Abstract
P14ARF (ARF; Alternative Reading Frame) is an extensively characterized tumor suppressor which, in response to oncogenic stimuli, mediates cell cycle arrest and apoptosis via p53-dependent and independent routes. ARF has been shown to be frequently lost through CpG island promoter methylation in a wide spectrum of human malignancies, such as colorectal, prostate, breast, and gastric cancers, while point mutations and deletions in the p14ARF locus have been linked with various forms of melanomas and glioblastomas. Although ARF has been mostly studied in the context of tumorigenesis, it has been also implicated in purely developmental processes, such as spermatogenesis, and mammary gland and ocular development, while it has been additionally involved in the regulation of angiogenesis. Moreover, ARF has been found to hold important roles in stem cell self-renewal and differentiation. As is often the case with tumor suppressors, ARF functions as a pleiotropic protein regulating a number of different mechanisms at the crossroad of development and tumorigenesis. Here, we provide an overview of the non-canonical functions of ARF in cancer and developmental biology, by dissecting the crosstalk of ARF signaling with key oncogenic and developmental pathways.
1. Introduction
The CDKN2A locus which is found on human chromosome 9p21 encodes two overlapping transcripts that produce two different proteins, p16INK4a and p14ARF (ARF) [1], both of which are established tumor suppressors. P16INK4a and ARF display no sequence identity, and function through distinct pathways. P16INK4a acts through inhibition of the Cyclin D-CDK4/6 complex, which maintains the Rb protein in its active form [2,3]. On the other hand, ARF acts as a sensor of various types of cellular stress, including oncogenic, heat-shock, and oxidative stress [4,5,6], to subsequently trigger either growth arrest or apoptotic mechanisms in a p53-dependent or independent fashion [7]. Interestingly, ARF has been considered to be an intermediate link between the Rb and p53 pathways, as Rb inactivation leads to increased ARF transcription, which in turn activates a p53-dependent checkpoint [5,8,9].
Early studies on the role of ARF in response to DNA damage had originally dismissed its contribution [10,11], mainly on account of the fact that p53 could still become activated in the absence of ARF, which was however elevated in response to oncogenic stimuli. These initial studies considered the DNA damage and oncogenic response as separate processes with different mediators and outcomes. However, it is now known that oncogene activation can trigger ARF signaling, readily contributing to senescence and cell cycle arrest [5,12,13,14,15,16,17,18]. Importantly, a link was established between ARF and the DNA double-strand break sensor Ataxia Telangiectasia Mutated (ATM), demonstrating that, following genotoxic stress in cancer cells, ATM negatively regulates ARF protein levels [15]. Of note, ARF was also shown to play a role in DNA single-strand break repair [19].
Regarding its established role in cancer biology, ARF was identified as a second line of defense against cancer following DNA damage response, with a higher threshold of oncogenic signals being potentially required for its activation [12]. ARF also holds a pivotal role in the Nucleotide Excision Repair (NER) pathway, facilitating the repair of UV-induced DNA lesions [20,21]. Moreover, loss of ARF was also shown to co-operate with BRAF mutations, resulting in increased UV-induced DNA damage and melanoma formation in BRAFV600E mice [22]. Although ARF is well-known for its role in stabilizing p53 levels, including those of mutant p53 [8,23], interestingly, increased ARF expression has been reported in some cases of lung cancer [24], cervical cancer [25], lymphomas [26], and cancer cell lines, such as HeLa and H1299, accompanied by p53 inactivation. Of note, in tumors such as thyroid carcinomas where ARF is upregulated, ARF is unusually found delocalized in the cytoplasm [27]. On the other hand, the role of ARF in the cytoplasm regulating cytoskeleton remodeling and cell adhesion processes has also been postulated [28], thus providing further insight into the pleiotropic roles of ARF in cancer.
In most cases of human cancers, both ARF and p16INK4a are lost, rendering it challenging to define their individual contribution to tumor suppression [9]. Alterations of the complete CDKN2A locus are identified in approximately 30% of human tumors, including glioblastoma, pancreatic cancer, adenocarcinoma, and melanoma [9,29]. The identification of chromatin remodeling events in the CDKN2A locus has recently started to delineate the genetic mechanisms governing expression of ARF and p16INK4a. A cis-element located beside the ARF promoter was recently found to down-regulate p16INK4a via long range interaction, thus providing a clear example of transcriptional regulation facilitated by chromatin folding [30].
Apart from the widely characterized canonical functions of ARF as a barrier to tumor progression, ARF was additionally implicated in other fundamental biological processes, including early development and morphogenesis. In this review, we attempt to discuss the non-canonical aspects of ARF biology by additionally highlighting important links between tumor suppression and developmental biology. We also aim to underline the interplay between ARF and key signaling pathways regulating major aspects of both cancer and stem cell biology.
2. The Role of ARF in Differentiation and Morphogenesis
ARF has been involved in numerous processes regulating cellular differentiation and organ development, such as ocular and mammary gland development, spermatogenesis, and angiogenesis. It was shown that Arf knockout results in the development of smaller eyes with no functional lenses in mice, due to incomplete maturation of the primary vitreous into the secondary vitreous [31]. ARF expression is required for hyaloid vascular system (HVS) involution, a critical process in ocular development, during which ARF prevents the accumulation of mural cells covering the blood vessels and maintains vessel stability through platelet-derived growth factor (PDGF) signaling, independently of p53 [32,33]. ARF was additionally shown to repress PDGF receptor synthesis through microRNA induction [9,34]. Of note, the phenotype of Arf -/- mice bears a strong resemblance with a human congenital ocular disorder termed Persistent Hyperplastic Primary Vitreous (PHPV), where patients also exhibit microphtalmia and lens degeneration [6,31].
ARF has been also found to determine the balance between cellular proliferation and apoptosis during murine mammary gland development [35]. Progesterone-dependent regulation of p19ARF (the p14ARF equivalent in mice) throughout pregnancy results in p19ARF induction, whereas loss of p19ARF leads to a delay in the early phase of involution and decrease in apoptosis through p21 downregulation. A second effect of p19ARF loss seems to be the immortalization of mammary epithelial cells [35]. The study by Yi et al. (2004) concluded that although p19ARF may be dispensable for mammary gland differentiation, its functional interplay with progesterone and p21 is critical for the control of the balance of cell proliferation versus apoptosis, possibly in a p53-dependent manner [35].
The process of spermatogenesis, through which spermatogonia lying on the basement membrane of seminiferous tubules are triggered to differentiate into spermatocytes, has been also found to be under ARF control. ARF is upregulated in mitotically active mouse spermatogonia, while Arf -/- male mice display testicular atrophy accompanied by p53-dependent apoptosis (anoikis) in prophase I, a paradox, given the anti-proliferative effects of ARF in somatic cells [32,36]. The functional link between ATM and ARF remains to be elucidated in male germ cell development, as ARF levels are found downregulated in spermatogonia in response to irradiation [15]. A pivotal role for ARF has been also demonstrated in facilitation of the meiotic divisions leading to spermatocyte maturation [36]. Interestingly, Mu et al. (2014) showed that spermatogonial stem cell self-renewal relies on Polycomb-repressive complex 2 (PRC2) which, through its activity in both mitotic and meiotic germ cells, represses soma-specific genes to facilitate spermatogonial differentiation in an ARF-independent mechanism [37].
ARF is not expressed in most fetal or young adult mouse tissues, however it is detected in the fetal yolk sac, a tissue derived from the extraembryonic endoderm (ExEn) [38]. ARF inactivation delays differentiation of the ExEn lineage in embryoid bodies originating either from embryonic stem cells or induced pluripotent stem cells, however, it does not affect the formation of other germ cell lineages [38]. It was shown that ARF functions via p53 to promote differentiation of the ExEn lineage [38].
