
Sphingosine Promotes Embryo Biomass in Upland Cotton: A Biochemical and Transcriptomic Analysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cotton Materials and In Vitro Ovule Culture
2.2. Lipid Extraction and Lipidomics
2.3. RNA-Sequencing (RNA-seq)
2.4. Bioinformatic Analysis
2.5. Heat Map Drawing
2.6. Semi-Quantitative PCR
3. Results
3.1. Sphingolipid Synthesis Inhibitor Reduced Cotton Embryo Biomass
3.2. Sphingosine Is the Main Sphingolipid Component in Upland Cotton Ovules/Embryos
3.3. SPT Enzyme Inhibitor Reduced Cotton Embryo Biomass
3.4. PHS Promoted Cotton Embryo Biomass
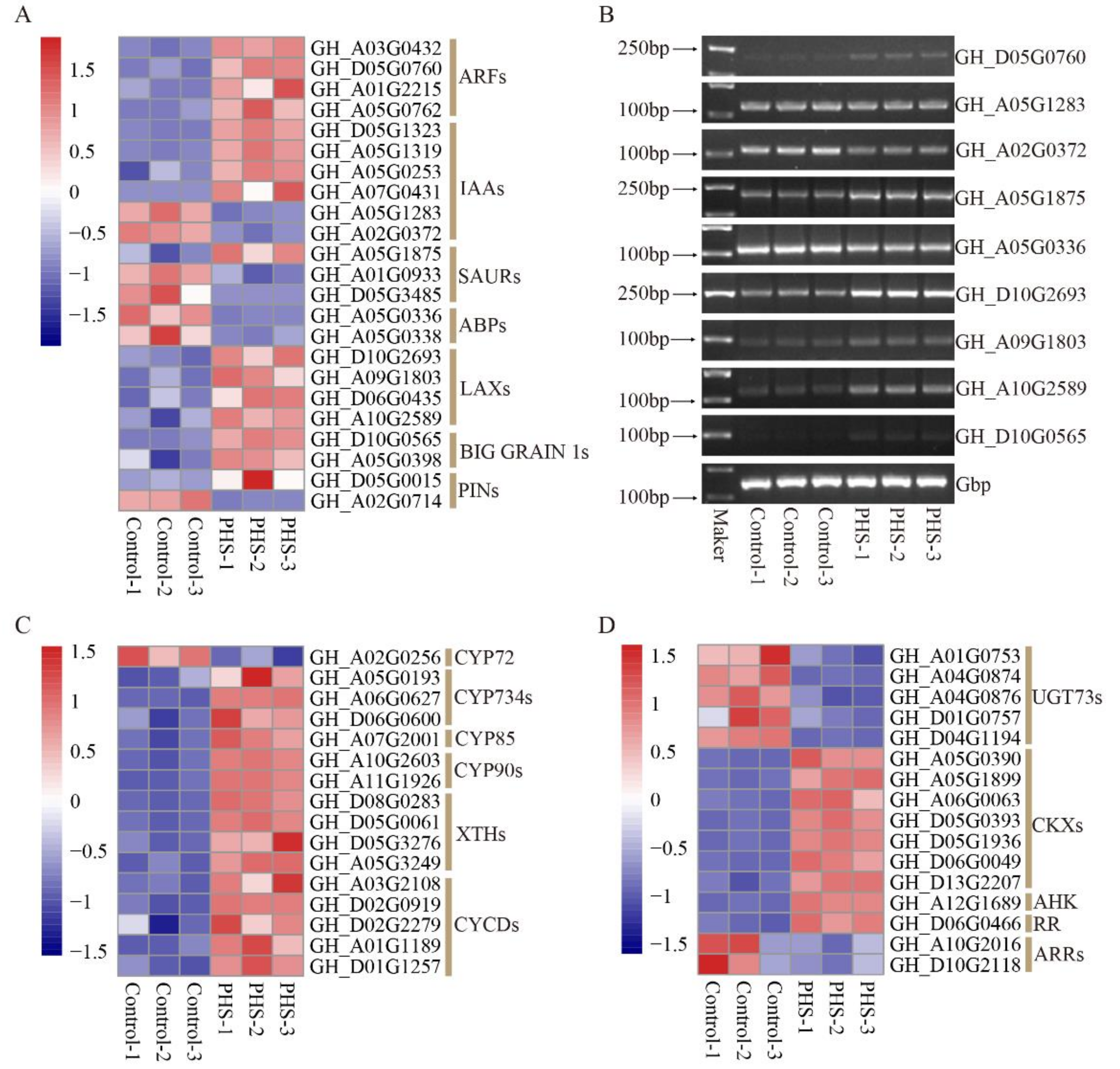
3.5. Transcriptomic Analysis of Embryos Treated with PHS
3.6. The DEGs of TFs Treated by PHS
3.7. The DEGs Related to Lipid Metabolism Treated by PHS
3.8. The DEGs Related to Plant Hormones Treated by PHS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lynch, D.V.; Dunn, T.M. An introduction to plant sphingolipids and a review of recent advances in understanding their metabolism and function. New Phytol. 2004, 161, 677–702. [Google Scholar] [CrossRef]
- Markham, J.E.; Lynch, D.V.; Napier, J.A.; Dunn, T.M.; Cahoon, E.B. Plant sphingolipids: Function follows form. Curr. Opin. Plant Biol. 2013, 16, 350–357. [Google Scholar] [CrossRef]
- Chen, M.; Markham, J.E.; Dietrich, C.R.; Jaworski, J.G.; Cahoon, E.B. Sphingolipid long-chain base hydroxylation is important for growth and regulation of sphingolipid content and composition in Arabidopsis. Plant Cell 2008, 20, 1862–1878. [Google Scholar] [CrossRef]
- Chen, M.; Markham, J.E.; Cahoon, E.B. Sphingolipid delta 8 unsaturation is important for glucosylceramide biosynthesis and low-temperature performance in Arabidopsis. Plant J. 2012, 69, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Markham, J.E.; Molino, D.; Gissot, L.; Bellec, Y.; Hematy, K.; Marion, J.; Belcram, K.; Palauqui, J.C.; Satiat-JeuneMaitre, B.; Faure, J.D. Sphingolipids Containing Very-Long-Chain Fatty Acids Define a Secretory Pathway for Specific Polar Plasma Membrane Protein Targeting in Arabidopsis. Plant Cell 2011, 23, 2362–2378. [Google Scholar] [CrossRef]
- Wang, L.; Liu, C.; Liu, Y.J.; Luo, M. Fumonisin B1-Induced Changes in Cotton Fiber Elongation Revealed by Sphingolipidomics and Proteomics. Biomolecules 2020, 10, 1258. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.T.; Marinho, H.S.; de Almeida, R.F.M. Sphingolipid hydroxylation in mammals, yeast and plants—An integrated view. Prog. Lipid Res. 2018, 71, 18–42. [Google Scholar] [CrossRef] [PubMed]
