
Alarming Cargo: The Role of Exosomes in Trauma-Induced Inflammation

 , ,
, ,
Abstract
1. Introduction
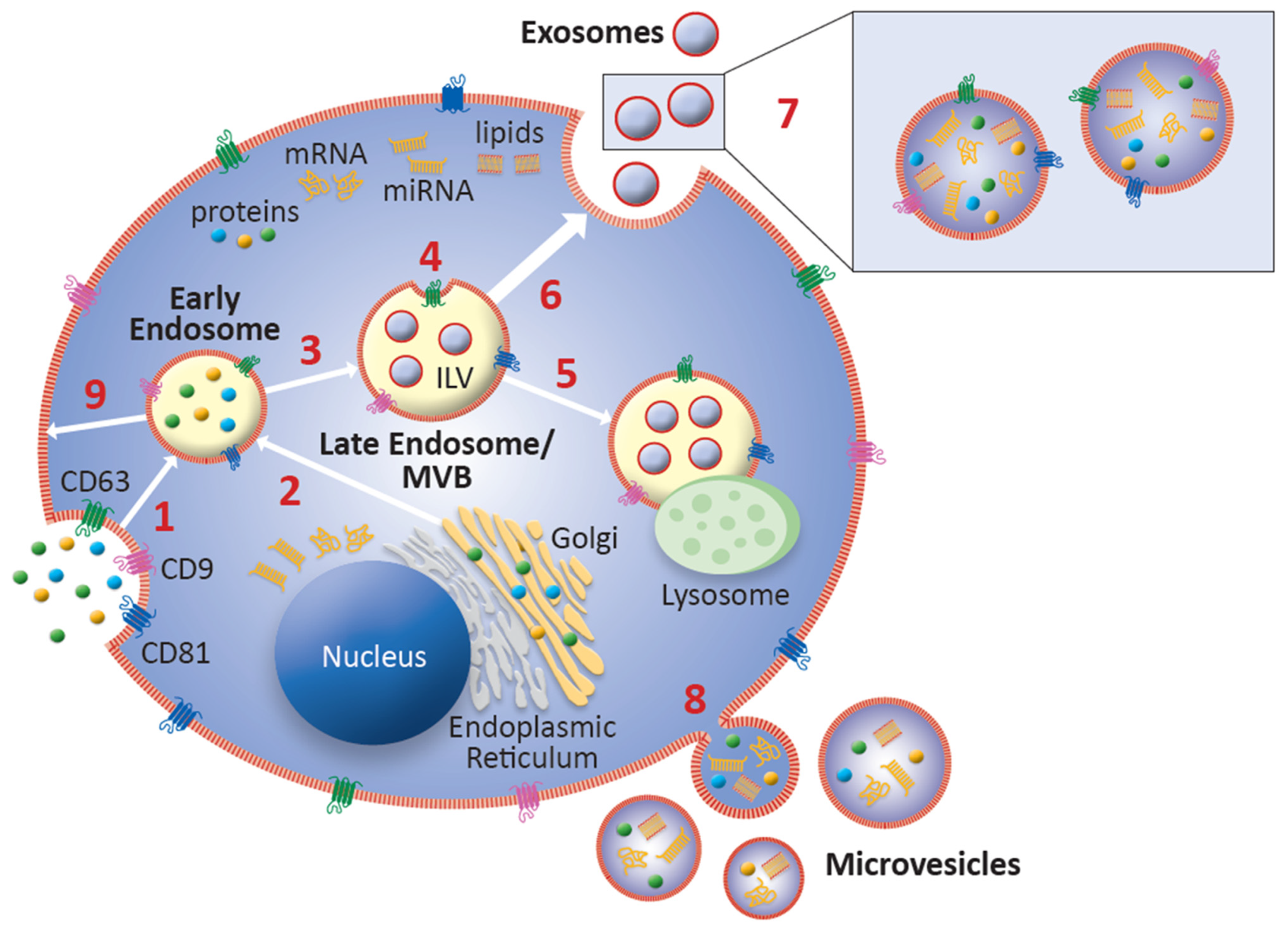
2. Biological Characteristics of Exosomes
2.1. Biogenesis and Intercellular Communication
2.2. Exosomal Cargo Overview
3. Inflammation and the Role of Exosomal Cargo
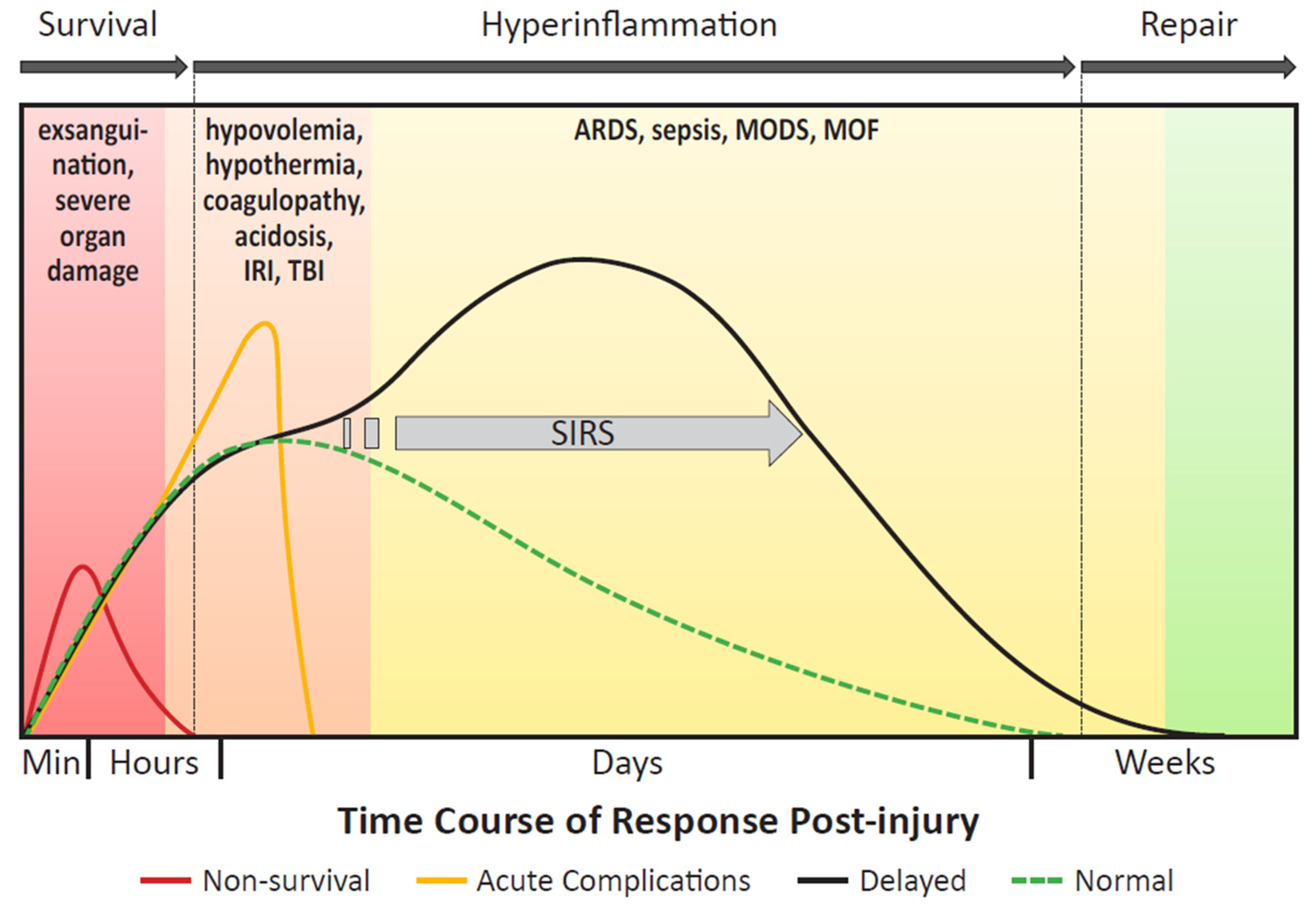
3.1. Inflammatory Response to Severe Trauma
3.2. Role of Exosomes in Regulating Inflammation
4. Exosomes and Their Cargo in Trauma-Associated Complications
4.1. Resuscitation and Ischemia-Reperfusion Injury
4.2. Systemic Inflammatory Response Syndrome
4.3. Acute Respiratory Distress Syndrome
4.4. Sepsis
4.5. Multi-Organ Dysfunction Syndrome and Multi-Organ Failure
4.6. Fracture Healing
4.7. Thermal Injury
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- DiMaggio, C.; Ayoung-Chee, P.; Shinseki, M.; Wilson, C.; Marshall, G.; Lee, D.C.; Wall, S.; Maulana, S.; Leon Pachter, H.; Frangos, S. Traumatic injury in the United States: In-patient epidemiology 2000–2011. Injury 2016, 47, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Hauser, C.J. Preclinical Models of Traumatic, Hemorrhagic Shock. Shock 2005, 24, 24–32. [Google Scholar] [CrossRef]
- Jin, H.; Liu, Z.; Xiao, Y.; Fan, X.; Yan, J.; Liang, H. Prediction of sepsis in trauma patients. Burns Trauma 2014, 2, 106–113. [Google Scholar] [CrossRef]
- Lenz, A.; Franklin, G.A.; Cheadle, W.G. Systemic inflammation after trauma. Injury 2007, 38, 1336–1345. [Google Scholar] [CrossRef]
- Lord, J.M.; Midwinter, M.J.; Chen, Y.-F.; Belli, A.; Brohi, K.; Kovacs, E.J.; Koenderman, L.; Kubes, P.; Lilford, R.J. The systemic immune response to trauma: An overview of pathophysiology and treatment. The Lancet 2014, 384, 1455–1465. [Google Scholar] [CrossRef]
- Chukwu-Lobelu, R.; Appukuttan, A.; Edwards, D.S.; Patel, H.D.L. Burn injuries from the london suicide bombings: A new classification of blast-related thermal injuries. Ann. Burns Fire Disasters 2017, 30, 256–260. [Google Scholar] [PubMed]
- Edwards, M.J.; Lustik, M.; Eichelberger, M.R.; Elster, E.; Azarow, K.; Coppola, C. Blast injury in children: An analysis from Afghanistan and Iraq, 2002–2010. J. Trauma Acute Care Surg. 2012, 73, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.S.; Clasper, J.C.; Patel, H.D. Heterotopic ossification in victims of the London 7/7 bombings. J. R Army Med. Corps 2015, 161, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Hasan, O.; Sheikh, S.; Fatima, A.; Abbas, A.; Zahid, N.; Baloch, N. Motor-vehicle Crash Patient Injury Patterns from a Level One Trauma Center in a Metropolitan City: A Cross-Sectional Study. Cureus 2019, 11, e4073. [Google Scholar] [CrossRef]
- Markogiannakis, H.; Sanidas, E.; Messaris, E.; Koutentakis, D.; Alpantaki, K.; Kafetzakis, A.; Tsiftsis, D. Motor vehicle trauma: Analysis of injury profiles by road-user category. Emerg Med. J. 2006, 23, 27–31. [Google Scholar] [CrossRef]
- Björnstig, U.; Bylund, P.O.; Albertsson, P.; Falkmer, T.; Björnstig, J.; Petzäll, J. Injury Events among Bus and Coach Occupants. IATSS Res. 2005, 29, 79–87. [Google Scholar] [CrossRef]
- Payal, P.; Sonu, G.; Anil, G.K.; Prachi, V. Management of polytrauma patients in emergency department: An experience of a tertiary care health institution of northern India. World J. Emerg. Med. 2013, 4, 15–19. [Google Scholar] [CrossRef]
- Sarani, B.; Hendrix, C.; Matecki, M.; Estroff, J.; Amdur, R.L.; Robinson, B.R.H.; Shapiro, G.; Gondek, S.; Mitchell, R.; Smith, E.R. Wounding Patterns Based on Firearm Type in Civilian Public Mass Shootings in the United States. J. Am. Coll. Surg. 2019, 228, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Hoencamp, R.; Vermetten, E.; Tan, E.C.; Putter, H.; Leenen, L.P.; Hamming, J.F. Systematic review of the prevalence and characteristics of battle casualties from NATO coalition forces in Iraq and Afghanistan. Injury 2014, 45, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Pierrakos, C.; Karanikolas, M.; Scolletta, S.; Karamouzos, V.; Velissaris, D. Acute respiratory distress syndrome: Pathophysiology and therapeutic options. J. Clin. Med. Res. 2012, 4, 7–16. [Google Scholar] [CrossRef]
- Salim, A.; Martin, M.; Constantinou, C.; Sangthong, B.; Brown, C.; Kasotakis, G.; Demetriades, D.; Belzberg, H. Acute respiratory distress syndrome in the trauma intensive care unit: Morbid but not mortal. Arch. Surg. 2006, 141, 655–658. [Google Scholar] [CrossRef][Green Version]
- Sauaia, A.; Moore, E.E.; Johnson, J.L.; Chin, T.L.; Banerjee, A.; Sperry, J.L.; Maier, R.V.; Burlew, C.C. Temporal trends of postinjury multiple-organ failure: Still resource intensive, morbid, and lethal. J. Trauma Acute Care Surg. 2014, 76, 582–592. [Google Scholar] [CrossRef]
- Valparaiso, A.P.; Vicente, D.A.; Bograd, B.A.; Elster, E.A.; Davis, T.A. Modeling acute traumatic injury. J. Surg. Res. 2015, 194, 220–232. [Google Scholar] [CrossRef]
- Xiao, W.; Mindrinos, M.N.; Seok, J.; Cuschieri, J.; Cuenca, A.G.; Gao, H.; Hayden, D.L.; Hennessy, L.; Moore, E.E.; Minei, J.P.; et al. A genomic storm in critically injured humans. J. Exp. Med. 2011, 208, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.M.; Redl, H.; Bahrami, S.; Schlag, G. The inflammatory basis of trauma/shock-associated multiple organ failure. Inflamm. Res. 1998, 47, 201–210. [Google Scholar] [CrossRef]
- Baue, A.E. MOF, MODS, and SIRS: What is in a name or an acronym? Shock 2006, 26, 438–449. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Dewar, D.; Moore, F.A.; Moore, E.E.; Balogh, Z. Postinjury multiple organ failure. Injury 2009, 40, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Osuka, A.; Ogura, H.; Ueyama, M.; Shimazu, T.; Lederer, J.A. Immune response to traumatic injury: Harmony and discordance of immune system homeostasis. Acute Med. Surg. 2014, 1, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Osborn, T.M.; Tracy, J.K.; Dunne, J.R.; Pasquale, M.; Napolitano, L.M. Epidemiology of sepsis in patients with traumatic injury. Crit. Care Med. 2004, 32, 2234–2240. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate immune responses to trauma. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune. Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.F.; Porterfield, N.; Pannell, D.; Davis, T.A.; Elster, E.A. Trauma is danger. J. Transl. Med. 2011, 9, 92. [Google Scholar] [CrossRef]