Keratinocyte differentiation is another developmental process implicating ARF signaling. It was found that p63 levels were downregulated while ARF levels increased at the early stages of human keratinocyte differentiation, potentially due to ARF-mediated p63 SUMOylation and subsequent inactivation [39]. Of note, serious skin and skeleton phenotypic defects accompanying the p63-null mouse are partially rectified upon ARF silencing, suggesting that in the absence of p63, aberrant upregulation of ARF may be incompatible with skin development [40].
In addition to its role in cell maturation and organ development, ARF was shown to hold an important role in the control of angiogenesis, a process concerning both normal and pathological conditions. Kawagishi et al. (2010) showed that ARF suppresses Vascular Endothelial Growth Factor A (VEGFA) expression through the p53 pathway in mouse cell lines [41]. Subsequent studies performed in cultured and xenotransplanted human cell lines lacking functional p53 expression showed that following ATM inhibition, an inverse relationship was established between ARF and VEGF expression [6]. Those studies implied that ARF stabilization through ATM inhibition in a p53-deficient background may comprise a potential anti-angiogenic approach against cancer. In line with those observations, ARF was shown to halt angiosarcoma development, potentially in a p53-dependent fashion [42]. Moreover, an increased number of CD31-positive cells in tumors derived from Arf -/- mice strongly suggested new blood vessel formation, while endothelial cells exposed to paracrine signaling from Arf -/- macrophages displayed increased mobility, indicating that ARF deficiency promotes both macrophage infiltration and angiogenesis [43].
3. ARF in Stem Cell Biology
CDKN2A expression increases with age, followed by a decline in the tissue regenerative potential. It has been suggested that during the transition from stemness to differentiation, the CDKN2A locus is remodeled to become responsive to stress and mitogenic signals emerging in the differentiation process [9]. The regenerative potential of tissues depends on the balance between stem cell quiescence and self-renewal, both of which are critical processes implicated in tissue homeostasis. Hence, stem cell exhaustion has been considered to be a major hallmark of aging [44,45]. Aging is characterized by accumulation of chronic stress-induced cellular damage, accompanied by higher incidence of tumorigenesis. The ARF/p53 pathway, which is dormant in several tissues throughout development and postnatal life, is progressively activated from adulthood to old age in a wide spectrum of tissues and species [44]. By introducing regulatory and coding sequences of human ARF into the zebrafish genome, it was shown that ARF increases during epimorphic fin regeneration after amputation, contributing to inhibition of the regeneration process [46]. However, inhibition of ARF alone was insufficient to allow regeneration [47].
A major mechanism through which ARF contributes to stem cell regulation is through p53 stabilization. While reduced p53 activity is linked to increased stem cell self-renewal, p53 hyperactivation in mouse models has resulted in limited regeneration potential attributed to premature exhaustion of stem cell niches [44]. Thus, depletion of hematopoietic stem cells (HSCs) accompanied by impaired hematopoiesis, disruption of mammary gland morphogenesis, reduction of the neural stem cell pools, and disrupted olfactory functions have been reported in p53 mutant mouse models with higher p53 activity than wild-type counterparts [48,49,50]. In line with those phenotypes, p53 induction in the mouse epidermis through Mdm2 ablation has resulted in compromised stem cell activity and premature skin aging [51]. In contrast, mice with an extra copy of Ink4a/Arf/p53 exhibit extended lifespan and delayed aging, linked to extended preservation of stem cell populations [44,52]. Hence, it has been proposed that moderate and regulated activation of the ARF/p53 pathway during aging yields slower proliferation capacities, likely contributing to stem cell quiescence and ameliorating stem cell aging, by simultaneously preventing the exhaustion of stem cell populations. In support of this notion, p53 or p21 loss in mouse models has resulted in exit from quiescence and long-term depletion of stem cell reservoirs at advanced ages [53,54,55].
The polycomb group gene BMI1, which is required for adult stem cell maintenance in many organs [56,57], was found to regulate cell proliferation and senescence through the CDKN2A locus [58]. Jacobs et al. (1999) demonstrated that in Bmi1-deficient mouse embryonic fibroblasts and lymphocytes undergoing premature senescence, the expression of both p16INK4a and p19ARF was markedly increased, while Bmi1 overexpression led to fibroblast immortalization and a decrease in p16INK4A and p19ARF levels [58]. Depletion of the Cdkn2a locus dramatically rescued the phenotypes observed in Bmi1-deficient mice, rendering Cdkn2a critical in vivo Bmi1 target [58]. In line with this observation, it was additionally shown that Bmi1 repressed Ink4a/Arf and Hox genes to allow stem cell self-renewal in rodents [59]. A recent study confirmed the previously reported links between Bmi1 and the Cdkn2a locus, as it demonstrated that the diminished self-renewal capacity of Bmi1-deficient innate-like B lymphocytes was rescued by additional deletion of the Cdkn2a locus [60].
4. ARF Crosstalk with Major Signaling Pathways
Apart from its canonical functions as a tumor suppressor, ARF and the ARF/p53 pathway were also found to be directly or indirectly involved in the regulation of other key signaling pathways, which hold important roles in development and tumorigenesis. Those non-canonical functions of ARF signaling were identified through the various types of interactions between ARF and other pathway components or effectors.
4.1. The Hippo Pathway
The Hippo pathway is an evolutionarily conserved signaling pathway which actively controls organ growth and development, tissue regeneration, and epithelial homeostasis. Several of those functions are elicited through the terminal effectors YAP and TAZ, which direct gene expression through transcription factor binding [61]. Deregulated Hippo pathway is linked with various diseases, including tumorigenesis, rendering its components promising therapeutic targets. The tumor suppressor RASSF1A, which is epigenetically silenced in the vast majority of sporadic human malignancies, is an upstream regulator of the Hippo pathway, driving the formation of pro-apoptotic YAP–p73 complexes at the expense of pro-proliferative YAP–TEAD complexes [62,63,64]. RASSF1A has been recently found to additionally act as a “molecular switch” required for the transition from pluripotency to differentiation in the pre-implantation mouse embryo, mouse embryonic stem cells, and induced pluripotent stem cells (iPSC) [65].
ARF has been previously shown to stabilize the epithelial-to-mesenchymal transition (EMT) marker SLUG in prostate cancer [66], while it also regulates the tumor microenvironment through matrix metalloproteinase-7 (MMP7) nuclear translocation [67]. A potential crosstalk between the Hippo pathway and ARF was discovered when ARF and YAP were found co-elevated in the Pten/p53-null mouse [68]. Subsequent work demonstrated that ARF may deregulate both Hippo and Wnt pathways in human prostate cancer cells [69]. Mechanistically, ARF was shown to deregulate the Hippo pathway by stabilizing cytoplasmic YAP and preventing its nuclear accumulation, potentially through ARF-mediated YAP SUMOylation. Tea-derived carbon nanodots were shown to interact with nuclear ARF to stabilize it, and consequently contribute to YAP cytoplasmic retention, thus providing a potential avenue for targeting the pro-proliferative effects of nuclear YAP complexes [69].
