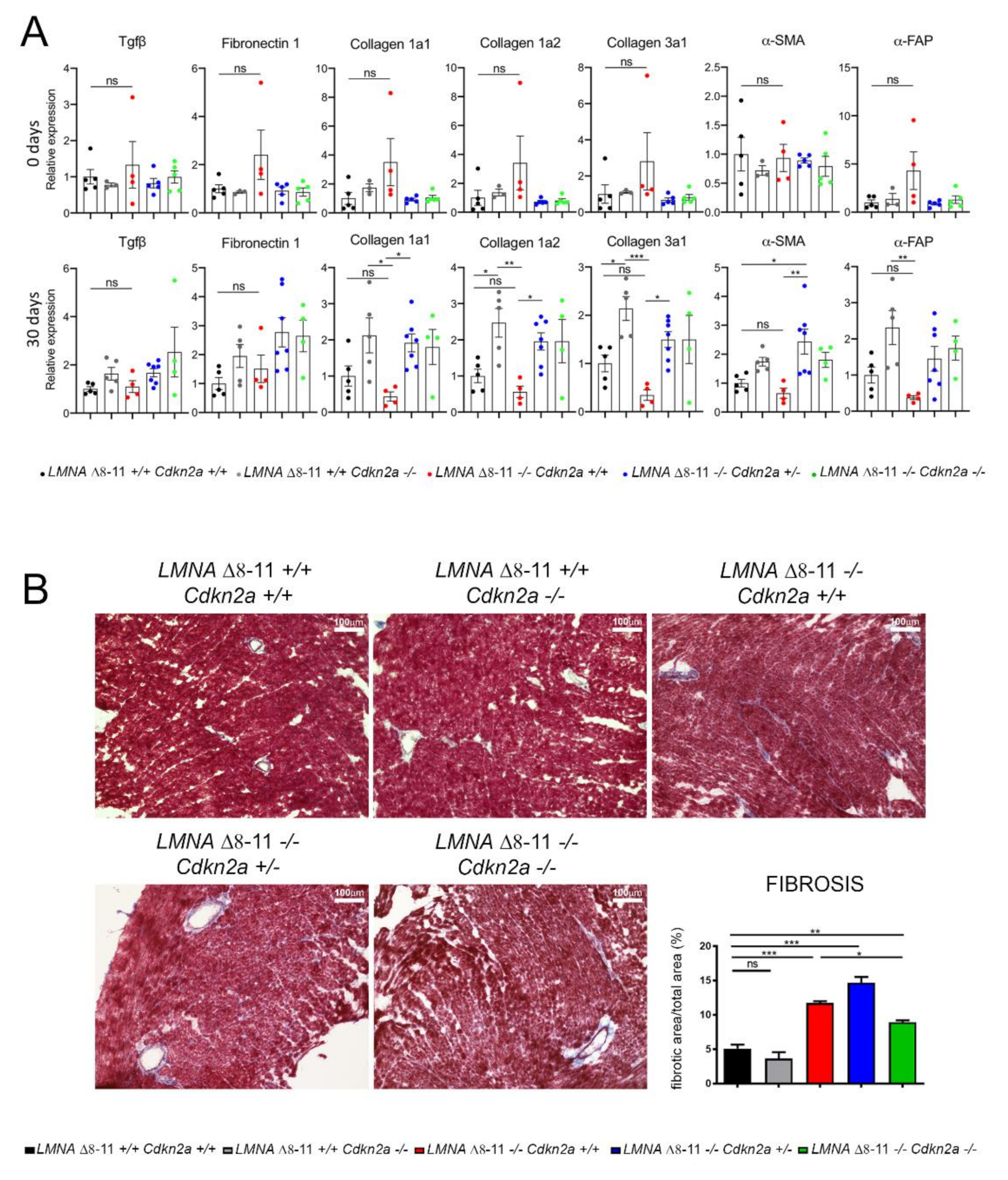
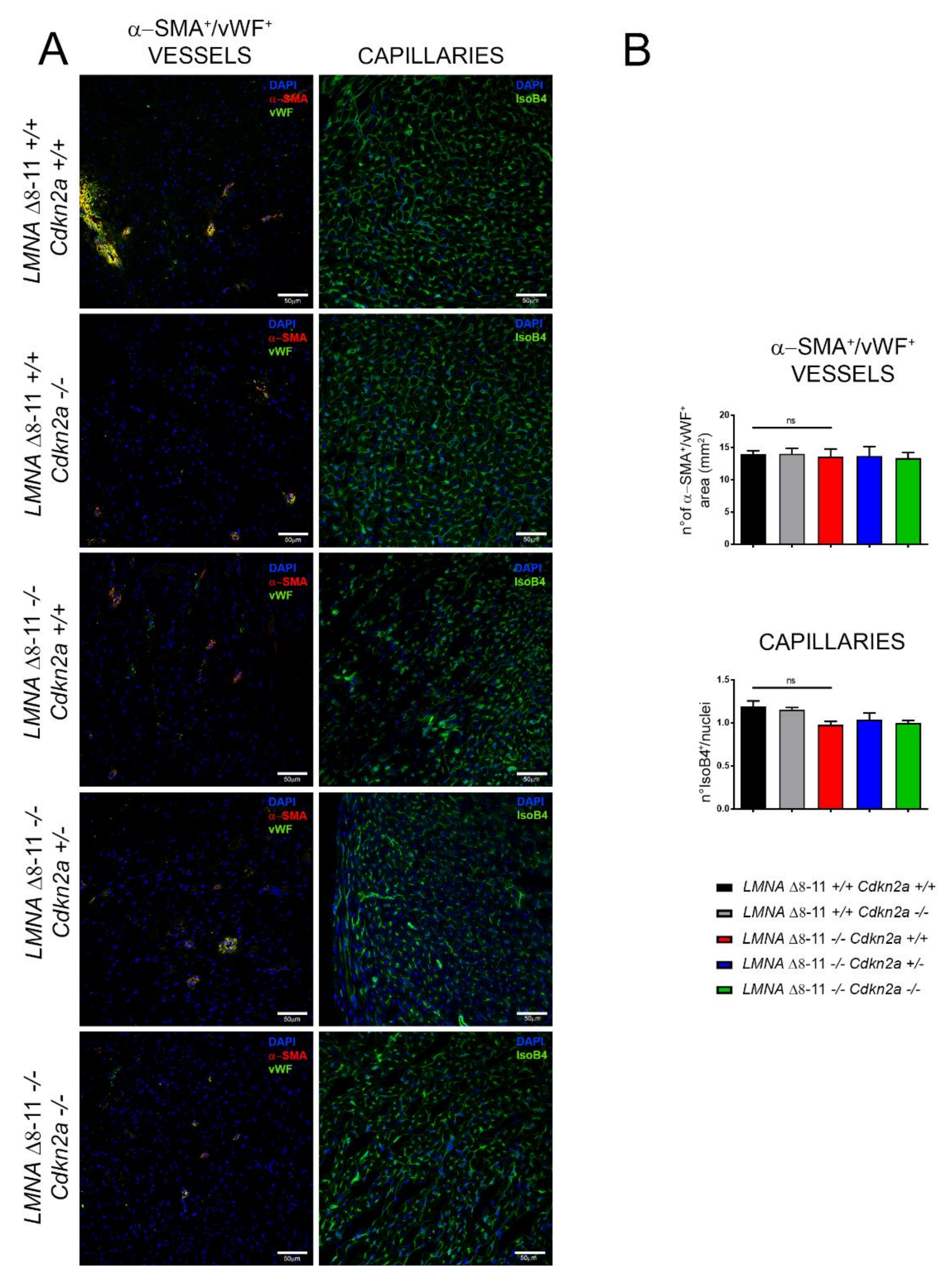
3.3. LMNA Δ8–11 −/− Mice Accumulate Fibrosis during Postnatal Heart Development
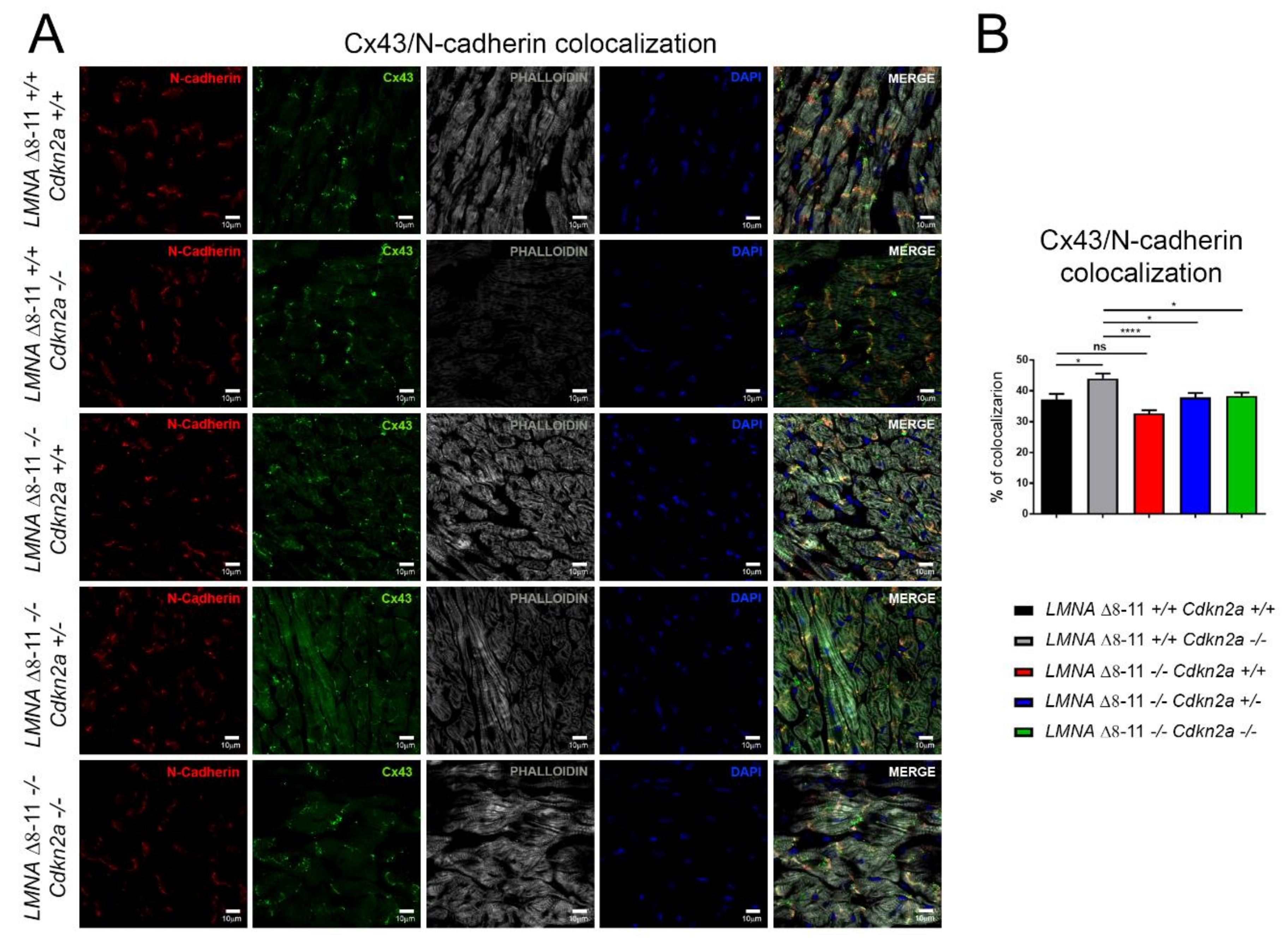
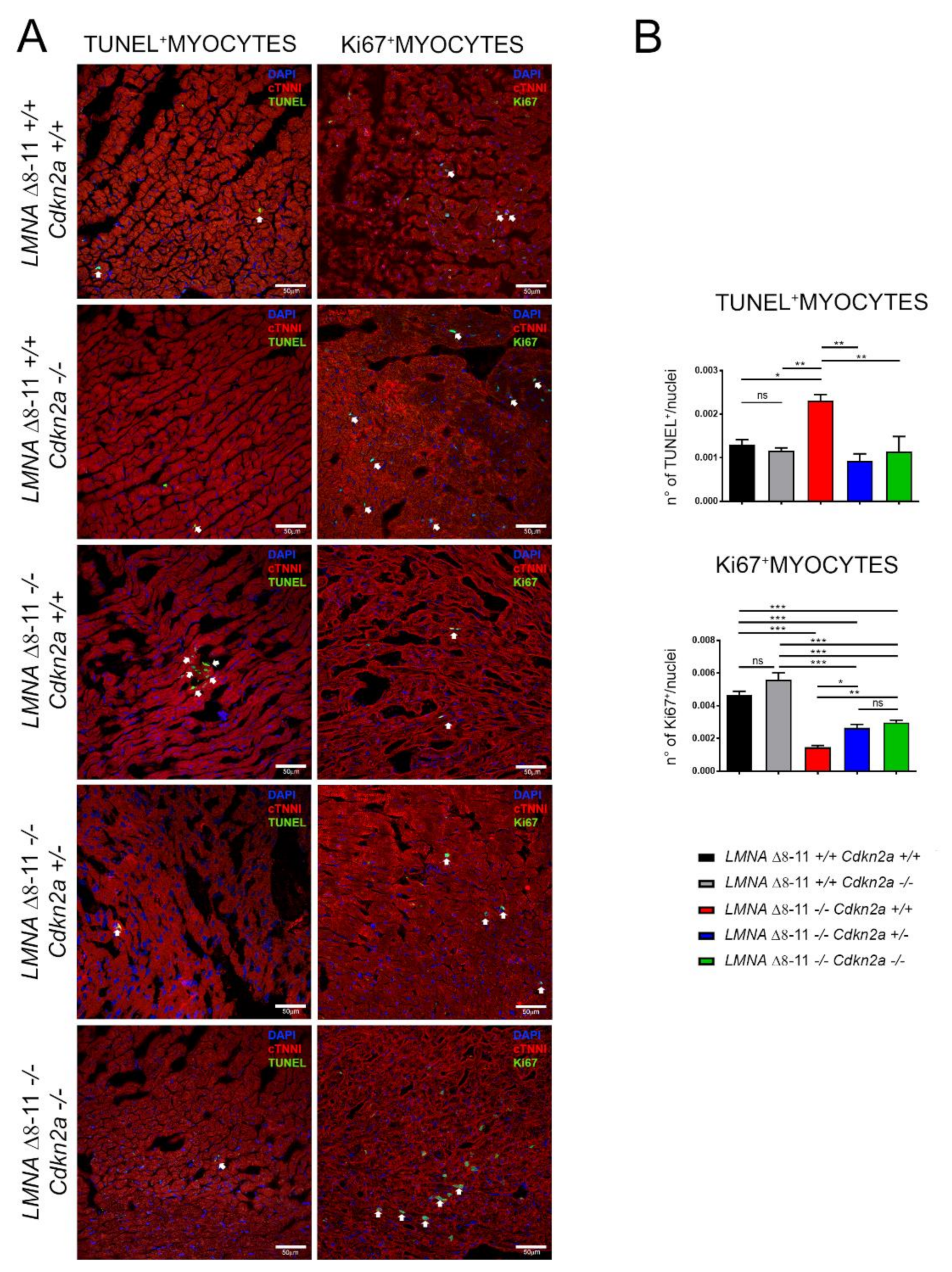
3.4. Alteration of LMNA Δ8–11 −/− Cardiac Tissue Is Partially Recovered in a Cdkn2a KO Background
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Serra, S.; Chetty, R. p16. J. Clin. Pathol. 2018, 71, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Sharpless, N.E. The Regulation of INK4/ARF in Cancer and Aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Ramon y Cajal, S. p16Ink4a overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Poi, M.J.; Tsai, M.-D. Regulatory Mechanisms of Tumor Suppressor P16 INK4A and Their Relevance to Cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef]
- Sharpless, N.E.; DePinho, R.A. The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 1999, 9, 22–30. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef]
- Heenan, P.R.; Wang, X.; Gooding, A.R.; Cech, T.R.; Perkins, T.T. Bending and looping of long DNA by Polycomb repressive complex 2 revealed by AFM imaging in liquid. Nucleic Acids Res. 2020, 48, 2969–2981. [Google Scholar] [CrossRef]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef]
- Lanzuolo, C.; Roure, V.; Dekker, J.; Bantignies, F.; Orlando, V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat. Cell Biol. 2007, 9, 1167–1174. [Google Scholar] [CrossRef]
- Gonzalez, S.; Serrano, M. A New Mechanism of Inactivation of the INK4/ARF Locus. Cell Cycle 2006, 5, 1382–1384. [Google Scholar] [CrossRef]
- Minami, R.; Muta, K.; Umemura, T.; Motomura, S.; Abe, Y.; Nishimura, J.; Nawata, H. p16INK4a induces differentiation and apoptosis in erythroid lineage cells. Exp. Hematol. 2003, 31, 355–362. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Sherr, C.J. Ink4-Arf locus in cancer and aging. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 731–741. [Google Scholar] [CrossRef]
- Gan, Q.; Huang, J.; Zhou, R.; Niu, J.; Zhu, X.; Wang, J.; Zhang, Z.; Tong, T. PPAR accelerates cellular senescence by inducing p16INK4 expression in human diploid fibroblasts. J. Cell Sci. 2008, 121, 2235–2245. [Google Scholar] [CrossRef]