- Rani, M.; Nicholson, S.E.; Zhang, Q.; Schwacha, M.G. Damage-associated molecular patterns (DAMPs) released after burn are associated with inflammation and monocyte activation. Burns 2017, 43, 297–303. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Greven, J.; Pfeifer, R.; Zhi, Q.; Pape, H.C. Update on the role of endothelial cells in trauma. Eur. J. Trauma Emerg. Surg. 2018, 44, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Keel, M.; Trentz, O. Pathophysiology of polytrauma. Injury 2005, 36, 691–709. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Aswad, H.; Forterre, A.; Wiklander, O.P.; Vial, G.; Danty-Berger, E.; Jalabert, A.; Lamaziere, A.; Meugnier, E.; Pesenti, S.; Ott, C.; et al. Exosomes participate in the alteration of muscle homeostasis during lipid-induced insulin resistance in mice. Diabetologia 2014, 57, 2155–2164. [Google Scholar] [CrossRef] [PubMed]
- Behera, J.; Tyagi, N. Exosomes: Mediators of bone diseases, protection, and therapeutics potential. Oncoscience 2018, 5, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Bonjoch, L.; Casas, V.; Carrascal, M.; Closa, D. Involvement of exosomes in lung inflammation associated with experimental acute pancreatitis. J. Pathol. 2016, 240, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Bucan, V.; Vaslaitis, D.; Peck, C.T.; Strauss, S.; Vogt, P.M.; Radtke, C. Effect of Exosomes from Rat Adipose-Derived Mesenchymal Stem Cells on Neurite Outgrowth and Sciatic Nerve Regeneration After Crush Injury. Mol. Neurobiol. 2019, 56, 1812–1824. [Google Scholar] [CrossRef]
- Cui, Y.; Luan, J.; Li, H.; Zhou, X.; Han, J. Exosomes derived from mineralizing osteoblasts promote ST2 cell osteogenic differentiation by alteration of microRNA expression. FEBS Lett. 2016, 590, 185–192. [Google Scholar] [CrossRef]
- Goldie, B.J.; Dun, M.D.; Lin, M.; Smith, N.D.; Verrills, N.M.; Dayas, C.V.; Cairns, M.J. Activity-associated miRNA are packaged in Map1b-enriched exosomes released from depolarized neurons. Nucleic Acids Res. 2014, 42, 9195–9208. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T.F.; Charles, P.D.; Gracia, T.; Hester, S.S.; Gatto, L.; Al-Lamki, R.; Floto, R.A.; Su, Y.; Skepper, J.N.; Lilley, K.S.; et al. Human urinary exosomes as innate immune effectors. J. Am. Soc. Nephrol. 2014, 25, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Wang, Y.; Dakhlallah, D.; Moldovan, L.; Agarwal, K.; Batte, K.; Shah, P.; Wisler, J.; Eubank, T.D.; Tridandapani, S.; et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 2013, 121, 984–995. [Google Scholar] [CrossRef]
- Smythies, L.E.; Smythies, J.R. Exosomes in the gut. Front. Immunol 2014, 5, 104. [Google Scholar] [CrossRef][Green Version]
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 140203. [Google Scholar] [CrossRef]
- Chan, B.D.; Wong, W.Y.; Lee, M.M.; Cho, W.C.; Yee, B.K.; Kwan, Y.W.; Tai, W.C. Exosomes in inflammation and inflammatory disease. Proteomics 2019, e1800149. [Google Scholar] [CrossRef]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.; Hole, P.; Carr, B.; Redman, C.W.; Harris, A.L.; Dobson, P.J.; et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine 2011, 7, 780–788. [Google Scholar] [CrossRef]
- Edgar, J.R. Q&A: What are exosomes, exactly? BMC Biol. 2016, 14, 46. [Google Scholar] [CrossRef]
- Urbanelli, L.; Magini, A.; Buratta, S.; Brozzi, A.; Sagini, K.; Polchi, A.; Tancini, B.; Emiliani, C. Signaling pathways in exosomes biogenesis, secretion and fate. Genes 2013, 4, 152–170. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; Brand, F., 3rd; Adamczak, S.; Lee, S.W.; Perez-Barcena, J.; Wang, M.Y.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W. Exosome-mediated inflammasome signaling after central nervous system injury. J. Neurochem 2016, 136 (Suppl. 1), 39–48. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Tan, J.; Miao, Y.; Zhang, Q. Potential role of exosomes in the pathophysiology, diagnosis, and treatment of hypoxic diseases. Am. J. Transl. Res. 2019, 11, 1184–1201. [Google Scholar]
- Fleshner, M.; Crane, C.R. Exosomes, DAMPs and miRNA: Features of Stress Physiology and Immune Homeostasis. Trends Immunol. 2017, 38, 768–776. [Google Scholar] [CrossRef]
- Williams, E.C.; Coimbra, R.; Chan, T.W.; Baird, A.; Eliceiri, B.P.; Costantini, T.W. Precious cargo: Modulation of the mesenteric lymph exosome payload after hemorrhagic shock. J. Trauma Acute Care Surg. 2019, 86, 52–61. [Google Scholar] [CrossRef]
- Wu, J.; Xu, D.; Luo, P. Differential protein expression in exosomal samples taken from trauma patients. Proteomics Clin. Appl. 2017, 11. [Google Scholar] [CrossRef]
- Console, L.; Scalise, M.; Indiveri, C. Exosomes in inflammation and role as biomarkers. Clin. Chim. Acta 2019, 488, 165–171. [Google Scholar] [CrossRef]
- Alexander, M.; Ramstead, A.G.; Bauer, K.M.; Lee, S.H.; Runtsch, M.C.; Wallace, J.; Huffaker, T.B.; Larsen, D.K.; Tolmachova, T.; Seabra, M.C.; et al. Rab27-Dependent Exosome Production Inhibits Chronic Inflammation and Enables Acute Responses to Inflammatory Stimuli. J. Immunol. 2017, 199, 3559–3570. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys Acta 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Yang, J.; Guo, S.; Zhao, G.; Wu, H.; Deng, G. Peripheral Circulating Exosome-Mediated Delivery of miR-155 as a Novel Mechanism for Acute Lung Inflammation. Mol. Ther. 2019, 27, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Urinary biomarkers of prostate cancer. Int. J. Urol. 2018, 25, 770–779. [Google Scholar] [CrossRef]
- Manek, R.; Moghieb, A.; Yang, Z.; Kumar, D.; Kobessiy, F.; Sarkis, G.A.; Raghavan, V.; Wang, K.K.W. Protein Biomarkers and Neuroproteomics Characterization of Microvesicles/Exosomes from Human Cerebrospinal Fluid Following Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 6112–6128. [Google Scholar] [CrossRef]
- Sarkar, F.H. The Role of Exosomal Cargo in the Regulation of the Biological Complexity of Pancreatic Cancer. Pancreat. Disord. Ther. 2015, 5, 4. [Google Scholar] [CrossRef]
- Ticlea, M.; Bratu, L.M.; Bodog, F.; Bedreag, O.H.; Rogobete, A.F.; Crainiceanu, Z.P. The Use of Exosomes as Biomarkers for Evaluating and Monitoring Critically Ill Polytrauma Patients with Sepsis. Biochem. Genet. 2017, 55, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Zhou, W.; Liu, L.; Fong, M.Y.; Champer, J.; Van Haute, D.; Chin, A.R.; Ren, X.; Gugiu, B.G.; Meng, Z.; et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-kappaB. Sci. Rep. 2014, 4, 5750. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, T.; Zheng, M.; Liu, Y.; Chen, Z. Exosomal proteins as potential markers of tumor diagnosis. J. Hematol. Oncol. 2017, 10, 175. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Saha, B.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. Increased number of circulating exosomes and their microRNA cargos are potential novel biomarkers in alcoholic hepatitis. J. Transl. Med. 2015, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Gao, Y.; Zhang, L.; Chen, Y.; Ge, W.; Tang, P. Involvement of serum-derived exosomes of elderly patients with bone loss in failure of bone remodeling via alteration of exosomal bone-related proteins. Aging Cell 2018, 17, e12758. [Google Scholar] [CrossRef]