The Forkhead Box (Fox) m1b (FOXM1) transcription factor, which is required for hepatocellular carcinoma (HCC) development, comprises an inhibitory ARF target [70]. Kalinichenko et al. (2004) demonstrated that a p19ARF peptide containing nine D-Arg was sufficient for inhibition of both Foxm1 transcription and Foxm1-dependent growth of osteosarcoma cells, rendering the (D-Arg)9-p19ARF 26–44 peptide a potential FOXM1 inhibitor in tumorigenesis [70]. Interestingly, Hippo pathway deregulation in cancer has been shown to drive FOXM1 activation through YAP–TEAD-dependent FOXM1 transcription, while FOXM1 additionally interacts directly with TEAD on target gene promoters, contributing to cell proliferation and tumor development [71]. Importantly, pharmacological FOXM1 inhibition limits tumor size in vivo, implying that FOXM1 can potentially serve as a therapeutic target in sarcomas [71].
Another potential link between the Hippo pathway and ARF signaling may derive from the fact that the RASSF1A scaffold protein has been shown to inhibit MDM2, similarly to ARF [72]. RASSF1A promotes MDM2 self-ubiquitination and p53 stabilization by disrupting the MDM2–DAXX–HAUSP complex in the nucleus [72]. Although the RASSF1A binding region on MDM2 is distinct from the ARF binding region [73], the E1A-regulated transcription factor E4F1 can physically interact with both ARF and RASSF1A [74,75], suggesting a potential involvement of RASSF1A on the ARF-MDM2 regulation overall. Besides its involvement in p53 stabilization, RASSF1A is also implicated in p53-mediated checkpoint activation in response to DNA damage [72]. Interestingly, RASSF1A was found to be directly activated by ATM upon DNA damage, thus prescribing the Hippo pathway with a pro-apoptotic role via p73 [76,77], whereas ATM was shown to inhibit ARF [15]. Given that RASSF1A is actively involved in YAP–p73-mediated apoptosis, ARF suppression by ATM may be a mechanism employed by cells to prevent ARF-mediated YAP inactivation, a meaningful scenario at least in the case of p53-deficient tumors.
4.2. The Wnt Pathway
The Wnt signal transduction cascade is a master regulator of biological processes throughout the development and adult life of all animals. Aberrant Wnt signaling underlies a wide range of pathological conditions in humans, including cancer [78]. The Wnt signaling cascade, which exerts its functions via β-catenin-dependent gene regulation, is an important player in maintaining stem cell self-renewal [78]. Mutations in key components of the Wnt cascade are responsible for the development of various types of cancer, such as melanomas, colorectal, and liver cancers [78,79]. Due to its major involvement in the regulation of stemness, Wnt pathway activation plays a critical role in patient-derived organoid formation, as it has been shown for several tissues such as colon, lung, and skin [80,81,82,83]. Inactivation of the Hippo pathway scaffold RASSF1A was found to be required for Wnt and Hippo pathway crosstalk resulting in maintenance of pluripotency, while RASSF1A uncouples Wnt from Hippo signaling to enable stem cell and embryonic differentiation towards all cell lineages, through p73 [65].
DNA promoter methylation of the p14ARF, RASSF1A, and APC1A genes is considered to be an independent prognostic factor in colorectal cancer patients [84]. Patients with a methylated p14ARF promoter display a significantly worse prognosis, and concurrent methylation of one or more genes from the above set has been linked with poor prognosis independent of tumor stage and differentiation status, suggesting that the pathways involved may synergize towards oncogenic progression and stem cell regulation [84,85] (Figure 1).
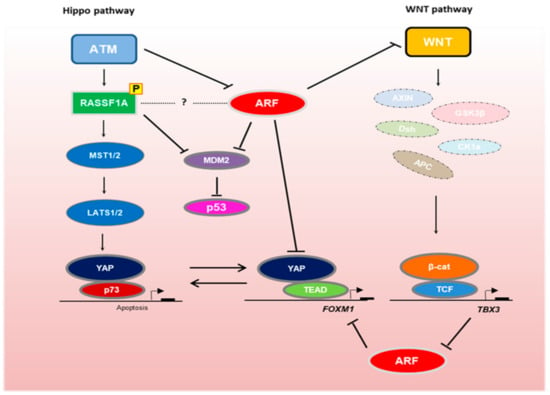
Figure 1.
Interplay between the Hippo, Wnt and ARF/p53 signaling pathways in development and tumorigenesis. The Hippo pathway scaffold RASSF1A is activated by ATM-mediated phosphorylation [76,77]. Although ATM inhibits ARF [15], both RASSF1A and ARF prevent MDM2-mediated p53 degradation. RASSF1A activation results in the formation of stable YAP–p73 transcriptional complexes, which drive stem cell differentiation and apoptosis [63,65]. In the absence of RASSF1A, YAP–TEAD complexes drive cellular proliferation via transcription of genes, such as FOXM1. FOXM1 is inhibited by ARF [70]. ARF also suppresses Wnt signaling [89,90], which upon activation destabilizes the β-catenin destruction complex, resulting in β-catenin nuclear translocation and TCF/LEF-mediated transcription of oncogenes, such as TBX3. TBX3 inhibits ARF to promote oncogenic proliferation [92].
Interestingly, the Wnt pathway was found to restrict embryonic stem cell self-renewal via activation of the CDKN2A locus [86]. The Wnt effector Tcf1 is recruited to the Cdkn2a locus of mouse embryonic stem cells (mESC), where it triggers transcription of both p16INK4a and p19ARF, which act as negative regulators of the cell cycle, thereby reducing proliferation rates without affecting the pluripotent status of the cells [86]. The anti-proliferative effect of the Wnt pathway in mESC contrasts with its documented mitogenic effects in somatic cells.
Damalas et al. (2001) showed that constitutively active β-catenin is capable of inducing an ARF/p53-dependent growth arrest and senescence-like phenotype in mouse embryonic fibroblasts; however, in the absence of either ARF or p53, β-catenin cooperates with Ras to elicit oncogenic transformation [87]. Despite demonstrating β-catenin-mediated activation of ARF and stabilization of p53, the study by Damalas et al. did not show subsequent induction of apoptosis. Other studies suggested that β-catenin likely activates survival pathways to repress the pro-apoptotic effects of the ARF/p53 pathway [16,88].
To identify downstream targets of ARF signaling, gene expression profiling was implemented in human melanoma cell lines with either wild-type or mutant ARF protein [89]. Pathway analysis of the differentially expressed genes between wild-type and mutant ARF cell lines demonstrated that Wnt pathway components may act downstream of ARF signaling, contributing to tumor development when ARF activity is compromised [89]. In line with this, an inverse relationship between ARF and Sprouty RTK Signaling Antagonist 4 (SPRY4), an evolutionarily conserved downstream target of Wnt signaling was observed, as SPRY4 levels increased in the absence of ARF [89,90]. Moreover, TBX3, which was also identified as a downstream target of the Wnt pathway and a pivotal mediator of β-catenin-dependent proliferation and survival in liver tumorigenesis [91], was also found to be upregulated in breast cancer where it represses ARF expression by interacting with histone deacetylases [92]. Of note, the use of HDAC inhibitors was able to rescue TBX3-mediated ARF repression in a dose-dependent fashion, thus providing a potential therapeutic window to disrupt Wnt-mediated ARF suppression [92].
A clear involvement of p19ARF, and not p16INK4a, in tumor escape from growth constraints such as chemotherapy, was demonstrated in a Wnt1-dependent mouse breast cancer model [93]. In order to identify genetic determinants directing tumor escape, a transgenic mouse model was employed, where targeted chemotherapy was simulated by blocking doxycycline-dependent Wnt1 transgene expression. Tumor escape was observed via reactivation of the Wnt pathway [93]. However, Cdkn2a loss, affecting both p16INK4a and p19ARF expression, resulted in tumor relapses with EMT features, without reactivation of the Wnt pathway. Interestingly, p19ARF loss alone enabled rapid EMT-driven tumor escape, while p16INK4a deficiency failed to accelerate relapse, indicating that deregulated ARF/p53 signaling may foster breast cancer relapse regardless of Wnt pathway activation [93].