- Bengal, E.; Perdiguero, E.; Serrano, A.L.; Muñoz-Cánoves, P. Rejuvenating stem cells to restore muscle regeneration in aging. F1000Research 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Gutarra, S.; García-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardí, M.; Ballestar, E.; González, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef]
- Carlson, M.E.; Hsu, M.; Conboy, I.M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008, 454, 528–532. [Google Scholar] [CrossRef]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef]
- Hatzistergos, K.E.; Williams, A.R.; Dykxhoorn, D.; Bellio, M.A.; Yu, W.; Hare, J.M. Tumor Suppressors RB1 and CDKN2a Cooperatively Regulate Cell-Cycle Progression and Differentiation During Cardiomyocyte Development and Repair. Circ. Res. 2019, 124, 1184–1197. [Google Scholar] [CrossRef]
- An, S.; Chen, Y.; Gao, C.; Qin, B.; Du, X.; Meng, F.; Qi, Y. Inactivation of INK4a and ARF induces myocardial proliferation and improves cardiac repair following ischemia.reperfusion. Mol. Med. Rep. 2015, 12, 5911–5916. [Google Scholar] [CrossRef]
- Lewis, J.L.; Chinswangwatanakul, W.; Zheng, B.; Marley, S.B.; Nguyen, D.X.; Cross, N.C.P.; Banerji, L.; Glassford, J.; Thomas, N.S.B.; Goldman, J.M.; et al. The influence of INK4 proteins on growth and self-renewal kinetics of hematopoietic progenitor cells. Blood 2001, 97, 2604–2610. [Google Scholar] [CrossRef]
- Bianchi, A.; Mozzetta, C.; Pegoli, G.; Lucini, F.; Valsoni, S.; Rosti, V.; Petrini, C.; Cortesi, A.; Gregoretti, F.; Antonelli, L.; et al. Dysfunctional polycomb transcriptional repression contributes to lamin A/C–dependent muscular dystrophy. J. Clin. Investig. 2020, 130, 2408–2421. [Google Scholar] [CrossRef]
- Pajcini, K.V.; Corbel, S.Y.; Sage, J.; Pomerantz, J.H.; Blau, H.M. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 2010, 7, 198–213. [Google Scholar] [CrossRef]
- Pegoli, G.; Lucini, F.; Mozzetta, C.; Lanzuolo, C. Single myofiber isolation and culture from a murine model of emery-dreifuss muscular dystrophy in early post-natal development. J. Vis. Exp. 2020. [Google Scholar] [CrossRef]
- Kandert, S.; Wehnert, M.; Müller, C.R.; Buendia, B.; Dabauvalle, M.-C. Impaired nuclear functions lead to increased senescence and inefficient differentiation in human myoblasts with a dominant p.R545C mutation in the LMNA gene. Eur. J. Cell Biol. 2009, 88, 593–608. [Google Scholar] [CrossRef]