- Kelemen, E.; Danis, J.; Göblös, A.; Bata-Csörgő, Z.; Széll, M. Exosomal long non-coding RNAs as biomarkers in human diseases. EJIFCC 2019, 30, 224–236. [Google Scholar]
- Kouwaki, T.; Okamoto, M.; Tsukamoto, H.; Fukushima, Y.; Oshiumi, H. Extracellular Vesicles Deliver Host and Virus RNA and Regulate Innate Immune Response. Int. J. Mol. Sci. 2017, 18, 666. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef]
- Heijnen, H.F.G.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exosomes Derived From Exocytosis of Multivesicular Bodies and alpha-Granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes--vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Timár, C.I.; Lőrincz, A.M.; Csépányi-Kömi, R.; Vályi-Nagy, A.; Nagy, G.; Buzás, E.I.; Iványi, Z.; Kittel, A.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Tanaka, Y.; Okada, R.; Takahashi, A. Biology of extracellular vesicles secreted from senescent cells as senescence-associated secretory phenotype factors. Geriatr. Gerontol. Int. 2020, 20, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; Mittelbrunn, M.; Sánchez-Madrid, F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol. Rev. 2013, 251, 125–142. [Google Scholar] [CrossRef]
- Ge, Q.; Zhou, Y.; Lu, J.; Bai, Y.; Xie, X.; Lu, Z. miRNA in plasma exosome is stable under different storage conditions. Molecules 2014, 19, 1568–1575. [Google Scholar] [CrossRef]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J.O. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef]
- Sanz-Rubio, D.; Martin-Burriel, I.; Gil, A.; Cubero, P.; Forner, M.; Khalyfa, A.; Marin, J.M. Stability of Circulating Exosomal miRNAs in Healthy Subjects. Sci. Rep. 2018, 8, 10306. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Umansky, S.R. Circulating cell-free microRNA as biomarkers for screening, diagnosis and monitoring of neurodegenerative diseases and other neurologic pathologies. Front. Cell Neurosci. 2013, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Raposo, G.; Thery, C. Exosome secretion: Molecular mechanisms and roles in immune responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Geis-Asteggiante, L.; Belew, A.T.; Clements, V.K.; Edwards, N.J.; Ostrand-Rosenberg, S.; El-Sayed, N.M.; Fenselau, C. Differential Content of Proteins, mRNAs, and miRNAs Suggests that MDSC and Their Exosomes May Mediate Distinct Immune Suppressive Functions. J. Proteome Res. 2018, 17, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Andreu, Z.; Yanez-Mo, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef]
- Batagov, A.O.; Kurochkin, I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3’-untranslated regions. Biol. Direct 2013, 8, 12. [Google Scholar] [CrossRef]
- Bagga, S.; Bracht, J.; Hunter, S.; Massirer, K.; Holtz, J.; Eachus, R.; Pasquinelli, A.E. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 2005, 122, 553–563. [Google Scholar] [CrossRef]
- Guo, L.; Lu, Z. The fate of miRNA* strand through evolutionary analysis: Implication for degradation as merely carrier strand or potential regulatory molecule? PLoS ONE 2010, 5, e11387. [Google Scholar] [CrossRef]
- Islam, A.; Ghimbovschi, S.; Zhai, M.; Swift, J.M. An Exploration of Molecular Correlates Relevant to Radiation Combined Skin-Burn Trauma. PLoS ONE 2015, 10, e0134827. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, X.; Cui, H.; Luo, X.; Tang, Y.; Chen, S.; Wu, L.; Shen, N. miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood 2010, 116, 5885–5894. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.C.; Singh, J.; Barik, S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol. Cell Pharmacol. 2011, 3, 83–92. [Google Scholar]
- Gu, H.; Wu, L.; Chen, H.; Huang, Z.; Xu, J.; Zhou, K.; Zhang, Y.; Chen, J.; Xia, J.; Yin, X. Identification of differentially expressed microRNAs in the bone marrow of osteoporosis patients. Am. J. Transl. Res. 2019, 11, 2940–2954. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; Baer, C.; Burdet, F.; Maderna, C.; Gilfillan, G.D.; Lyle, R.; Ibberson, M.; De Palma, M. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014, 8, 1432–1446. [Google Scholar] [CrossRef]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Yousefi, H.; Maheronnaghsh, M.; Molaei, F.; Mashouri, L.; Reza Aref, A.; Momeny, M.; Alahari, S.K. Long noncoding RNAs and exosomal lncRNAs: Classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene 2020, 39, 953–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ao, L.; Yang, J. Long non-coding RNAs in diseases related to inflammation and immunity. Ann. Transl. Med. 2019, 7, 494. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.A.; Moss, L.D.; Lee, J.Y.; Tajiri, N.; Acosta, S.; Hudson, C.; Parag, S.; Cooper, D.R.; Borlongan, C.V.; Bickford, P.C. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J. Neuroinflammation 2018, 15, 204. [Google Scholar] [CrossRef] [PubMed]
- Sobrino, J.; Shafi, S. Timing and Causes of Death After Injuries. Bayl. Univ. Med Cent. Proc. 2013, 26, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, R.; Teuben, M.; Andruszkow, H.; Barkatali, B.M.; Pape, H.-C. Mortality Patterns in Patients with Multiple Trauma: A Systematic Review of Autopsy Studies. PLoS ONE 2016, 11, e0148844. [Google Scholar] [CrossRef]
- Thompson, K.B.; Krispinsky, L.T.; Stark, R.J. Late immune consequences of combat trauma: A review of trauma-related immune dysfunction and potential therapies. Mil. Med. Res. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Burk, A.M.; Martin, M.; Flierl, M.A.; Rittirsch, D.; Helm, M.; Lampl, L.; Bruckner, U.; Stahl, G.L.; Blom, A.M.; Perl, M.; et al. Early complementopathy after multiple injuries in humans. Shock 2012, 37, 348–354. [Google Scholar] [CrossRef]
- Frith, D.; Goslings, J.C.; Gaarder, C.; Maegele, M.; Cohen, M.J.; Allard, S.; Johansson, P.I.; Stanworth, S.; Thiemermann, C.; Brohi, K. Definition and drivers of acute traumatic coagulopathy: Clinical and experimental investigations. J. Thromb. Haemost. 2010, 8, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, J.A.; Potter, B.K.; Polfer, E.M.; Safford, S.D.; Elster, E.A. Do inflammatory markers portend heterotopic ossification and wound failure in combat wounds? Clin. Orthop Relat. Res. 2014, 472, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.N.; Forsberg, J.A.; Potter, B.K.; Hawksworth, J.S.; Brown, T.S.; Andersen, R.; Dunne, J.R.; Tadaki, D.; Elster, E.A. Inflammatory cytokine and chemokine expression is associated with heterotopic ossification in high-energy penetrating war injuries. J. Orthop. Trauma 2012, 26, e204–e213. [Google Scholar] [CrossRef]