Cdkn2ab knockout mice, deficient for three reading frames (p15INK4b, p19ARF, and p16INK4a), developed a wider spectrum of tumors than Cdkn2a -/- mice, with a higher proportion of skin carcinomas [94]. It was recently shown that the Wnt7b 129P2 allele was sufficient for promotion of oncogenic transformation of Cdkn2ab -/- cell lines via CDK6 activity, an identified Wnt target [95,96]. The recent study by Krimpenfort et al. (2019) suggested that loss of p15INK4b activity, which plays a fundamental role in the hair follicle cell cycle by keeping Wnt-mediated CDK6 activation in check, potentially leads to aberrant progenitor cell migration and proliferation. As p16INK4a and p19ARF present a barrier to cells that respond to these oncogenic signals, concomitant loss of p16INK4a and p19ARF may be a predisposition to skin tumorigenesis [95].
4.3. The Notch Pathway
The highly conserved Notch signaling pathway operates in various contexts across which the outcomes can widely differ [97,98]. Notch is a cell surface receptor, formed by intracellular proteolytic cleavage of a single polypeptide chain, whose two deriving subunits undergo dimerization and transport to the plasma membrane [97]. Upon binding of Notch ligands (members of the Delta/Jagged family), the Notch receptor is extracellularly cleaved by an ADAM protease, while intracellular cleavage by γ-secretase results in the release of the Notch Intracellular Domain (NICD) into the cytoplasm, where it enters the nucleus to regulate transcription of target genes [97]. Notch pathway activation can lead to different outcomes depending on the cell type and developmental stage, as it was found to promote differentiation, stem cell maintenance, or tumorigenesis, depending on the set of target genes deployed in each case [99]. Extrinsic signals may, therefore, be important for navigating Notch activity.
Two of the hallmarks of T-cell acute lymphoblastic leukemia (T-ALL) are aberrant Notch signaling and loss of the CDKN2A locus [100,101]. Interestingly, it has been shown that the intracellular domain of Notch1 results in Arf activation in T-cells, serving as a tumor suppressive mechanism [102]. Arf expression is, however, accompanied by strong selective pressure for deletion of the Cdkn2a locus in T-ALL, thereby leading to the survival of clones bypassing tumor suppression [102]. Subsequent research demonstrated that the Cdkn2a locus is epigenetically silenced in cultured T-ALL initiating cells [103]. However, in T-cell precursors targeted by aberrant Notch1 signaling for transformation, the epigenetic silencing of the Cdkn2a locus was reversed. This enables Arf expression, and again, provides selective pressure for the survival of clones with Cdkn2a loss [103]. Thus, the epigenetic status of Cdkn2a regulates the developmental stages of T-cell maturation where aberrant Notch1 function and Cdkn2a inactivation promote T-ALL [103].
The LIM-only transcription factor (LMO2) gene is subjected to chromosomal translocations in T-cell tumors, leading to abnormally increased LMO2 expression in approximately 9% of all T-ALL cases [100,104,105]. In murine T-cell malignancies, Arf inactivation promotes thymocyte self-renewal and sets the ground for the emergence of aggressive tumors [106]. The Arf promoter is not directly occupied by either Lmo2 or Notch, implying that Notch activation may be a subsequent event in T-cell oncogenesis, and Lmo2 induction functions together with Arf loss to facilitate primitive thymocyte self-renewal [106]. Importantly, aberrant Notch activation and Arf silencing independently cooperate with Lmo2 upregulation to induce T-ALL [106]. Along the same lines, Lmo2 induction promotes the growth of astrocytes isolated from Cdkn2a -/- mice and acquisition of glioma stem cell phenotypes [107]. Intriguingly, and in keeping with the role of ARF in suppressing angiogenesis [6], Lmo2 is capable of inducing angiogenesis in a Cdkn2a -/- background [107].
A critical functional link between the Notch and ARF signaling pathways is the Polycomb group gene BMI1, which actively promotes cellular proliferation and stem cell self-renewal by repressing the CDKN2A locus [58] (Figure 2). BMI1 has been found to be a downstream Notch target, and its loss in mouse intestinal stem cells (ISC) is accompanied by reduced proliferation of the ISC compartment, p16INK4a/p19ARF accumulation, and differentiation towards the goblet lineage mimicking Notch loss-of-function phenotypes [108]. Bruggeman et al. (2005) attempted to explore whether p16INK4a or p19ARF was more critical for eliciting Bmi1 activity, and demonstrated that although p19ARF comprises a general Bmi1 target, p16INK4a derepression plays a role only in a subset of cell types, such as neural stem cells, for the manifestation of Bmi1 -/- phenotypes [109]. The results of the study implied differential cell-type specific functions of p16INK4a versus p19ARF in Bmi-1-mediated cell cycle control [109]. Of note, BMI1 was also shown to promote tumorigenesis independently of ARF in human liver cancer [110]. The tumorigenic activity of BMI1 also appeared to be independent of ARF signaling in human ovarian cancer cells, where genetic or pharmacological BMI1 inhibition impaired clonal growth without affecting expression of the CDKN2A locus or ARF protein stability [111]. The proposed mechanism involved induction of autophagy through ATP depletion, as targeting of BMI1 triggered the PINK1-PARK2-dependent mitochondrial pathway, thereby leading to necroptosis [111].
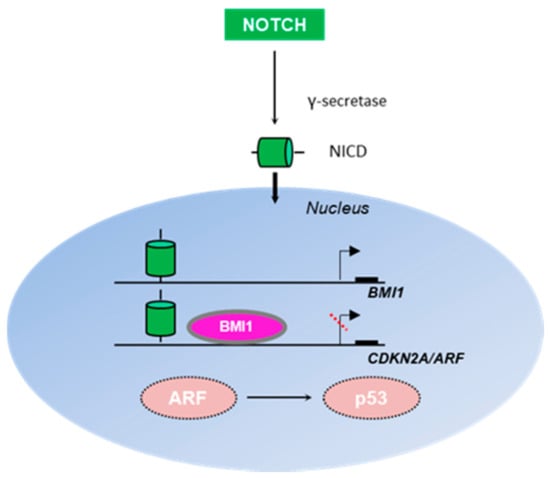
Figure 2.
Interplay between the Notch and ARF/p53 signaling pathways. Notch is a transmembrane receptor, which upon cleavage by γ-secretase produces an intracellular domain (NICD) capable of translocating to the nucleus and directly regulating gene transcription. BMI1 is activated by Notch to regulate cellular proliferation and stem cell self-renewal, especially in neural tissue [108]. Both Notch and Bmi1 directly occupy the Arf promoter to repress Arf expression [108], thus perturbing Arf-mediated p53 stabilization.
Notch was additionally reported to suppress p53 in murine T-cell lymphomagenesis through repression of Arf expression, whereas p53 was activated upon Notch signaling perturbation by γ-secretase inhibitors [112]. A subsequent study demonstrated that Notch represses Arf through direct Arf promoter occupancy, where the Polycomb Repressive Complex 2 (PRC2) was recruited in an LSD1-dependent fashion, allowing histone modifications to ultimately terminate Arf transcription (Figure 2) [113]. In contrast, Notch stimulation has been shown to activate p53 in muscle stem cells to promote muscle regeneration via Hey transcription factors [114]. However, retardation of Notch signaling was observed in aged animals, leading to p53 suppression independent of either p16INK4a or p19ARF, thus culminating in a decline in the regenerative potential of muscle cells [114].