- Cohen, T.V.; Gnocchi, V.F.; Cohen, J.E.; Aditi, P.; Liu, H.; Ellis, J.A.; Foisner, R.; Stewart, C.L.; Zammit, P.S.; Partridge, T.A. Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of lap2α. Hum. Mol. Genet. 2013, 22, 2852–2869. [Google Scholar] [CrossRef]
- Bonne, G.; Quijano-roy, S. Emery-Dreifuss Muscular Dystrophy, Laminopathies, and Other Nuclear Envelopathies. Handb. Clin. Neurol. 2013, 113, 1367–1376. [Google Scholar] [CrossRef]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Bécane, H.-M.; Hammouda, E.-H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.-A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef]
- Bianchi, A.; Manti, P.G.; Lucini, F.; Lanzuolo, C. Mechanotransduction, nuclear architecture and epigenetics in Emery Dreifuss Muscular Dystrophy: Tous pour un, un pour tous. Nucleus 2018, 9, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S.; et al. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Marullo, F.; Cesarini, E.; Antonelli, L.; Gregoretti, F.; Oliva, G.; Lanzuolo, C. Nucleoplasmic Lamin A/C and Polycomb group of proteins: An evolutionarily conserved interplay. Nucleus 2016, 7, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Sebestyén, E.; Marullo, F.; Lucini, F.; Petrini, C.; Bianchi, A.; Valsoni, S.; Olivieri, I.; Antonelli, L.; Gregoretti, F.; Oliva, G.; et al. SAMMY-seq reveals early alteration of heterochromatin and deregulation of bivalent genes in Hutchinson-Gilford Progeria Syndrome. Nat. Commun. 2020, 11, 6274. [Google Scholar] [CrossRef]
- Lu, J.T.; Muchir, A.; Nagy, P.L.; Worman, H.J. LMNA cardiomyopathy: Cell biology and genetics meet clinical medicine. Dis. Model. Mech. 2011, 4, 562–568. [Google Scholar] [CrossRef]
- Becane, H.-M.; Bonne, G.; Varnous, S.; Muchir, A.; Ortega, V.; Hammouda, E.H.; Urtizberea, J.-A.; Lavergne, T.; Fardeau, M.; Eymard, B.; et al. High Incidence of Sudden Death with Conduction System and Myocardial Disease Due to Lamins A and C Gene Mutation. Pacing Clin. Electrophysiol. 2000, 23, 1661–1666. [Google Scholar] [CrossRef]
- Karkkainen, S. A novel mutation, Ser143Pro, in the lamin A/C gene is common in finnish patients with familial dilated cardiomyopathy. Eur. Heart J. 2004, 25, 885–893. [Google Scholar] [CrossRef]