- Beasley, C.A.; Ting, I.P. The effects of plant growth substances on in vitro fiber development from fertilized cotton ovules. Am. J. Bot. 1973, 60, 130–139. [Google Scholar] [CrossRef]
- Welti, R.; Li, W.Q.; Li, M.Y.; Sang, Y.M.; Biesiada, H.; Zhou, H.E.; Rajashekar, C.B.; Williams, T.D.; Wang, X.M. Profiling membrane lipids in plant stress responses—Role of phospholipase D alpha in freezing-induced lipid changes in Arabidopsis. J. Biol. Chem. 2002, 277, 31994–32002. [Google Scholar] [CrossRef]
- Huang, D.Q.; Sun, Y.B.; Ma, Z.M.; Ke, M.Y.; Cui, Y.; Chen, Z.C.; Chen, C.F.; Ji, C.Y.; Tran, T.M.; Yang, L.; et al. Salicylic acid-mediated plasmodesmal closure via Remorin-dependent lipid organization. Proc. Natl. Acad. Sci. USA 2020, 117, 8659. [Google Scholar] [CrossRef]
- Markham, J.E.; Jaworski, J.G. Rapid measurement of sphingolipids from Arabidopsis thaliana by reversed-phase high-performance liquid chromatography coupled to electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.D.; Fang, L.; Zhang, Z.Y.; Ma, W.; Niu, Y.C.; Ju, L.Z.; Deng, J.Q.; Zhao, T.; Lian, J.M.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with Deseq2. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Angenent, G.C.; Franken, J.; Busscher, M.; Vandijken, A.; Vanwent, J.L.; Dons, H.J.M.; Vantunen, A.J. A Novel Class of Mads Box Genes Is Involved in Ovule Development in Petunia. Plant Cell 1995, 7, 1569–1582. [Google Scholar]
- Duan, P.G.; Ni, S.; Wang, J.M.; Zhang, B.L.; Xu, R.; Wang, Y.X.; Chen, H.Q.; Zhu, X.D.; Li, Y.H. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat. Plants 2016, 2, 15203. [Google Scholar] [CrossRef]
- Ehlers, K.; Bhide, A.S.; Tekleyohans, D.G.; Wittkop, B.; Snowdon, R.J.; Becker, A. The MADS Box Genes ABS, SHP1, and SHP2 Are Essential for the Coordination of Cell Divisions in Ovule and Seed Coat Development and for Endosperm Formation in Arabidopsis thaliana. PLoS ONE 2016, 11, e0165075. [Google Scholar] [CrossRef]
- Kunieda, T.; Mitsuda, N.; Ohme-Takagi, M.; Takeda, S.; Aida, M.; Tasaka, M.; Kondo, M.; Nishimura, M.; Hara-Nishimura, I. NAC Family Proteins NARS1/NAC2 and NARS2/NAM in the Outer Integument Regulate Embryogenesis in Arabidopsis. Plant Cell 2008, 20, 2631–2642. [Google Scholar] [CrossRef]