- Kleinveld, D.J.; Tuip-de Boer, A.M.; Hollmann, M.W.; Juffermans, N.P. Early increase in anti-inflammatory biomarkers is associated with the development of multiple organ dysfunction syndrome in severely injured trauma patients. Trauma Surg Acute Care Open 2019, 4, e000343. [Google Scholar] [CrossRef]
- Alazawi, W.; Pirmadjid, N.; Lahiri, R.; Bhattacharya, S. Inflammatory and Immune Responses to Surgery and Their Clinical Impact. Ann. Surg. 2016, 264, 73–80. [Google Scholar] [CrossRef]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.F. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Crit. Care 2009, 13, R174. [Google Scholar] [CrossRef]
- Giannoudis, P.V.; Mallina, R.; Harwood, P.; Perry, S.; Sante, E.D.; Pape, H.C. Pattern of release and relationship between HMGB-1 and IL-6 following blunt trauma. Injury 2010, 41, 1323–1327. [Google Scholar] [CrossRef]
- Deitch, E.A. Multiple organ failure. Pathophysiology and potential future therapy. Ann. Surg. 1992, 216, 117–134. [Google Scholar] [CrossRef]
- Gebhard, F.; Pfetsch, H.; Steinbach, G.; Strecker, W.; Kinzl, L.; Bruckner, U.B. Is interleukin 6 an early marker of injury severity following major trauma in humans? Arch. Surg. 2000, 135, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.; Raposo, F.; Fonseca, S.; Valente, L.; Duarte, F.; Goncalves, M.; Tuna, D.; Paiva, J.A. Measurement of cytokines and adhesion molecules in the first 72 h after severe trauma: Association with severity and outcome. Dis. Markers 2015, 2015, 747036. [Google Scholar] [CrossRef]
- Dekker, A.B.; Krijnen, P.; Schipper, I.B. Predictive value of cytokines for developing complications after polytrauma. World J. Crit. Care Med. 2016, 5, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.M.; Prince, J.M.; Yang, R.; Mollen, K.P.; Liao, H.; Watson, G.A.; Fink, M.P.; Vodovotz, Y.; Billiar, T.R. Systemic inflammation and remote organ damage following bilateral femur fracture requires Toll-like receptor 4. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R970–R976. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Massin, F.; Alauzet, C.; Montemont, C.; Lozniewski, A.; Bollaert, P.E.; Levy, B. Effects of the TREM-1 pathway modulation during mesenteric ischemia-reperfusion in rats. Crit. Care Med. 2008, 36, 504–510. [Google Scholar] [CrossRef]
- Mishra, K.P.; Jain, S.; Ganju, L.; Singh, S.B. Hypoxic Stress Induced TREM-1 and Inflammatory Chemokines in Human Peripheral Blood Mononuclear Cells. Indian J. Clin. Biochem. 2014, 29, 133–138. [Google Scholar] [CrossRef][Green Version]
- Tran, T.P.; Tu, H.; Liu, J.; Muelleman, R.L.; Li, Y.L. Mitochondria-derived superoxide links to tourniquet-induced apoptosis in mouse skeletal muscle. PLoS ONE 2012, 7, e43410. [Google Scholar] [CrossRef][Green Version]
- Bertheloot, D.; Latz, E. HMGB1, IL-1alpha, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Schiraldi, M.; Raucci, A.; Munoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tao, T.; Raftery, M.J.; Youssef, P.; Di Girolamo, N.; Geczy, C.L. Proinflammatory properties of the human S100 protein S100A12. J. Leukoc. Biol. 2001, 69, 986–994. [Google Scholar]
- Beninson, L.A.; Fleshner, M. Exosomes: An emerging factor in stress-induced immunomodulation. Semin. Immunol. 2014, 26, 394–401. [Google Scholar] [CrossRef]
- Li, H.; Liu, J.; Yao, J.; Zhong, J.; Guo, L.; Sun, T. Fracture initiates systemic inflammatory response syndrome through recruiting polymorphonuclear leucocytes. Immunol. Res. 2016, 64, 1053–1059. [Google Scholar] [CrossRef]
- Schwacha, M.G.; Rani, M.; Zhang, Q.; Nunez-Cantu, O.; Cap, A.P. Mitochondrial damage-associated molecular patterns activate gammadelta T-cells. Innate Immun. 2014, 20, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Warner, N.; Viani, K.; Nunez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, M.H.; Abdelwahab, S.F.; Wan, J.; Cai, W.; Huixuan, W.; Jianjun, C.; Kumar, K.D.; Vasudevan, A.; Sadek, A.; Su, Z.; et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J. Transl. Med. 2020, 18, 58. [Google Scholar] [CrossRef]
- Ward, N.S.; Casserly, B.; Ayala, A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin. Chest Med. 2008, 29, 617–625. [Google Scholar] [CrossRef]
- Vourc’h, M.; Roquilly, A.; Asehnoune, K. Trauma-Induced Damage-Associated Molecular Patterns-Mediated Remote Organ Injury and Immunosuppression in the Acutely Ill Patient. Front. Immunol. 2018, 9, 1330. [Google Scholar] [CrossRef]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef]
- Kudo, S.; Mizuno, K.; Hirai, Y.; Shimizu, T. Clearance and tissue distribution of recombinant human interleukin 1 beta in rats. Cancer Res. 1990, 50, 5751–5755. [Google Scholar]
- Fantuzzi, G.; Ku, G.; Harding, M.W.; Livingston, D.J.; Sipe, J.D.; Kuida, K.; Flavell, R.A.; Dinarello, C.A. Response to local inflammation of IL-1 beta-converting enzyme- deficient mice. J. Immunol. 1997, 158, 1818–1824. [Google Scholar]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Fattori, E.; Cappelletti, M.; Costa, P.; Sellitto, C.; Cantoni, L.; Carelli, M.; Faggioni, R.; Fantuzzi, G.; Ghezzi, P.; Poli, V. Defective inflammatory response in interleukin 6-deficient mice. J. Exp. Med. 1994, 180, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Kuribayashi, T. Elimination half-lives of interleukin-6 and cytokine-induced neutrophil chemoattractant-1 synthesized in response to inflammatory stimulation in rats. Lab. Anim. Res. 2018, 34, 80–83. [Google Scholar] [CrossRef]
- Jiang, W.G.; Sanders, A.J.; Ruge, F.; Harding, K.G. Influence of interleukin-8 (IL-8) and IL-8 receptors on the migration of human keratinocytes, the role of PLC-gamma and potential clinical implications. Exp. Ther. Med. 2012, 3, 231–236. [Google Scholar] [CrossRef]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Ni Choileain, N.; Redmond, H.P. Cell response to surgery. Arch. Surg. 2006, 141, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Chijioke, O.; Munz, C. Dendritic cell derived cytokines in human natural killer cell differentiation and activation. Front. Immunol. 2013, 4, 365. [Google Scholar] [CrossRef] [PubMed]
- Sobo-Vujanovic, A.; Munich, S.; Vujanovic, N.L. Dendritic-cell exosomes cross-present Toll-like receptor-ligands and activate bystander dendritic cells. Cell Immunol. 2014, 289, 119–127. [Google Scholar] [CrossRef]