- De Backer, J.; Van Beeumen, K.; Loeys, B.; Duytschaever, M. Expanding the phenotype of sudden cardiac death—An unusual presentation of a family with a Lamin A/C mutation. Int. J. Cardiol. 2010, 138, 97–99. [Google Scholar] [CrossRef]
- Van der Kooi, A.J.; Ledderhof, T.M.; DeVoogt, W.G.; Res, J.C.J.; Bouwsma, G.; Troost, D.; Busch, H.F.M.; Becker, A.E.; DeVisser, M. A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann. Neurol. 1996, 39, 636–642. [Google Scholar] [CrossRef]
- Van Der Kooi, A.J.; Van Meegen, M.; Ledderhof, T.M.; McNally, E.M.; De Visser, M.; Bolhuis, P.A. Genetic localization of a newly recognized autosomal dominant limb- girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11-21. Am. J. Hum. Genet. 1997, 60, 891–895. [Google Scholar]
- Rudnik-Schöneborn, S.; Botzenhart, E.; Eggermann, T.; Senderek, J.; Schoser, B.G.H.; Schröder, R.; Wehnert, M.; Wirth, B.; Zerres, K. Mutations of the LMNA gene can mimic autosomal dominant proximal spinal muscular atrophy. Neurogenetics 2007, 8, 137–142. [Google Scholar] [CrossRef]
- Boriani, G.; Wollmann, C.; Biffi, M.; Kuhl, M.; Schuchert, A.; Sperzel, J.; Stiller, S.; Gasparini, G.; Bocker, D. Evaluation of a Dual Chamber Implantable Cardioverter Defibrillator for the Treatment of Atrial and Ventricular Arrhythmias. Pacing Clin. Electrophysiol. 2003, 26, 461–465. [Google Scholar] [CrossRef]
- Sanna, T.; Dello Russo, A.; Toniolo, D.; Vytopil, M.; Pelargonio, G.; De Martino, G.; Ricci, E.; Silvestri, G.; Giglio, V.; Messano, L.; et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur. Heart J. 2003. [Google Scholar] [CrossRef]
- Sakata, K.; Shimizu, M.; Ino, H.; Yamaguchi, M.; Terai, H.; Fujino, N.; Hayashi, K.; Kaneda, T.; Inoue, M.; Oda, Y.; et al. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation 2005, 111, 3352–3358. [Google Scholar] [CrossRef]
- Astejada, M.N.; Goto, K.; Nagano, A.; Ura, S.; Noguchi, S.; Nonaka, I.; Nishino, I.; Hayashi, Y.K. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol. 2007, 26, 159–164. [Google Scholar]