- Ohto, M.; Fischer, R.L.; Goldberg, R.B.; Nakamura, K.; Harada, J.J. Control of seed mass by Apetala2. Proc. Natl. Acad. Sci. USA 2005, 102, 3123–3128. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.D.; Shantharaj, D.; Kang, X.J.; Ni, M. Transcriptional and hormonal signaling control of Arabidopsis seed development. Curr. Opin. Plant Biol. 2010, 13, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chakrabarti, M.; Taitano, N.; Okazaki, Y.; Saito, K.; Al-Abdallat, A.; van der Knaap, E. Differential expression of SlKLUH controlling fruit and seed weight is associated with changes in lipid metabolism and photosynthesis-related genes. J. Exp. Bot. 2021, 72, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yao, N.; Song, L.T.; Luo, S.; Lu, H.; Greenberg, L.T. Ceramides modulate programmed cell death in plants. Genes Dev. 2003, 17, 2636–2641. [Google Scholar] [CrossRef]
- Bi, F.C.; Liu, Z.; Wu, J.X.; Liang, H.; Xi, X.L.; Fang, C.; Sun, T.J.; Yin, J.; Dai, G.Y.; Rong, C.; et al. Loss of Ceramide Kinase in Arabidopsis Impairs Defenses and Promotes Ceramide Accumulation and Mitochondrial H2O2 Bursts. Plant Cell 2014, 26, 3449–3467. [Google Scholar] [CrossRef]
- Wu, J.X.; Li, J.; Liu, Z.; Yin, J.; Chang, Z.Y.; Rong, C.; Wu, J.L.; Bi, F.C.; Yao, N. The Arabidopsis ceramidase AtACER functions in disease resistance and salt tolerance. Plant J. 2015, 81, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Wu, J.X.; Sahu, S.K.; Zeng, H.Y.; Huang, L.Q.; Liu, Z.; Xiao, S.; Yao, N. Loss of alkaline ceramidase inhibits autophagy in Arabidopsis and plays an important role during environmental stress response. Plant Cell Environ. 2018, 41, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Iswanto, A.B.B.; Shon, J.C.; Liu, K.H.; Vu, M.H.; Kumar, R.; Kim, J.Y. Sphingolipids Modulate Secretion of Glycosylphosphatidylinositol-Anchored Plasmodesmata Proteins and Callose Deposition. Plant Physiol. 2020, 184, 407–420. [Google Scholar] [CrossRef]
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2004, 1682, 128. [Google Scholar] [CrossRef]
- Francoz, E.; Ranocha, P.; Nguyen-Kim, H.; Jamet, E.; Burlat, V.; Dunand, C. Roles of cell wall peroxidases in plant development. Phytochemistry 2015, 112, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kondou, Y.; Nakazawa, M.; Kawashima, M.; Ichikawa, T.; Yoshizumi, T.; Suzuki, K.; Ishikawa, A.; Koshi, T.; Matsui, R.; Muto, S.; et al. Retarded Growth of Embryo1, a new basic helix-loop-helix protein, expresses in endosperm to control embryo growth. Plant Physiol. 2008, 147, 1924–1935. [Google Scholar] [CrossRef]