- Tibúrcio, R.; Nunes, S.; Nunes, I.; Rosa Ampuero, M.; Silva, I.B.; Lima, R.; Machado Tavares, N.; Brodskyn, C. Molecular Aspects of Dendritic Cell Activation in Leishmaniasis: An Immunobiological View. Front. Immunol. 2019, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol. 2000 2015, 69, 142–159. [Google Scholar] [CrossRef] [PubMed]
- Stoecklein, V.M.; Osuka, A.; Lederer, J.A. Trauma equals danger--damage control by the immune system. J. Leukoc. Biol. 2012, 92, 539–551. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Ni Choileain, N.; MacConmara, M.; Zang, Y.; Murphy, T.J.; Mannick, J.A.; Lederer, J.A. Enhanced regulatory T cell activity is an element of the host response to injury. J. Immunol. 2006, 176, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Lukas, D.; Clausen, B.E.; Yogev, N. Dendritic cells as gatekeepers of tolerance. Semin. Immunopathol. 2017, 39, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Llopiz, D.; Ruiz, M.; Infante, S.; Villanueva, L.; Silva, L.; Hervas-Stubbs, S.; Alignani, D.; Guruceaga, E.; Lasarte, J.J.; Sarobe, P. IL-10 expression defines an immunosuppressive dendritic cell population induced by antitumor therapeutic vaccination. Oncotarget 2017, 8, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Vulpis, E.; Soriani, A.; Cerboni, C.; Santoni, A.; Zingoni, A. Cancer Exosomes as Conveyors of Stress-Induced Molecules: New Players in the Modulation of NK Cell Response. Int. J. Mol. Sci. 2019, 20, 611. [Google Scholar] [CrossRef]
- Collett, G.P.; Redman, C.W.; Sargent, I.L.; Vatish, M. Endoplasmic reticulum stress stimulates the release of extracellular vesicles carrying danger-associated molecular pattern (DAMP) molecules. Oncotarget 2018, 9, 6707–6717. [Google Scholar] [CrossRef]
- Jian, B.; Hsieh, C.H.; Chen, J.; Choudhry, M.; Bland, K.; Chaudry, I.; Raju, R. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochim. Biophys. Acta 2008, 1782, 621–626. [Google Scholar] [CrossRef]
- Gupta, S.; Knowlton, A.A. HSP60 trafficking in adult cardiac myocytes: Role of the exosomal pathway. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, H3052–H3056. [Google Scholar] [CrossRef]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl Acad. Sci. USA 2017, 114, E10255. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.; Choksawangkarn, W.; Edwards, N.; Ostrand-Rosenberg, S.; Fenselau, C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J. Proteome Res. 2014, 13, 836–843. [Google Scholar] [CrossRef]
- Beninson, L.A.; Brown, P.N.; Loughridge, A.B.; Saludes, J.P.; Maslanik, T.; Hills, A.K.; Woodworth, T.; Craig, W.; Yin, H.; Fleshner, M. Acute stressor exposure modifies plasma exosome-associated heat shock protein 72 (Hsp72) and microRNA (miR-142-5p and miR-203). PLoS ONE 2014, 9, e108748. [Google Scholar] [CrossRef]
- Carrière, J.; Bretin, A.; Darfeuille-Michaud, A.; Barnich, N.; Nguyen, H.T. Exosomes Released from Cells Infected with Crohn’s Disease-associated Adherent-Invasive Escherichia coli Activate Host Innate Immune Responses and Enhance Bacterial Intracellular Replication. Inflamm. Bowel. Dis. 2016, 22, 516–528. [Google Scholar] [CrossRef]
- Alivernini, S.; Gremese, E.; McSharry, C.; Tolusso, B.; Ferraccioli, G.; McInnes, I.B.; Kurowska-Stolarska, M. MicroRNA-155-at the Critical Interface of Innate and Adaptive Immunity in Arthritis. Front. Immunol. 2018, 8, 1932. [Google Scholar] [CrossRef]
- Mahesh, G.; Biswas, R. MicroRNA-155: A Master Regulator of Inflammation. J. Interferon Cytokine Res. 2019, 39, 321–330. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef]
- Kerr, N.; Garcia-Contreras, M.; Abbassi, S.; Mejias, N.H.; Desousa, B.R.; Ricordi, C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome Proteins in Serum and Serum-Derived Extracellular Vesicles as Biomarkers of Stroke. Front. Mol. Neurosci 2018, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, H.; Peng, H.; Huyan, T.; Cacalano, N.A. Exosomes: Versatile Nano Mediators of Immune Regulation. Cancers 2019, 11, 1557. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Liu, L.; A, X.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- Wong, W.Y.; Lee, M.M.; Chan, B.D.; Kam, R.K.; Zhang, G.; Lu, A.P.; Tai, W.C. Proteomic profiling of dextran sulfate sodium induced acute ulcerative colitis mice serum exosomes and their immunomodulatory impact on macrophages. Proteomics 2016, 16, 1131–1145. [Google Scholar] [CrossRef]
- Das, S.; Halushka, M.K. Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc. Pathol. 2015, 24, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Shelke, G.V.; Yin, Y.; Jang, S.C.; Lässer, C.; Wennmalm, S.; Hoffmann, H.J.; Li, L.; Gho, Y.S.; Nilsson, J.A.; Lötvall, J. Endosomal signalling via exosome surface TGFbeta-1. J. Extracell Vesicles 2019, 8, 1650458. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, T.S.; Jo, E.K. MiR-146 and miR-125 in the regulation of innate immunity and inflammation. BMB Rep. 2016, 49, 311–318. [Google Scholar] [CrossRef]
- Bhatt, K.; Lanting, L.L.; Jia, Y.; Yadav, S.; Reddy, M.A.; Magilnick, N.; Boldin, M.; Natarajan, R. Anti-Inflammatory Role of MicroRNA-146a in the Pathogenesis of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2277–2288. [Google Scholar] [CrossRef]
- Xie, Y.; Chu, A.; Feng, Y.; Chen, L.; Shao, Y.; Luo, Q.; Deng, X.; Wu, M.; Shi, X.; Chen, Y. MicroRNA-146a: A Comprehensive Indicator of Inflammation and Oxidative Stress Status Induced in the Brain of Chronic T2DM Rats. Front. Pharmacol. 2018, 9, 478. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Dou, H.; Li, X.; Zhao, X.; Li, Y.; Liu, D.; Ji, J.; Liu, F.; Ding, L.; Ni, Y.; et al. Exosomal miR-146a Contributes to the Enhanced Therapeutic Efficacy of Interleukin-1beta-Primed Mesenchymal Stem Cells Against Sepsis. Stem Cells 2017, 35, 1208–1221. [Google Scholar] [CrossRef]
- Roy, S.; Sen, C.K. miRNA in wound inflammation and angiogenesis. Microcirculation 2012, 19, 224–232. [Google Scholar] [CrossRef]
- Nejad, C.; Stunden, H.J.; Gantier, M.P. A guide to miRNAs in inflammation and innate immune responses. FEBS J. 2018, 285, 3695–3716. [Google Scholar] [CrossRef]
- Venkat, P.; Cui, C.; Chopp, M.; Zacharek, A.; Wang, F.; Landschoot-Ward, J.; Shen, Y.; Chen, J. MiR-126 Mediates Brain Endothelial Cell Exosome Treatment-Induced Neurorestorative Effects After Stroke in Type 2 Diabetes Mellitus Mice. Stroke 2019, 50, 2865–2874. [Google Scholar] [CrossRef]