5. Discussion
ARF is a tumor suppressor protein with an established role in important aspects of tumorigenesis, such as the DNA damage response. Apart from its well-known function as a p53 stabilizer, ARF is also implicated in p53-independent mechanisms to promote cell growth arrest [9]. Intriguingly, ARF signaling was also found upregulated in certain tumor types, such as in Burkitt’s lymphoma or the majority of p53 mutant tumors [9,26,115]. To conclusively determine the effects of ARF signaling, ARF still needs to be investigated separately from p16, as the two transcripts of the CDKN2A locus appear to have differing roles in processes such as neural stem cell self-renewal and differentiation [109].
In this review, we sought to summarize key findings regarding non-canonical roles of ARF signaling in cancer and developmental biology. In addition, given that development and tumorigenesis are often governed by similar mechanisms, we provide an overview of the crosstalk between ARF alone or the ARF/p53 pathway with major signaling pathways, with pivotal roles in both contexts. The accumulated evidence suggests that ARF is a pleiotropic protein with different regulators and interactors depending on the cell type, developmental stage, and mutation load. By highlighting the interplay between ARF and other pathway components, new links may appear between important signal transduction processes, thus offering the possibility to explore novel therapeutic approaches aimed both at rectifying developmental defects and restricting tumor growth. For example, novel functions identified for the Hippo scaffold RASSF1A in regulating the Wnt pathway [64,65], or the observation that Notch signaling negatively correlates with RASSF1A expression in human tumors such as in the lung and breast [116], in conjunction with RASSF1A- and ARF-mediated p53 regulation, provide a potentially meaningful link between RASSF1A–ARF that deserves to be explored. Along those lines, the Notch transcriptional target BMI1 is a major regulator of ARF expression in development [109], known to hold a neuroprotective role against degenerative disorders such as Alzheimer’s disease [117,118,119]. Hence, the Notch–BMI1–ARF axis constitutes an important clinical target in this field that could be potentially therapeutically exploited.
Author Contributions
Conceptualization, A.P. and V.G.; Writing—original draft preparation, N.L., A.A., A.P., and V.G.; Writing—review and editing, K.B., A.P., and V.G.; Supervision, A.P. and V.G.; Funding acquisition, V.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), under the grant agreement No 775, entitled “Hippo pathway in genome and epigenome maintenance–HIPPO”. This work is also supported by the HFRI and the GSRT, under the grant agreement No 3782, entitled “Interrogating the Pathophysiological Consequences of deregulated Replication Licensing–PaCoReL”, the National Public Investment Program of the Ministry of Development and Investment/General Secretariat for Research and Technology, in the framework of the Flagship Initiative to address SARS-CoV-2 (2020ΣΕ01300001) and H. Pappas’ donation.
Acknowledgments
The authors gratefully acknowledge Georgia Velimezi for her contribution in the initial stages of this work.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef]
- Tsantoulis, P.; Gorgoulis, V. Involvement of E2F transcription factor family in cancer. Eur. J. Cancer 2005, 41, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.G.; Hannon, G.J.; Beach, D.C. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nat. Cell Biol. 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Damalas, A.; Velimezi, G.; Kalaitzakis, A.; Liontos, M.; Papavassiliou, A.G.; Gorgoulis, V.; Angelidis, C. Loss of p14ARF confers resistance to heat shock- and oxidative stress-mediated cell death by upregulating β-catenin. Int. J. Cancer 2010, 128, 1989–1995. [Google Scholar] [CrossRef]
- De Stanchina, E.; McCurrach, M.E.; Zindy, F.; Shieh, S.-Y.; Ferbeyre, G.; Samuelson, A.V.; Prives, C.; Roussel, M.F.; Sherr, C.J.; Lowe, S.W. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes Dev. 1998, 12, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Kotsinas, A.; Papanagnou, P.; Evangelou, K.; Trigas, G.C.; Kostourou, V.; Townsend, P.; Gorgoulis, V.G. ARF: A versatile DNA damage response ally at the crossroads of development and tumorigenesis. Front. Genet. 2014, 5, 236. [Google Scholar] [CrossRef]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.M.; Palmero, I.; Ryan, K.M.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef]
- Kamijo, T.; Van De Kamp, E.; Chong, M.J.; Zindy, F.; Diehl, J.A.; Sherr, C.J.; McKinnon, P.J. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res. 1999, 59, 2464–2469. [Google Scholar]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor Suppression at the Mouse INK4a Locus Mediated by the Alternative Reading Frame Product p19 ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Evangelou, K.; Bartkova, J.; Kotsinas, A.; Pateras, I.S.; Liontos, M.; Velimezi, G.; Kosar, M.; Liloglou, T.; Trougakos, I.P.; Dyrskjot, L.; et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 2013, 20, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The ARF tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Ramsey, M.R.; Balasubramanian, P.; Castrillon, D.H.; DePinho, R.A. The differential impact of p16INK4a or p19ARF deficiency on cell growth and tumorigenesis. Oncogene 2004, 23, 379–385. [Google Scholar] [CrossRef]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Kotsinas, A.; Papanagnou, P.; Galanos, P.; Schramek, D.; Townsend, P.; Penninger, J.M.; Bartek, J.; Gorgoulis, V.G. MKK7 and ARF. Cell Cycle 2014, 13, 1227–1236. [Google Scholar] [CrossRef]
- Orlando, G.; Khoronenkova, S.V.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. ARF induction in response to DNA strand breaks is regulated by PARP1. Nucleic Acids Res. 2013, 42, 2320–2329. [Google Scholar] [CrossRef]
- Dominguez-Brauer, C.; Chen, Y.-J.; Brauer, P.M.; Pimkina, J.; Raychaudhuri, P. ARF stimulates XPC to trigger nucleotide excision repair by regulating the repressor complex of E2F4. EMBO Rep. 2009, 10, 1036–1042. [Google Scholar] [CrossRef]
- Sarkar-Agrawal, P.; Vergilis, I.; Sharpless, N.E.; Depinho, R.A.; Rünger, T.M. Impaired Processing of DNA Photoproducts and Ultraviolet Hypermutability With Loss of p16INK4a or p19ARF. J. Natl. Cancer Inst. 2004, 96, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Sheng, J.; Hu, M.G.; Haluska, F.G.; Cui, R.; Xu, Z.; Tsichlis, P.N.; Hu, G.-F.; Hinds, P.W. Loss of ARF Sensitizes Transgenic BRAFV600E Mice to UV-Induced Melanoma via Suppression of XPC. Cancer Res. 2013, 73, 4337–4348. [Google Scholar] [CrossRef]