- Garcia, D.; Fitz Gerald, J.N.; Berger, F. Maternal control of integument cell elongation and zygotic control of endosperm growth are coordinated to determine seed size in Arabidopsis. Plant Cell 2005, 17, 52–60. [Google Scholar] [CrossRef]
- Wang, A.H.; Garcia, D.; Zhang, H.Y.; Feng, K.; Chaudhury, A.; Berger, F.; Peacock, W.J.; Dennis, E.S.; Luo, M. The VQ motif protein IKU1 regulates endosperm growth and seed size in Arabidopsis. Plant J. 2010, 63, 670–679. [Google Scholar] [CrossRef]
- Luo, M.; Dennis, E.S.; Berger, F.; Peacock, W.J.; Chaudhury, A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 17531–17536. [Google Scholar] [CrossRef]
- Na, G.; Mu, X.P.; Grabowski, P.; Schmutz, J.; Lu, C.F. Enhancing microRNA167A expression in seed decreases the alpha-linolenic acid content and increases seed size in Camelina sativa. Plant J. 2019, 98, 346–358. [Google Scholar] [CrossRef]
- Singh, A.K.; Fu, D.Q.; El-Habbak, M.; Navarre, D.; Ghabrial, S.; Kachroo, A. Silencing Genes Encoding Omega-3 Fatty Acid Desaturase Alters Seed Size and Accumulation of Bean pod mottle virus in Soybean. Mol. Plant Microbe. Interact. 2011, 24, 506–515. [Google Scholar] [CrossRef]
- Chen, M.X.; Wang, Z.; Zhu, Y.N.; Li, Z.L.; Hussain, N.; Xuan, L.J.; Guo, W.L.; Zhang, G.P.; Jiang, L.X. The Effect of Transparent Testa2 on Seed Fatty Acid Biosynthesis and Tolerance to Environmental Stresses during Young Seedling Establishment in Arabidopsis. Plant Physiol. 2012, 160, 1023–1036. [Google Scholar] [CrossRef]
- Schruff, M.C.; Spielman, M.; Tiwari, S.; Adams, S.; Fenby, N.; Scott, R.J. The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development 2006, 133, 251–261. [Google Scholar] [CrossRef]
- Liu, J.; Hua, W.; Hu, Z.Y.; Yang, H.L.; Zhang, L.; Li, R.J.; Deng, L.B.; Sun, X.C.; Wang, X.F.; Wang, H.Z. Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proc. Natl. Acad. Sci. USA 2015, 112, E5123–E5132. [Google Scholar] [CrossRef]
- Sun, Y.W.; Wang, C.M.; Wang, N.; Jiang, X.Y.; Mao, H.Z.; Zhu, C.X.; Wen, F.J.; Wang, X.H.; Lu, Z.J.; Yue, G.H.; et al. Manipulation of Auxin Response Factor 19 affects seed size in the woody perennial Jatropha curcas. Sci. Rep. 2017, 7, 40844. [Google Scholar] [CrossRef]
- Liu, L.C.; Tong, H.N.; Xiao, Y.H.; Che, R.H.; Xu, F.; Hu, B.; Liang, C.Z.; Chu, J.F.; Li, J.Y.; Chu, C.C. Activation of Big Grain1 significantly improves grain size by regulating auxin transport in rice. Proc. Natl. Acad. Sci. USA 2015, 112, 11102–11107. [Google Scholar] [CrossRef]