- Alexander, M.; Hu, R.; Runtsch, M.C.; Kagele, D.A.; Mosbruger, T.L.; Tolmachova, T.; Seabra, M.C.; Round, J.L.; Ward, D.M.; O’Connell, R.M. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 2015, 6, 7321. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lei, Y.; Wu, M.; Li, N. Regulation of Macrophage Activation and Polarization by HCC-Derived Exosomal lncRNA TUC339. Int. J. Mol. Sci. 2018, 19, 2958. [Google Scholar] [CrossRef]
- Chen, F.; Wang, N.; Tan, H.Y.; Guo, W.; Zhang, C.; Feng, Y. The functional roles of exosomes-derived long non-coding RNA in human cancer. Cancer Biol. Ther. 2019, 20, 583–592. [Google Scholar] [CrossRef]
- Naderi-Meshkin, H.; Lai, X.; Amirkhah, R.; Vera, J.; Rasko, J.E.J.; Schmitz, U. Exosomal lncRNAs and cancer: Connecting the missing links. Bioinformatics 2019, 35, 352–360. [Google Scholar] [CrossRef]
- Chen, Z.; Dong, W.H.; Qiu, Z.M.; Li, Q.G. The Monocyte-Derived Exosomal CLMAT3 Activates the CtBP2-p300-NF-kappaB Transcriptional Complex to Induce Proinflammatory Cytokines in ALI. Mol. Ther. Nucleic. Acids 2020, 21, 1100–1110. [Google Scholar] [CrossRef]
- Buschow, S.I.; Nolte-’t Hoen, E.N.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Théry, C.; Duban, L.; Segura, E.; Veron, P.; Lantz, O.; Amigorena, S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef]
- van der Vlist, E.J.; Arkesteijn, G.J.; van de Lest, C.H.; Stoorvogel, W.; Nolte-’t Hoen, E.N.; Wauben, M.H. CD4(+) T cell activation promotes the differential release of distinct populations of nanosized vesicles. J. Extracell Vesicles 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Li, Z.; Ling, W.; Zhu, D.; Feng, Z.; Kong, L. Exosomes Derived from Dendritic Cells Attenuate Liver Injury by Modulating the Balance of Treg and Th17 Cells After Ischemia Reperfusion. Cell Physiol. Biochem. 2018, 46, 740–756. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Rodriguez, M.C.; Pindado, J.; Merino, E.; Merida, I.; Izquierdo, M. Diacylglycerol kinase alpha regulates the secretion of lethal exosomes bearing Fas ligand during activation-induced cell death of T lymphocytes. J. Biol. Chem. 2005, 280, 28439–28450. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.G.; Gentile, L.F.; Lopez, M.C.; Ungaro, R.; Liu, H.; Xiao, W.; Seok, J.; Mindrinos, M.N.; Ang, D.; Baslanti, T.O.; et al. Development of a genomic metric that can be rapidly used to predict clinical outcome in severely injured trauma patients. Crit. Care Med. 2013, 41, 1175–1185. [Google Scholar] [CrossRef]
- Hawksworth, J.S.; Stojadinovic, A.; Gage, F.A.; Tadaki, D.K.; Perdue, P.W.; Forsberg, J.; Davis, T.A.; Dunne, J.R.; Denobile, J.W.; Brown, T.S.; et al. Inflammatory biomarkers in combat wound healing. Ann. Surg. 2009, 250, 1002–1007. [Google Scholar] [CrossRef]
- Crouser, E.D.; Julian, M.W.; Bicer, S.; Ghai, V.; Kim, T.K.; Maier, L.A.; Gillespie, M.; Hamzeh, N.Y.; Wang, K. Circulating exosomal microRNA expression patterns distinguish cardiac sarcoidosis from myocardial ischemia. PLoS ONE 2021, 16, e0246083. [Google Scholar] [CrossRef]
- Minghua, W.; Zhijian, G.; Chahua, H.; Qiang, L.; Minxuan, X.; Luqiao, W.; Weifang, Z.; Peng, L.; Biming, Z.; Lingling, Y.; et al. Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring miR-24. Cell Death. Dis. 2018, 9, 320. [Google Scholar] [CrossRef]
- Emanueli, C.; Shearn, A.I.; Laftah, A.; Fiorentino, F.; Reeves, B.C.; Beltrami, C.; Mumford, A.; Clayton, A.; Gurney, M.; Shantikumar, S.; et al. Coronary Artery-Bypass-Graft Surgery Increases the Plasma Concentration of Exosomes Carrying a Cargo of Cardiac MicroRNAs: An Example of Exosome Trafficking Out of the Human Heart with Potential for Cardiac Biomarker Discovery. PLoS ONE 2016, 11, e0154274. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Lee, B.R.; Park, K.H.; Nihalani, D.; Yoon, J.H.; Ikeda, M.; Kwon, S.H. miRNA profiling of urinary exosomes to assess the progression of acute kidney injury. Sci. Rep. 2019, 9, 4692. [Google Scholar] [CrossRef]
- Cheng, M.; Yang, J.; Zhao, X.; Zhang, E.; Zeng, Q.; Yu, Y.; Yang, L.; Wu, B.; Yi, G.; Mao, X.; et al. Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat. Commun. 2019, 10, 959. [Google Scholar] [CrossRef]
- Eldh, M.; Ekström, K.; Valadi, H.; Sjöstrand, M.; Olsson, B.; Jernås, M.; Lötvall, J. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS ONE 2010, 5, e15353. [Google Scholar] [CrossRef]
- Yang, J.C.; Lin, M.W.; Rau, C.S.; Jeng, S.F.; Lu, T.H.; Wu, Y.C.; Chen, Y.C.; Tzeng, S.L.; Wu, C.J.; Hsieh, C.H. Altered exosomal protein expression in the serum of NF-kappaB knockout mice following skeletal muscle ischemia-reperfusion injury. J. Biomed. Sci. 2015, 22, 40. [Google Scholar] [CrossRef]
- Wu, X.; Wu, C.; Gu, W.; Ji, H.; Zhu, L. Serum Exosomal MicroRNAs Predict Acute Respiratory Distress Syndrome Events in Patients with Severe Community-Acquired Pneumonia. Biomed. Res. Int. 2019, 2019, 3612020. [Google Scholar] [CrossRef]
- Kim, T.H.; Hong, S.B.; Lim, C.M.; Koh, Y.; Jang, E.Y.; Huh, J.W. The Role of Exosomes in Bronchoalveloar Lavage from Patients with Acute Respiratory Distress Syndrome. J. Clin. Med. 2019, 8, 1148. [Google Scholar] [CrossRef]
- Moon, H.G.; Cao, Y.; Yang, J.; Lee, J.H.; Choi, H.S.; Jin, Y. Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated inflammatory responses via ROCK1 pathway. Cell Death Dis. 2015, 6, e2016. [Google Scholar] [CrossRef] [PubMed]
- Hashemian, S.M.; Pourhanifeh, M.H.; Fadaei, S.; Velayati, A.A.; Mirzaei, H.; Hamblin, M.R. Non-coding RNAs and Exosomes: Their Role in the Pathogenesis of Sepsis. Mol. Ther. Nucleic. Acids. 2020, 21, 51–74. [Google Scholar] [CrossRef] [PubMed]
- Appiah, M.G.; Park, E.J.; Darkwah, S.; Kawamoto, E.; Akama, Y.; Gaowa, A.; Kalsan, M.; Ahmad, S.; Shimaoka, M. Intestinal Epithelium-Derived Luminally Released Extracellular Vesicles in Sepsis Exhibit the Ability to Suppress TNF-a and IL-17A Expression in Mucosal Inflammation. Int. J. Mol. Sci. 2020, 21, 8445. [Google Scholar] [CrossRef]
- Wu, S.C.; Yang, J.C.; Rau, C.S.; Chen, Y.C.; Lu, T.H.; Lin, M.W.; Tzeng, S.L.; Wu, Y.C.; Wu, C.J.; Hsieh, C.H. Profiling circulating microRNA expression in experimental sepsis using cecal ligation and puncture. PLoS ONE 2013, 8, e77936. [Google Scholar] [CrossRef]
- Real, J.M.; Ferreira, L.R.P.; Esteves, G.H.; Koyama, F.C.; Dias, M.V.S.; Bezerra-Neto, J.E.; Cunha-Neto, E.; Machado, F.R.; Salomao, R.; Azevedo, L.C.P. Exosomes from patients with septic shock convey miRNAs related to inflammation and cell cycle regulation: New signaling pathways in sepsis? Crit. Care 2018, 22, 68. [Google Scholar] [CrossRef] [PubMed]