- Hamilton, G.; Abraham, A.G.; Morton, J.; Sampson, O.; Pefani, D.E.; Khoronenkova, S.; Grawenda, A.; Papaspyropoulos, A.; Jamieson, N.; McKay, C.; et al. AKT regulates NPM dependent ARF localization and p53mut stability in tumors. Oncotarget 2014, 5, 6142–6167. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, H.; Long, J.; Ye, S. Clinicopathological significance of p14ARF expression in lung cancer: A meta-analysis. OncoTargets Ther. 2017, 10, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Masuda, N.; Oyama, T.; Nakajima, T. Overexpression of p16 and p14ARF is associated with human papillomavirus infection in cervical squamous cell carcinoma and dysplasia. Pathol. Int. 2002, 52, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aguilera, A.; Sánchez-Beato, M.; García, J.F.; Prieto, I.; Pollán, M.; Piris, M.A. p14ARF nuclear overexpression in aggressive B-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood 2002, 99, 1411–1418. [Google Scholar] [CrossRef]
- Ferru, A.; Fromont, G.; Gibelin, H.; Guilhot, J.; Savagner, F.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. The status of CDKN2A alpha (p16INK4A) and beta (p14ARF) transcripts in thyroid tumour progression. Br. J. Cancer 2006, 95, 1670–1677. [Google Scholar] [CrossRef]
- Vivo, M.; Fontana, R.; Ranieri, M.; Capasso, G.; Angrisano, T.; Pollice, A.; Calabrò, V.; La Mantia, G. p14ARF interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017, 36, 4913–4928. [Google Scholar] [CrossRef]
- Maggi, L.B.; Winkeler, C.L.; Miceli, A.P.; Apicelli, A.J.; Brady, S.N.; Kuchenreuther, M.J.; Weber, J.D. ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta 2014, 1842, 831–839. [Google Scholar] [CrossRef]
- Zhang, Y.; Hyle, J.; Wright, S.; Shao, Y.; Zhao, X.; Zhang, H.; Li, C. Acis-element within the ARFlocus mediates repression ofp16INK4Aexpression via long-range chromatin interactions. Proc. Natl. Acad. Sci. USA 2019, 116, 26644–26652. [Google Scholar] [CrossRef]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The Arftumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl. Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef] [PubMed]
- Gromley, A.; Churchman, M.L.; Zindy, F.; Sherr, C.J. Transient expression of the Arf tumor suppressor during male germ cell and eye development in Arf-Cre reporter mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6285–6290. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.L.A.; Thornton, J.D.; Martin, A.C.; Rehg, J.E.; Bertwistle, D.; Zindy, F.; Skapek, S.X. Arf-dependent regulation of Pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 2005, 24, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.S.; Mei, J.; Liu, J.; Skapek, S.X. miR-34a is essential for p19Arf-driven cell cycle arrest. Cell Cycle 2014, 13, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Shepard, A.; Kittrell, F.; Mulac-Jericevic, B.; Medina, D.; Said, T.K. p19ARFDetermines the Balance between Normal Cell Proliferation Rate and Apoptosis during Mammary Gland Development. Mol. Biol. Cell 2004, 15, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Churchman, M.L.; Roig, I.; Jasin, M.; Keeney, S.; Sherr, C.J. Expression of Arf Tumor Suppressor in Spermatogonia Facilitates Meiotic Progression in Male Germ Cells. PLoS Genet. 2011, 7, e1002157. [Google Scholar] [CrossRef]
- Mu, W.; Starmer, J.; Fedoriw, A.M.; Yee, D.; Magnuson, T. Repression of the soma-specific transcriptome by Polycomb-repressive complex 2 promotes male germ cell development. Genes Dev. 2014, 28, 2056–2069. [Google Scholar] [CrossRef]
- Li, C.; Finkelstein, D.; Sherr, C.J. Arf tumor suppressor and miR-205 regulate cell adhesion and formation of extraembryonic endoderm from pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1112–E1121. [Google Scholar] [CrossRef]
- Vivo, M.; Di Costanzo, A.; Fortugno, P.; Pollice, A.; Calabrò, V.; La Mantia, G. Downregulation of ΔNp63α in keratinocytes by p14ARF-mediated SUMO-conjugation and degradation. Cell Cycle 2009, 8, 3545–3551. [Google Scholar] [CrossRef]
- Su, X.; Cho, M.S.; Gi, Y.-J.; Ayanga, B.A.; Sherr, C.J.; Flores, E.R. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009, 28, 1904–1915. [Google Scholar] [CrossRef]
- Kawagishi, H.; Nakamura, H.; Maruyama, M.; Mizutani, S.; Sugimoto, K.; Takagi, M.; Sugimoto, M. ARF Suppresses Tumor Angiogenesis through Translational Control of VEGFA mRNA. Cancer Res. 2010, 70, 4749–4758. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.E.; Gurley, K.E.; Moser, R.D.; Kemp, C.J. ARF suppresses hepatic vascular neoplasia in a carcinogen-exposed murine model. J. Pathol. 2012, 227, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, L.; Herranz, S.; Higueras, M.A.; Luque, A.; Hortelano, S. Tumor suppressor ARF regulates tissue microenvironment and tumor growth through modulation of macrophage polarization. Oncotarget 2016, 7, 66835–66850. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Garcia, E.; Moreno, M.; Moreno-Cugnon, L.; Matheu, A. Increased Arf/p53 activity in stem cells, aging and cancer. Aging Cell 2017, 16, 219–225. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Hesse, R.G.; Kouklis, G.K.; Ahituv, N.; Pomerantz, J.H. The human ARF tumor suppressor senses blastema activity and suppresses epimorphic tissue regeneration. eLife 2015, 4, e07702. [Google Scholar] [CrossRef]
- Lee, S.; Hesse, R.; Tamaki, S.; Garland, C.B.; Pomerantz, J.H. Human ARF Specifically Inhibits Epimorphic Regeneration in the Zebrafish Heart. Genes 2020, 11, 666. [Google Scholar] [CrossRef]
- Dumble, M.; Moore, L.; Chambers, S.M.; Geiger, H.; Van Zant, G.; Goodell, M.A.; Donehower, L.A. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood 2006, 109, 1736–1742. [Google Scholar] [CrossRef]
- Gatza, C.E.; Dumble, M.; Kittrell, F.; Edwards, D.G.; Dearth, R.K.; Lee, A.V.; Xu, J.; Medina, D.; Donehower, L.A. Altered mammary gland development in the p53+/m mouse, a model of accelerated aging. Dev. Biol. 2008, 313, 130–141. [Google Scholar] [CrossRef]
- Medrano, S.; Burns-Cusato, M.; Atienza, M.B.; Rahimi, D.; Scrable, H. Regenerative capacity of neural precursors in the adult mammalian brain is under the control of p53. Neurobiol. Aging 2009, 30, 483–497. [Google Scholar] [CrossRef]
- Gannon, H.S.; Donehower, L.A.; Lyle, S.; Jones, S.N. Mdm2–p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev. Biol. 2011, 353, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-García, E.; Arrizabalaga, O.; Serrano, M.; Lovell-Badge, R.; Matheu, A. Increased gene dosage ofInk4/Arfandp53delays age-associated central nervous system functional decline. Aging Cell 2015, 14, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Rodrigues, N.; Shen, H.; Yang, Y.-G.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic Stem Cell Quiescence Maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar] [CrossRef] [PubMed]
- Kippin, T.E.; Martens, D.J.; Van Der Kooy, D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005, 19, 756–767. [Google Scholar] [CrossRef]