- Carboni, N.; Mura, M.; Mercuri, E.; Marrosu, G.; Manzi, R.C.; Cocco, E.; Nissardi, V.; Isola, F.; Mateddu, A.; Solla, E.; et al. Cardiac and muscle imaging findings in a family with X-linked Emery–Dreifuss muscular dystrophy. Neuromuscul. Disord. 2012, 22, 152–158. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Becane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Arimura, T.; Helbling-Leclerc, A.; Massart, C.; Varnous, S.; Niel, F.; Lacène, E.; Fromes, Y.; Toussaint, M.; Mura, A.M.; Kelle, D.I.; et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005, 14, 155–169. [Google Scholar] [CrossRef]
- Vignier, N.; Mougenot, N.; Bonne, G.; Muchir, A. Effect of genetic background on the cardiac phenotype in a mouse model of Emery-Dreifuss muscular dystrophy. Biochem. Biophys. Rep. 2019, 19, 100664. [Google Scholar] [CrossRef]
- Le Dour, C.; Macquart, C.; Sera, F.; Homma, S.; Bonne, G.; Morrow, J.P.; Worman, H.J.; Muchir, A. Decreased WNT/β-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 2017, 26, ddw389. [Google Scholar] [CrossRef]
- Muchir, A.; Wu, W.; Choi, J.C.; Iwata, S.; Morrow, J.; Homma, S.; Worman, H.J. Abnormal p38 mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2012, 21, 4325–4333. [Google Scholar] [CrossRef]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117. [Google Scholar] [CrossRef]
- Muchir, A.; Wu, W.; Worman, H.J. Mitogen-Activated Protein Kinase Inhibitor Regulation of Heart Function and Fibrosis in Cardiomyopathy Caused by Lamin A/C Gene Mutation. Trends Cardiovasc. Med. 2010, 20, 217–221. [Google Scholar] [CrossRef]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef]
- Wu, W.; Shan, J.; Bonne, G.; Worman, H.J.; Muchir, A. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim. Biophys. Acta 2010, 1802, 632–638. [Google Scholar] [CrossRef]
- Wu, W.; Muchir, A.; Shan, J.; Bonne, G.; Worman, H.J. Mitogen-Activated Protein Kinase Inhibitors Improve Heart Function and Prevent Fibrosis in Cardiomyopathy Caused by Mutation in Lamin A/C Gene. Circulation 2011, 123, 53–61. [Google Scholar] [CrossRef]
- Wu, W.; Chordia, M.D.; Hart, B.P.; Kumarasinghe, E.S.; Ji, M.K.; Bhargava, A.; Lawlor, M.W.; Shin, J.; Sera, F.; Homma, S.; et al. Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by lamin A/C gene mutation. Bioorg. Med. Chem. 2017, 25, 1004–1013. [Google Scholar] [CrossRef][Green Version]