- Reithmair, M.; Buschmann, D.; Marte, M.; Kirchner, B.; Hagl, D.; Kaufmann, I.; Pfob, M.; Chouker, A.; Steinlein, O.K.; Pfaffl, M.W.; et al. Cellular and extracellular miRNAs are blood-compartment-specific diagnostic targets in sepsis. J. Cell Mol. Med. 2017, 21, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, T.; Bah, I.; Kumbhare, A.; Youssef, D.; Yao, Z.Q.; McCall, C.E.; Gazzar, M.E. Long Non-Coding RNA Hotairm1 Promotes S100A9 Support of MDSC Expansion during Sepsis. J. Clin. Cell Immunol. 2020, 11, 600. [Google Scholar] [PubMed]
- Gambim, M.H.; do Carmo Ade, O.; Marti, L.; Veríssimo-Filho, S.; Lopes, L.R.; Janiszewski, M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: Experimental evidence for a novel mechanism of septic vascular dysfunction. Crit. Care 2007, 11, R107. [Google Scholar] [CrossRef]
- Gao, K.; Jin, J.; Huang, C.; Li, J.; Luo, H.; Li, L.; Huang, Y.; Jiang, Y. Exosomes Derived From Septic Mouse Serum Modulate Immune Responses via Exosome-Associated Cytokines. Front. Immunol. 2019, 10, 1560. [Google Scholar] [CrossRef]
- Tran, T.P.; Tu, H.; Pipinos, I.I.; Muelleman, R.L.; Albadawi, H.; Li, Y.L. Tourniquet-induced acute ischemia-reperfusion injury in mouse skeletal muscles: Involvement of superoxide. Eur. J. Pharmacol. 2011, 650, 328–334. [Google Scholar] [CrossRef][Green Version]
- Rushing, G.D.; Britt, L.D. Reperfusion injury after hemorrhage: A collective review. Ann. Surg. 2008, 247, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Krausz, M.M. Initial resuscitation of hemorrhagic shock. World J. Emerg. Surg. 2006, 1, 14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saini, H.K.; Xu, Y.J.; Zhang, M.; Liu, P.P.; Kirshenbaum, L.A.; Dhalla, N.S. Role of tumour necrosis factor-alpha and other cytokines in ischemia-reperfusion-induced injury in the heart. Exp. Clin. Cardiol. 2005, 10, 213–222. [Google Scholar]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro, E.S.O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef]
- Kojima, M.; Gimenes-Junior, J.A.; Langness, S.; Morishita, K.; Lavoie-Gagne, O.; Eliceiri, B.; Costantini, T.W.; Coimbra, R. Exosomes, not protein or lipids, in mesenteric lymph activate inflammation: Unlocking the mystery of post-shock multiple organ failure. J. Trauma Acute Care Surg. 2017, 82, 42–50. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-mediated production of exosomes during hypoxia is protective in renal tubular cells. Am. J. Physiol. Renal. Physiol. 2017, 313, F906–F913. [Google Scholar] [CrossRef]
- Yu, X.; Deng, L.; Wang, D.; Li, N.; Chen, X.; Cheng, X.; Yuan, J.; Gao, X.; Liao, M.; Wang, M.; et al. Mechanism of TNF-alpha autocrine effects in hypoxic cardiomyocytes: Initiated by hypoxia inducible factor 1alpha, presented by exosomes. J. Mol. Cell Cardiol. 2012, 53, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef]
- Peng, P.; Li, Z.; Liu, X. Reduced Expression of miR-23a Suppresses A20 in TLR-stimulated Macrophages. Inflammation 2015, 38, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.D.; Khorram, O. miR-200c regulates IL8 expression by targeting IKBKB: A potential mediator of inflammation in leiomyoma pathogenesis. PLoS ONE 2014, 9, e95370. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Liu, G.; Qi, X.; Cao, X. MicroRNA-24 inhibits the proliferation and migration of endothelial cells in patients with atherosclerosis by targeting importin-alpha3 and regulating inflammatory responses. Exp. Ther. Med. 2018, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xu, Z.; Yuan, M.; Zhang, Y.; Zhao, B.; Wang, J.; Zhang, A.; Li, G. MicroRNA-16 suppresses the activation of inflammatory macrophages in atherosclerosis by targeting PDCD4. Int. J. Mol. Med. 2016, 37, 967–975. [Google Scholar] [CrossRef]
- Mica, L.; Furrer, E.; Keel, M.; Trentz, O. Predictive ability of the ISS, NISS, and APACHE II score for SIRS and sepsis in polytrauma patients. Eur. J. Trauma Emerg. Surg. 2012, 38, 665–671. [Google Scholar] [CrossRef]
- Marik, P.E.; Taeb, A.M. SIRS, qSOFA and new sepsis definition. J. Thorac. Dis. 2017, 9, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Angele, M.K.; Chaudry, I.H. Surgical trauma and immunosuppression: Pathophysiology and potential immunomodulatory approaches. Langenbecks Arch. Surg. 2005, 390, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.F.; Cuenca, A.G.; Efron, P.A.; Ang, D.; Bihorac, A.; McKinley, B.A.; Moldawer, L.L.; Moore, F.A. Persistent inflammation and immunosuppression: A common syndrome and new horizon for surgical intensive care. J. Trauma Acute Care Surg. 2012, 72, 1491–1501. [Google Scholar] [CrossRef]
- Xu, P.; Wang, F.; Zhou, X.L.; Li, L.; Xiong, D.; Yong, Y.Q.; Zhao, Y.; Jiang, W.X. Systemic Inflammatory Response and Multiple Organ Dysfunctions Following Crush Injury: A New Experimental Model in Rabbits. Inflammation 2018, 41, 240–248. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Kornblith, L.Z.; Robles, A.J.; Conroy, A.S.; Redick, B.J.; Howard, B.M.; Hendrickson, C.M.; Moore, S.; Nelson, M.F.; Moazed, F.; Callcut, R.A.; et al. Predictors of postinjury acute respiratory distress syndrome: Lung injury persists in the era of hemostatic resuscitation. J. Trauma Acute Care Surg. 2019, 87, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Annane, D.; Chrousos, G.P.; Marik, P.E.; Sinclair, S.E. Activation and regulation of systemic inflammation in ARDS: Rationale for prolonged glucocorticoid therapy. Chest 2009, 136, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Abdul Roda, M.; Xu, X.; Rezonzew, G.; Viera, L.; et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell 2019, 176, 113–126.e15. [Google Scholar] [CrossRef]
- Nedeva, C.; Menassa, J.; Puthalakath, H. Sepsis: Inflammation Is a Necessary Evil. Front. Cell Dev. Biol 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Tamura, T.; Sawatsubashi, Y. Sepsis and disseminated intravascular coagulation. J. Intensive Care 2016, 4, 23. [Google Scholar] [CrossRef]
- Faix, J.D. Biomarkers of sepsis. Crit. Rev. Clin. Lab. Sci. 2013, 50, 23–36. [Google Scholar] [CrossRef]
- Terrasini, N.; Lionetti, V. Exosomes in Critical Illness. Crit. Care Med. 2017, 45, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Appiah, M.G.; Myint, P.K.; Gaowa, A.; Kawamoto, E.; Shimaoka, M. Exosomes in Sepsis and Inflammatory Tissue Injury. Curr. Pharm. Des. 2019, 25, 4486–4495. [Google Scholar] [CrossRef]