- Meletis, K.; Wirta, V.; Hede, S.-M.; Nistér, M.; Lundeberg, J.; Frisén, J. p53 suppresses the self-renewal of adult neural stem cells. Development 2005, 133, 363–369. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.-K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nat. Cell Biol. 2003, 425, 962–967. [Google Scholar] [CrossRef]
- Park, I.-K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nat. Cell Biol. 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Jacobs, J.J.L.; Kieboom, K.; Marino, S.; DePinho, R.A.; Van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nat. Cell Biol. 1999, 397, 164–168. [Google Scholar] [CrossRef]
- Biehs, B.; Hu, J.K.-H.; Strauli, N.B.; Sangiorgi, E.; Jung, H.; Heber, R.-P.; Ho, S.; Goodwin, A.F.; Dasen, J.S.; Capecchi, M.R.; et al. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat. Cell Biol. 2013, 15, 846–852. [Google Scholar] [CrossRef]
- Kobayashi, M.; Lin, Y.; Mishra, A.; Shelly, C.; Gao, R.; Reeh, C.W.; Wang, P.Z.; Xi, R.; Liu, Y.; Wenzel, P.; et al. Bmi1 Maintains the Self-Renewal Property of Innate-like B Lymphocytes. J. Immunol. 2020, 204, 3262–3272. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Bergot, E.; Zalcman, G.; Levallet, G. RASSF1A, puppeteer of cellular homeostasis, fights tumorigenesis, and metastasis—An updated review. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Van Der Weyden, L.; Papaspyropoulos, A.; Poulogiannis, G.; Rust, A.G.; Rashid, M.; Adams, D.J.; Arends, M.J.; O’Neill, E. Loss of Rassf1a Synergizes with Deregulated Runx2 Signaling in Tumorigenesis. Cancer Res. 2012, 72, 3817–3827. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, N.; Scrace, S.; Soto, M.S.; Grawenda, A.M.; Bradley, L.; Pankova, D.; Papaspyropoulos, A.; Yee, K.S.; Buffa, F.M.; Goding, C.R.; et al. Alternate RASSF1 Transcripts Control SRC Activity, E-Cadherin Contacts, and YAP-Mediated Invasion. Curr. Biol. 2015, 25, 3019–3034. [Google Scholar] [CrossRef]
- Papaspyropoulos, A.; Bradley, L.; Thapa, A.; Leung, C.Y.; Toskas, K.; Koennig, D.; Pefani, D.-E.; Raso, C.; Grou, C.; Hamilton, G.; et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates E-cadherin repression via p19Arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Goodwin, J.S.; Sathyanarayana, S.A.; Pratap, S.; Chen, Z. MMP7 interacts with ARF in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016, 7, 47609–47619. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.L.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nat. Cell Biol. 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Xie, Y.; Sun, Q.; Nurkesh, A.A.; Lu, J.; Kauanova, S.; Feng, J.; Tursynkhan, D.; Yang, Q.; Kassymbek, A.; Karibayev, M.; et al. Dysregulation of YAP by ARF Stimulated with Tea-derived Carbon Nanodots. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Kalinichenko, V.V.; Major, M.L.; Wang, X.; Petrovic, V.; Kuechle, J.; Yoder, H.M.; Dennewitz, M.B.; Shin, B.; Datta, A.; Raychaudhuri, P.; et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004, 18, 830–850. [Google Scholar] [CrossRef]
- Eisinger-Mathason, T.S.K.; Mucaj, V.; Biju, K.M.; Nakazawa, M.S.; Gohil, M.; Cash, T.P.; Yoon, S.S.; Skuli, N.; Park, K.M.; Gerecht, S.; et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E3402–E3411. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Song, S.J.; Kim, S.Y.; Oh, H.J.; Lim, D.-S. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2–DAXX–HAUSP complex. EMBO J. 2008, 27, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF Promotes MDM2 Degradation and Stabilizes p53: ARF-INK4a Locus Deletion Impairs Both the Rb and p53 Tumor Suppression Pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Fenton, S.L.; Dallol, A.; Agathanggelou, A.; Hesson, L.; Ahmed-Choudhury, J.; Baksh, S.; Sardet, C.; Dammann, R.; Minna, J.D.; Downward, J.; et al. Identification of the E1A-Regulated Transcription Factor p120E4F as an Interacting Partner of the RASSF1A Candidate Tumor Suppressor Gene. Cancer Res. 2004, 64, 102–107. [Google Scholar] [CrossRef]
- Rizos, H.; Diefenbach, E.; Badhwar, P.; Woodruff, S.; Becker, T.M.; Rooney, R.J.; Kefford, R.F. Association of p14ARFwith the p120E4FTranscriptional Repressor Enhances Cell Cycle Inhibition. J. Biol. Chem. 2002, 278, 4981–4989. [Google Scholar] [CrossRef]
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. ATM regulates a RASSF1A-dependent DNA damage response. Curr. Biol. 2009, 19, 2020–2025. [Google Scholar] [CrossRef]
- Pefani, D.E.; Tognoli, M.L.; Ercan, D.P.; Gorgoulis, V.; O’Neill, E. MST2 kinase suppresses rDNA transcription in response to DNA damage by phosphorylating nucleolar histone H2B. EMBO J. 2018, 37, 98760. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nat. Cell Biol. 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Ommen, D.D.Z.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, 100300. [Google Scholar] [CrossRef] [PubMed]
- Wiener, D.J.; Basak, O.; Asra, P.; Boonekamp, K.E.; Kretzschmar, K.; Papaspyropoulos, A.; Clevers, H. Establishment and characterization of a canine keratinocyte organoid culture system. Vet. Dermatol. 2018, 29, e126–e375. [Google Scholar] [CrossRef] [PubMed]
- Papaspyropoulos, A.; Tsolaki, M.; Foroglou, N.; Pantazaki, A.A. Modeling and Targeting Alzheimer’s Disease With Organoids. Front. Pharmacol. 2020, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, T.K.; Löf-Öhlin, Z.M.; Sun, X.-F. DNA methylation of the p14ARF, RASSF1A and APC1A genes as an independent prognostic factor in colorectal cancer patients. Int. J. Oncol. 2012, 42, 127–133. [Google Scholar] [CrossRef]
- Olschwang, S.; Hamelin, R.; Laurent-Puig, P.; Thuille, B.; De Rycke, Y.; Li, Y.-J.; Muzeau, F.; Girodet, J.; Salmon, R.-J.; Thomas, G. Alternative genetic pathways in colorectal carcinogenesis. Proc. Natl. Acad. Sci. USA 1997, 94, 12122–12127. [Google Scholar] [CrossRef]
- De Jaime-Soguero, A.; Aulicino, F.; Ertaylan, G.; Griego, A.; Cerrato, A.; Tallam, A.; Del Sol, A.; Cosma, M.P.; Lluis, F. Wnt/Tcf1 pathway restricts embryonic stem cell cycle through activation of the Ink4/Arf locus. PLoS Genet. 2017, 13, e1006682. [Google Scholar] [CrossRef]
- Damalas, A.; Kahan, S.; Shtutman, M.; Ben-Ze’Ev, A.; Oren, M. Deregulated β-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. EMBO J. 2001, 20, 4912–4922. [Google Scholar] [CrossRef]
- You, Z.; Saims, D.; Chen, S.; Zhang, Z.; Guttridge, D.C.; Guan, K.-L.; MacDougald, O.A.; Brown, A.M.C.; Evan, G.; Kitajewski, J.; et al. Wnt signaling promotes oncogenic transformation by inhibiting c-Myc–induced apoptosis. J. Cell Biol. 2002, 157, 429–440. [Google Scholar] [CrossRef]