- Simmons, C.R.; Weers, B.P.; Reimann, K.S.; Abbitt, S.E.; Frank, M.J.; Wang, W.Y.; Wu, J.R.; Shen, B.; Habben, J.E. Maize BIG GRAIN1 homolog overexpression increases maize grain yield. Plant Biotechnol. J. 2020, 18, 2304–2315. [Google Scholar] [CrossRef]
- Choe, S.; Fujioka, S.; Noguchi, T.; Takatsuto, S.; Yoshida, S.; Feldmann, K.A. Overexpression of DWARF4 in the brassinosteroid biosynthetic pathway results in increased vegetative growth and seed yield in Arabidopsis. Plant J. 2001, 26, 573–582. [Google Scholar] [CrossRef]
- Wu, C.Y.; Trieu, A.; Radhakrishnan, P.; Kwok, S.F.; Harris, S.; Zhang, K.; Wang, J.L.; Wan, J.M.; Zhai, H.Q.; Takatsuto, S.; et al. Brassinosteroids regulate grain filling in rice. Plant Cell 2008, 20, 2130–2145. [Google Scholar] [CrossRef]
- Sahni, S.; Prasad, B.D.; Liu, Q.; Grbic, V.; Sharpe, A.; Singh, S.P.; Krishna, P. Overexpression of the brassinosteroid biosynthetic gene DWF4 in Brassica napus simultaneously increases seed yield and stress tolerance. Sci. Rep. 2016, 6, 28298. [Google Scholar] [CrossRef]
- Hu, Y.X.; Bao, F.; Li, J.Y. Promotive effect of brassinosteroids on cell division involves a distinct CycD3-induction pathway in Arabidopsis. Plant J. 2000, 24, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Sornay, E.; Forzani, C.; Forero-Vargas, M.; Dewitte, W.; Murray, J.A.H. Activation of CYCD7;1 in the central cell and early endosperm overcomes cell-cycle arrest in the Arabidopsis female gametophyte, and promotes early endosperm and embryo development. Plant J. 2015, 84, 41–55. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Suo, X.; Liu, Y.; Liu, C.; Luo, M. Sphingosine Promotes Embryo Biomass in Upland Cotton: A Biochemical and Transcriptomic Analysis. Biomolecules 2021, 11, 525. https://doi.org/10.3390/biom11040525
Wang L, Suo X, Liu Y, Liu C, Luo M. Sphingosine Promotes Embryo Biomass in Upland Cotton: A Biochemical and Transcriptomic Analysis. Biomolecules. 2021; 11(4):525. https://doi.org/10.3390/biom11040525
Chicago/Turabian StyleWang, Li, Xiaodong Suo, Yujie Liu, Chen Liu, and Ming Luo. 2021. "Sphingosine Promotes Embryo Biomass in Upland Cotton: A Biochemical and Transcriptomic Analysis" Biomolecules 11, no. 4: 525. https://doi.org/10.3390/biom11040525
APA StyleWang, L., Suo, X., Liu, Y., Liu, C., & Luo, M. (2021). Sphingosine Promotes Embryo Biomass in Upland Cotton: A Biochemical and Transcriptomic Analysis. Biomolecules, 11(4), 525. https://doi.org/10.3390/biom11040525