- Fröhlich, M.; Lefering, R.; Probst, C.; Paffrath, T.; Schneider, M.M.; Maegele, M.; Sakka, S.G.; Bouillon, B.; Wafaisade, A.; Committee on Emergency Medicine, I.C.; et al. Epidemiology and risk factors of multiple-organ failure after multiple trauma: An analysis of 31,154 patients from the TraumaRegister DGU. J. Trauma Acute Care Surg. 2014, 76, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, D.J.; Moore, E.E.; Johnson, J.L.; Sauaia, A.; Cothren, C.C.; Moore, J.B.; Burch, J.M. Multiple organ dysfunction during resuscitation is not postinjury multiple organ failure. Arch. Surg. 2004, 139, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.; Yoo, H.; Lee, J.Y.; Park, J.; Suh, G.Y.; Jeon, K. Association of plasma exosomes with severity of organ failure and mortality in patients with sepsis. J. Cell Mol. Med. 2020, 24, 9439–9445. [Google Scholar] [CrossRef]
- Murao, A.; Brenner, M.; Aziz, M.; Wang, P. Exosomes in Sepsis. Front. Immunol. 2020, 11, 2140. [Google Scholar] [CrossRef]
- Gao, M.; Gao, W.; Papadimitriou, J.M.; Zhang, C.; Gao, J.; Zheng, M. Exosomes-the enigmatic regulators of bone homeostasis. Bone Res. 2018, 6, 36. [Google Scholar] [CrossRef]
- Furuta, T.; Miyaki, S.; Ishitobi, H.; Ogura, T.; Kato, Y.; Kamei, N.; Miyado, K.; Higashi, Y.; Ochi, M. Mesenchymal Stem Cell-Derived Exosomes Promote Fracture Healing in a Mouse Model. Stem Cells Transl. Med. 2016, 5, 1620–1630. [Google Scholar] [CrossRef]
- Zhou, R.; O’Hara, S.P.; Chen, X.M. MicroRNA regulation of innate immune responses in epithelial cells. Cell Mol. Immunol. 2011, 8, 371–379. [Google Scholar] [CrossRef]
- Kaddoura, I.; Abu-Sittah, G.; Ibrahim, A.; Karamanoukian, R.; Papazian, N. Burn injury: Review of pathophysiology and therapeutic modalities in major burns. Ann. Burns Fire Disasters 2017, 30, 95–102. [Google Scholar]
- Williams, F.N.; Herndon, D.N.; Jeschke, M.G. The hypermetabolic response to burn injury and interventions to modify this response. Clin. Plast Surg. 2009, 36, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Yang, W.; Pan, Z.; Zhang, Y.; Li, X.; Lakshmanan, S. Differential proteomics analysis of serum exosomein burn patients. Saudi J. Biol. Sci. 2020, 27, 2215–2220. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Yang, Q.; Wang, Q.; Shi, C.; Wang, D.; Armato, U.; Prà, I.D.; Chiarini, A. Mesenchymal stromal cells-exosomes: A promising cell-free therapeutic tool for wound healing and cutaneous regeneration. Burns Trauma 2019, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, L.; Yang, J.; Yu, Y.; Chai, J.; Wang, L.; Ma, L.; Yin, H. Exosome Derived from Human Umbilical Cord Mesenchymal Stem Cell Mediates MiR-181c Attenuating Burn-induced Excessive Inflammation. EBioMedicine 2016, 8, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, M.; Gong, A.; Zhang, X.; Wu, X.; Zhu, Y.; Shi, H.; Wu, L.; Zhu, W.; Qian, H.; et al. HucMSC-Exosome Mediated-Wnt4 Signaling Is Required for Cutaneous Wound Healing. Stem Cells 2015, 33, 2158–2168. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trauma Associated Condition | Bioactive Material | Exosomal Cargo | Literature |
|---|---|---|---|
| IRI | miRNA | miR-23a | Crouser et al., 2021 [197] |
| miR-24 | Minghua et al., 2018 [198] | ||
| miR-1, miR-133a, miR-24, miR-210, miR-133b | Emanueli et al., 2016 [199] | ||
| miR-16-5p, miR-24-3p, miR-200c-3p, miR-9a-5p, miR-141-3p, miR-200a-3p, miR-429 | Sonoda et al., 2019 [200] | ||
| miR-1, miR-208, miR-499, miR-133 | Chen et al., 2019 [201] | ||
| mRNA | Upregulated: Vsig1, Top1, Ccbp2, 0610010K06Rik, Krit1, D230019N24Rik, Amy2a1, Lba1, Zfp385c, 2700057C20Rik, Ptar1, Smad3, 2810002D19Rik, Phf6, Hsd17b11, 6720457D02Rik, Yipf7, Mep1a, Sox15, 4930473M17Rik | Eldh et al., 2010 [202] | |
| Downregulated: Ctnna1, Pigq, Cct2, Rfc4, Gnas, Ttc3, Laptm5, Gabarapl1, Ipo4, Dnpep, Lmna, Ssr3, Qars, Gsn, Arap3, Med22, Csnk1d, Coro7, Lasp1, Ric8 | |||
| Protein | C3 propeptide, PK-120 precursor, alpha amylase one precursor, beta-enolase isoform 1, adenylosuccinate synthetase isozyme 1 | Yang et al., 2015 [203] | |
| Hsp70 | Zheng et al., 2018 [193] | ||
| ARDS | miRNA | Upregulated: miR-146a, miR-27a, miR-126, miR-155 | Wu et al., 2019 [204] |
| Downregulated: miR-223, miR-181b | |||
| miR-155 | Jiang et al., 2019 [57] | ||
| lncRNA | Upregulated: AOC4P, BCAR4 | Chen et al., 2020 [189] | |
| Downregulated: CLMAT3, MIAT | |||
| Protein | caspase 12, caspase 9, RIP3, microtubule associated proteins 1A/1B light chain B3 | Kim et al., 2019 [205] | |
| caspase 3 | Moon et al., 2015 [206] | ||
| Sepsis | miRNA | miR-155, miR-146a | Alexander et al., 2015 [185] |
| miR-27a | Hashemian et al., 2020 [207] | ||
| miR-19a, miR-21a, miR-22, miR-27a, miR-103-2, miR-107, miR-126a, miR-146b, miR-182, miR-200b, miR-203, miR-762 | Appiah et al., 2020 [208] | ||
| miR-16, miR-17, miR-20a, miR-20b, miR-26a, miR-26b | Wu et al., 2013 [209] | ||
| Upregulated: let-7b-5p, let-7c-5p, miR-122-5p, miR-1227-3p, miR-125b-5p, miR-1260a, miR-1262, miR-1267, miR-1290, miR-1298-5p, miR-1300, miR-140-3p, miR-16-5p, miR-1825, miR-192-5p, miR-193a-5p, miR-194-5p, miR-195-5p, miR-19a-3p, miR-25-3p, miR-30a-5p, miR-320a, miR-320b, miR-363-3p, miR-486-5p, miR-518d-3p, miR-519b-3p, miR-520d-3p, miR-532-3p, miR-548a-3p miR-548c-3p, miR-597-5p, miR-618, miR-625-3p, miR-636, miR-645, miR-720, miR-758-3p, miR-770-5p, miR-885-5p, miR-886-5p, miR-92a-3p, miR-99b-3p | Real et al., 2018 [210] | ||
| Downregulated: miR-127-3p, miR-146a-5p, miR-151a-3p, miR-186-5p, miR-18a-5p, miR-199a-3p, miR-221-3p, miR-26a-5p, miR-28-5p, miR-301a-3p, miR-328, miR-331-3p, miR-335-5p, miR-339-3p, miR-340-5p, miR-340-3p, miR-590-3p, miR-628-5p, miR-744-5p | |||
| miR-16, miR-17, miR-20a, miR-20b, miR-26a, miR-26b | Wu et al., 2013 [209] | ||
| miR-27b, miR-125b, miR-21-5p, miR-30a-5p, miR-100-5p, miR-122-5p, miR-193a-5p | Reithmair et al., 2017 [211] | ||
| lncRNA | Hotairm1 | Alkhateeb et al., 2020 [212] | |
| Protein | NADPH, NO synthase | Gambim et al., 2007 [213] | |
| IL-1β, IL-2, IL-6, TNF-α, IL-4, IL-10, CCL2, CCL3 | Gao et al., 2019 [214] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walsh, S.A.; Hoyt, B.W.; Rowe, C.J.; Dey, D.; Davis, T.A. Alarming Cargo: The Role of Exosomes in Trauma-Induced Inflammation. Biomolecules 2021, 11, 522. https://doi.org/10.3390/biom11040522
Walsh SA, Hoyt BW, Rowe CJ, Dey D, Davis TA. Alarming Cargo: The Role of Exosomes in Trauma-Induced Inflammation. Biomolecules. 2021; 11(4):522. https://doi.org/10.3390/biom11040522
Chicago/Turabian StyleWalsh, Sarah A., Benjamin W. Hoyt, Cassie J. Rowe, Devaveena Dey, and Thomas A. Davis. 2021. "Alarming Cargo: The Role of Exosomes in Trauma-Induced Inflammation" Biomolecules 11, no. 4: 522. https://doi.org/10.3390/biom11040522
APA StyleWalsh, S. A., Hoyt, B. W., Rowe, C. J., Dey, D., & Davis, T. A. (2021). Alarming Cargo: The Role of Exosomes in Trauma-Induced Inflammation. Biomolecules, 11(4), 522. https://doi.org/10.3390/biom11040522