- Packer, L.M.; Pavey, S.; Boyle, G.M.; Stark, M.S.; Ayub, A.L.; Rizos, H.; Hayward, N.K. Gene expression profiling in melanoma identifies novel downstream effectors ofp14ARF. Int. J. Cancer 2007, 121, 784–790. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. FGF signaling inhibitor, SPRY4, is evolutionarily conserved target of WNT signaling pathway in progenitor cells. Int. J. Mol. Med. 2006, 17, 529–532. [Google Scholar] [CrossRef]
- Renard, C.-A.; Labalette, C.; Armengol, C.; Cougot, D.; Wei, Y.; Cairo, S.; Pineau, P.; Neuveut, C.; De Reyniès, A.; Dejean, A.; et al. Tbx3 Is a Downstream Target of the Wnt/β-Catenin Pathway and a Critical Mediator of β-Catenin Survival Functions in Liver Cancer. Cancer Res. 2007, 67, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, W.; Barrientos, T.; Esmailpour, T.; Lin, L.; Carpenter, P.M.; Osann, K.; Anton-Culver, H.; Huang, T. TBX3 Is Overexpressed in Breast Cancer and Represses p14ARF by Interacting with Histone Deacetylases. Cancer Res. 2008, 68, 693–699. [Google Scholar] [CrossRef]
- Debies, M.T.; Gestl, S.A.; Mathers, J.L.; Mikse, O.R.; Leonard, T.L.; Moody, S.E.; Chodosh, L.A.; Cardiff, R.D.; Gunther, E.J. Tumor escape in a Wnt1-dependent mouse breast cancer model is enabled by p19Arf/p53 pathway lesions but not p16Ink4a loss. J. Clin. Investig. 2008, 118, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Krimpenfort, P.; Ijpenberg, A.; Song, J.-Y.; Van Der Valk, M.; Nawijn, M.C.; Zevenhoven, J.; Berns, A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nat. Cell Biol. 2007, 448, 943–946. [Google Scholar] [CrossRef]
- Krimpenfort, P.; Snoek, M.; Lambooij, J.-P.; Song, J.-Y.; Van Der Weide, R.; Bhaskaran, R.; Teunissen, H.; Adams, D.J.; De Wit, E.; Berns, A. A natural WNT signaling variant potently synergizes with Cdkn2ab loss in skin carcinogenesis. Nat. Commun. 2019, 10, 1425. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Komiya, M.; Matsumura, K.; Negishi, L.; Suda, S.; Okuno, M.; Yokota, N.; Osada, T.; Nagashima, T.; Hiyoshi, M.; et al. MYU, a Target lncRNA for Wnt/c-Myc Signaling, Mediates Induction of CDK6 to Promote Cell Cycle Progression. Cell Rep. 2016, 16, 2554–2564. [Google Scholar] [CrossRef] [PubMed]
- Falo-Sanjuan, J.; Bray, S.J. Decoding the Notch signal. Dev. Growth Differ. 2020, 62, 4–14. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Muskavitch, M.A. Notch: The Past, the Present, and the Future. Gastrulation Embryonic Pattern Form 2010, 92, 1–29. [Google Scholar]
- Krejci, A.; Bernard, F.; Housden, B.E.; Collins, S.; Bray, S.J. Direct Response to Notch Activation: Signaling Crosstalk and Incoherent Logic. Sci. Signal. 2009, 2, ra1. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.-H.; Downing, J.R.; Gilliland, D.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P., IV; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Volanakis, E.J.; Williams, R.T.; Sherr, C.J. Stage-specific Arf tumor suppression in Notch1-induced T-cell acute lymphoblastic leukemia. Blood 2009, 114, 4451–4459. [Google Scholar] [CrossRef] [PubMed]
- Volanakis, E.J.; Boothby, M.R.; Sherr, C.J. Epigenetic regulation of the Ink4a-Arf (Cdkn2a) tumor suppressor locus in the initiation and progression of Notch1-driven T cell acute lymphoblastic leukemia. Exp. Hematol. 2013, 41, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Boehm, T.; Foroni, L.; Kaneko, Y.; Perutz, M.F.; Rabbitts, T.H. The rhombotin family of cysteine-rich LIM-domain oncogenes: Distinct members are involved in T-cell translocations to human chromosomes 11p15 and 11p13. Proc. Natl. Acad. Sci. USA 1991, 88, 4367–4371. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Loos, U.; Ludwig, W.D. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene 1991, 6, 1887–1893. [Google Scholar]
- Treanor, L.M.; Volanakis, E.J.; Zhou, S.; Lu, T.; Sherr, C.J.; Sorrentino, B.P. Functional interactions between Lmo2, the Arf tumor suppressor, and Notch1 in murine T-cell malignancies. Blood 2011, 117, 5453–5462. [Google Scholar] [CrossRef][Green Version]
- Kim, S.-H.; Kim, E.-J.; Hitomi, M.; Oh, S.-Y.; Jin, X.; Jeon, H.-M.; Beck, S.; Kim, J.-K.; Park, C.G.; Chang, S.-Y.; et al. The LIM-only transcription factor LMO2 determines tumorigenic and angiogenic traits in glioma stem cells. Cell Death Differ. 2015, 22, 1517–1525. [Google Scholar] [CrossRef]
- López-Arribillaga, E.; Rodilla, V.; Pellegrinet, L.; Guiu, J.; Iglesias, M.; Román, A.-C.; Gutarra, S.; González, S.; Muñoz-Cánoves, P.; Fernandez-Salguero, P.M.; et al. Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of Notch. Development 2014, 142, 41–50. [Google Scholar] [CrossRef]
- Bruggeman, S.W.; Valk-Lingbeek, M.E.; Van Der Stoop, P.P.; Jacobs, J.J.L.; Kieboom, K.; Tanger, E.; Hulsman, D.; Leung, C.; Arsenijevic, Y.; Marino, S.; et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef]
- Xu, C.-R.; Lee, S.; Ho, C.; Bommi, P.; Huang, S.-A.; Cheung, S.T.; Dimri, G.P.; Chen, X. Bmi1 Functions as an Oncogene Independent of Ink4A/Arf Repression in Hepatic Carcinogenesis. Mol. Cancer Res. 2009, 7, 1937–1945. [Google Scholar] [CrossRef]
- Dey, A.; Mustafi, S.B.; Saha, S.; Dwivedi, S.K.D.; Mukherjee, P.; Bhattacharya, R. Inhibition of BMI1 induces autophagy-mediated necroptosis. Autophagy 2016, 12, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Beverly, L.J.; Felsher, D.W.; Capobianco, A.J. Suppression of p53 by Notch in Lymphomagenesis: Implications for Initiation and Regression. Cancer Res. 2005, 65, 7159–7168. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ranganathan, P.; Tzimas, C.; Weaver, K.L.; Jin, K.; Astudillo, L.; Zhou, W.; Zhu, X.; Li, B.; Robbins, D.J.; et al. Notch Represses Transcription by PRC2 Recruitment to the Ternary Complex. Mol. Cancer Res. 2017, 15, 1173–1183. [Google Scholar] [CrossRef]
- Liu, L.; Charville, G.W.; Cheung, T.H.; Yoo, B.; Santos, P.J.; Schroeder, M.; Rando, T.A. Impaired Notch Signaling Leads to a Decrease in p53 Activity and Mitotic Catastrophe in Aged Muscle Stem Cells. Cell Stem Cell 2018, 23, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Pefani, D.E.; Pankova, D.; Abraham, A.G.; Grawenda, A.M.; Vlahov, N.; Scrace, S.; O’Neill, E. TGF-β Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol. Cell 2016, 63, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Chatoo, W.; El Hajjar, J.; David, J.; Ferreira, J.; Bernier, G. Bmi1 Is Down-Regulated in the Aging Brain and Displays Antioxidant and Protective Activities in Neurons. PLoS ONE 2012, 7, e31870. [Google Scholar] [CrossRef]
- Hogan, R.; Flamier, A.; Nardini, E.; Bernier, G. The Role of BMI1 in Late-Onset Sporadic Alzheimer’s Disease. Genes 2020, 11, 825. [Google Scholar] [CrossRef]
- Tzekaki, E.E.; Papaspyropoulos, A.; Tsolaki, M.; Lazarou, E.; Kozori, M.; Pantazaki, A.A. Restoration of BMI1 levels after the administration of early harvest extra virgin olive oil as a therapeutic strategy against Alzheimer’s disease. Exp. Gerontol. 2021, 144, 111178. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).