- Muchir, A.; Wu, W.; Sera, F.; Homma, S.; Worman, H.J. Mitogen-activated protein kinase kinase 1/2 inhibition and angiotensin II converting inhibition in mice with cardiomyopathy caused by lamin A/C gene mutation. Biochem. Biophys. Res. Commun. 2014, 452, 958–961. [Google Scholar] [CrossRef]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H.; et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 2019, 572, 335–340. [Google Scholar] [CrossRef]
- Chen, S.N.; Lombardi, R.; Karmouch, J.; Tsai, J.-Y.; Czernuszewicz, G.; Taylor, M.R.G.; Mestroni, L.; Coarfa, C.; Gurha, P.; Marian, A.J. DNA Damage Response/TP53 Pathway Is Activated and Contributes to the Pathogenesis of Dilated Cardiomyopathy Associated With LMNA (Lamin A/C) Mutations. Circ. Res. 2019, 124, 856–873. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. Lamin A/C Cardiomyopathy: Implications for Treatment. Curr. Cardiol. Rep. 2019, 21, 160. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of a-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C–deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef]
- Wolf, C.M.; Wang, L.; Alcalai, R.; Pizard, A.; Burgon, P.G.; Ahmad, F.; Sherwood, M.; Branco, D.M.; Wakimoto, H.; Fishman, G.I.; et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell. Cardiol. 2008, 44, 293–303. [Google Scholar] [CrossRef]
- Serrano, M.; Lee, H.W.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef]
- Silva, M.C.; Magalhães, T.A.; Meira, Z.M.A.; Rassi, C.H.R.E.; Andrade, A.C.D.S.; Gutierrez, P.S.; Azevedo, C.F.; Gurgel-Giannetti, J.; Vainzof, M.; Zatz, M.; et al. Myocardial Fibrosis Progression in Duchenne and Becker Muscular Dystrophy. JAMA Cardiol. 2017, 2, 190. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef]
- Madej-Pilarczyk, A. Clinical aspects of emery-dreifuss muscular dystrophy. Nucleus 2018, 9, 314–320. [Google Scholar] [CrossRef]
- Himelman, E.; Lillo, M.A.; Nouet, J.; Patrick Gonzalez, J.; Zhao, Q.; Xie, L.H.; Li, H.; Liu, T.; Wehrens, X.H.T.; Lampe, P.D.; et al. Prevention of connexin-43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J. Clin. Investig. 2020, 130, 1713–1717. [Google Scholar] [CrossRef]
- Macquart, C.; Jüttner, R.; Morales Rodriguez, B.; Le Dour, C.; Lefebvre, F.; Chatzifrangkeskou, M.; Schmitt, A.; Gotthardt, M.; Bonne, G.; Muchir, A. Microtubule cytoskeleton regulates Connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef]
- Duffy, H.S. The molecular mechanisms of gap junction remodeling. Hear. Rhythm 2012. [Google Scholar] [CrossRef]
- Hertig, C.M.; Eppenberger-Eberhardt, M.; Koch, S.; Eppenberger, H.M. N-cadherin in adult rat cardiomyocytes in culture. I. Functional role of N-cadherin and impairment of cell-cell contact by a truncated N-cadherin mutant. J. Cell Sci. 1996, 109, 1–10. [Google Scholar]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisén, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef]
- Weinberger, F.; Eschenhagen, T. Heart regeneration: From mouse to human. Curr. Opin. Physiol. 2020, 14, 7–12. [Google Scholar] [CrossRef]
- Cui, M.; Wang, Z.; Chen, K.; Shah, A.M.; Tan, W.; Duan, L.; Sanchez-Ortiz, E.; Li, H.; Xu, L.; Liu, N.; et al. Dynamic Transcriptional Responses to Injury of Regenerative and Non-regenerative Cardiomyocytes Revealed by Single-Nucleus RNA Sequencing. Dev. Cell 2020, 53, 102–116. [Google Scholar] [CrossRef]
- Pillers, D.A.M.; Von Bergen, N.H. Emery–Dreifuss muscular dystrophy: A test case for precision medicine. Appl. Clin. Genet. 2016. [Google Scholar] [CrossRef]
- Blagova, O.; Nedostup, A.; Shumakov, D.; Poptsov, V.; Shestak, A.; Zaklyasminskaya, E. Dilated cardiomyopathy with severe arrhythmias in Emery–Dreifuss muscular dystrophy from ablation to heart transplantation. J. Atr. Fibrillation 2016. [Google Scholar] [CrossRef]
- Russo, V.; Rago, A.; Politano, L.; Papa, A.A.; Di Meo, F.; Russo, M.G.; Golino, P.; Calabrò, R.; Nigro, G. Increased dispersion of ventricular repolarization in emery dreifuss muscular dystrophy patients. Med. Sci. Monit. 2012, 18, 643–647. [Google Scholar] [CrossRef]
- Bialer, M.G.; Mcdaniel, N.L.; Kelly, T.E. Progression of cardiac disease in emery-dreifuss muscular dystrophy. Clin. Cardiol. 1991, 14, 411–416. [Google Scholar] [CrossRef]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660. [Google Scholar] [CrossRef]
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, Proliferation, and Growth of the Mammalian Heart. Mol. Ther. 2018, 26, 1599–1609. [Google Scholar] [CrossRef]
- Kubben, N.; Voncken, J.W.; Konings, G.; van Weeghel, M.; van den Hoogenhof, M.M.G.; Gijbels, M.; van Erk, A.; Schoonderwoerd, K.; van den Bosch, B.; Dahlmans, V.; et al. Post-natal myogenic and adipogenic developmental. Nucleus 2011, 2, 195–207. [Google Scholar] [CrossRef]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, R.; Campbell, K.F.; Carvalho, J.L.; Ruiz, E.C.; Kim, G.C.; Schmuck, E.G.; Raval, A.N.; da Rocha, A.M.; Herron, T.J.; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 2019, 10, 2238. [Google Scholar] [CrossRef]
- Furtado, M.B.; Nim, H.T.; Boyd, S.E.; Rosenthal, N.A. View from the heart: Cardiac fibroblasts in development, scarring and regeneration. Development 2016, 143, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Lajiness, J.D.; Conway, S.J. The Dynamic Role of Cardiac Fibroblasts in Development and Disease. J. Cardiovasc. Transl. Res. 2012, 5, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, H.; Song, X.; Clifton, A.C.; Xiao, J. The potential role of senescence in limiting fibrosis caused by aging. J. Cell. Physiol. 2020, 235, 4046–4059. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | Mouse | |
|---|---|---|
| First heartbeat | 3 weeks | E7.5 |
| Four chambers visible | 1.5 months | E9.5 |
| Decline in proliferation | 2 months | E11.5 |
| Lamin A/C expression | Not addressed | E12.5 |
| Stop proliferation | 1 week after birth | 12 days after birth |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pegoli, G.; Milan, M.; Manti, P.G.; Bianchi, A.; Lucini, F.; Santarelli, P.; Bearzi, C.; Rizzi, R.; Lanzuolo, C. Role of Cdkn2a in the Emery–Dreifuss Muscular Dystrophy Cardiac Phenotype. Biomolecules 2021, 11, 538. https://doi.org/10.3390/biom11040538
Pegoli G, Milan M, Manti PG, Bianchi A, Lucini F, Santarelli P, Bearzi C, Rizzi R, Lanzuolo C. Role of Cdkn2a in the Emery–Dreifuss Muscular Dystrophy Cardiac Phenotype. Biomolecules. 2021; 11(4):538. https://doi.org/10.3390/biom11040538
Chicago/Turabian StylePegoli, Gloria, Marika Milan, Pierluigi Giuseppe Manti, Andrea Bianchi, Federica Lucini, Philina Santarelli, Claudia Bearzi, Roberto Rizzi, and Chiara Lanzuolo. 2021. "Role of Cdkn2a in the Emery–Dreifuss Muscular Dystrophy Cardiac Phenotype" Biomolecules 11, no. 4: 538. https://doi.org/10.3390/biom11040538
APA StylePegoli, G., Milan, M., Manti, P. G., Bianchi, A., Lucini, F., Santarelli, P., Bearzi, C., Rizzi, R., & Lanzuolo, C. (2021). Role of Cdkn2a in the Emery–Dreifuss Muscular Dystrophy Cardiac Phenotype. Biomolecules, 11(4), 538. https://doi.org/10.3390/biom11040538