
A Molecular Modeling Approach to Identify Potential Antileishmanial Compounds Against the Cell Division Cycle (cdc)-2-Related Kinase 12 (CRK12) Receptor of Leishmania donovani

 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval
2.2. Obtaining the Structure of LdCRK12 and Human CDK9
2.2.1. Template Search and Selection
2.2.2. Structure Prediction Using Modeller
2.2.3. Structure Prediction Using I-TASSER
2.2.4. Structure Prediction Using Robetta
2.3. Structural Validation
2.4. Prediction of Binding Sites
2.5. Preparation of Proteins and Ligand Libraries
2.6. Virtual Screening
2.7. Characterisation of Mechanism of Binding
2.8. Pharmacological Profiling
2.9. Quality Evaluation of Shortlisted Molecules
2.10. MD Simulations of Proteins and Protein–Ligand Complexes
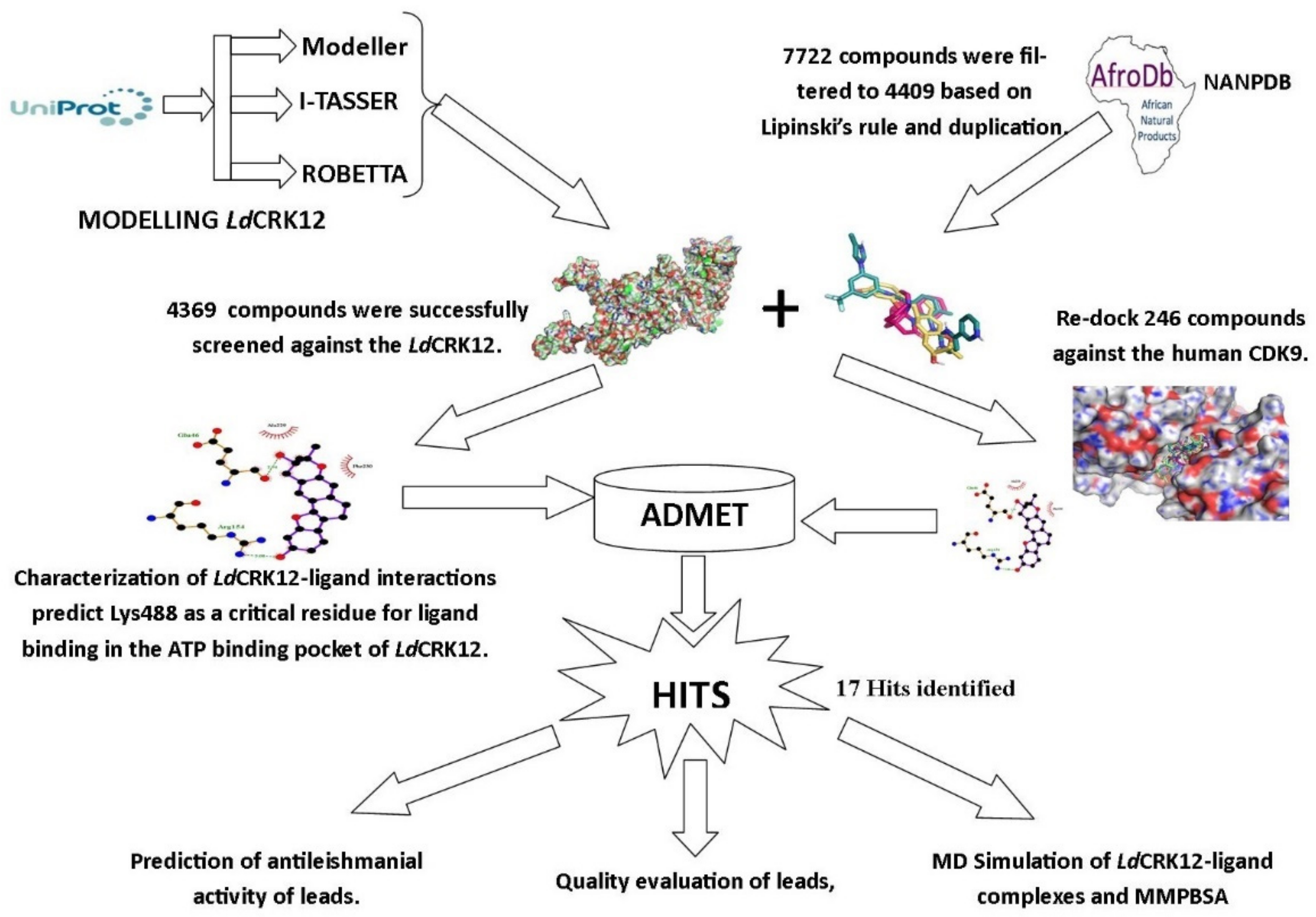
3. Results and Discussion
3.1. Modelling the Structure of LdCRK12
3.1.1. Template Search
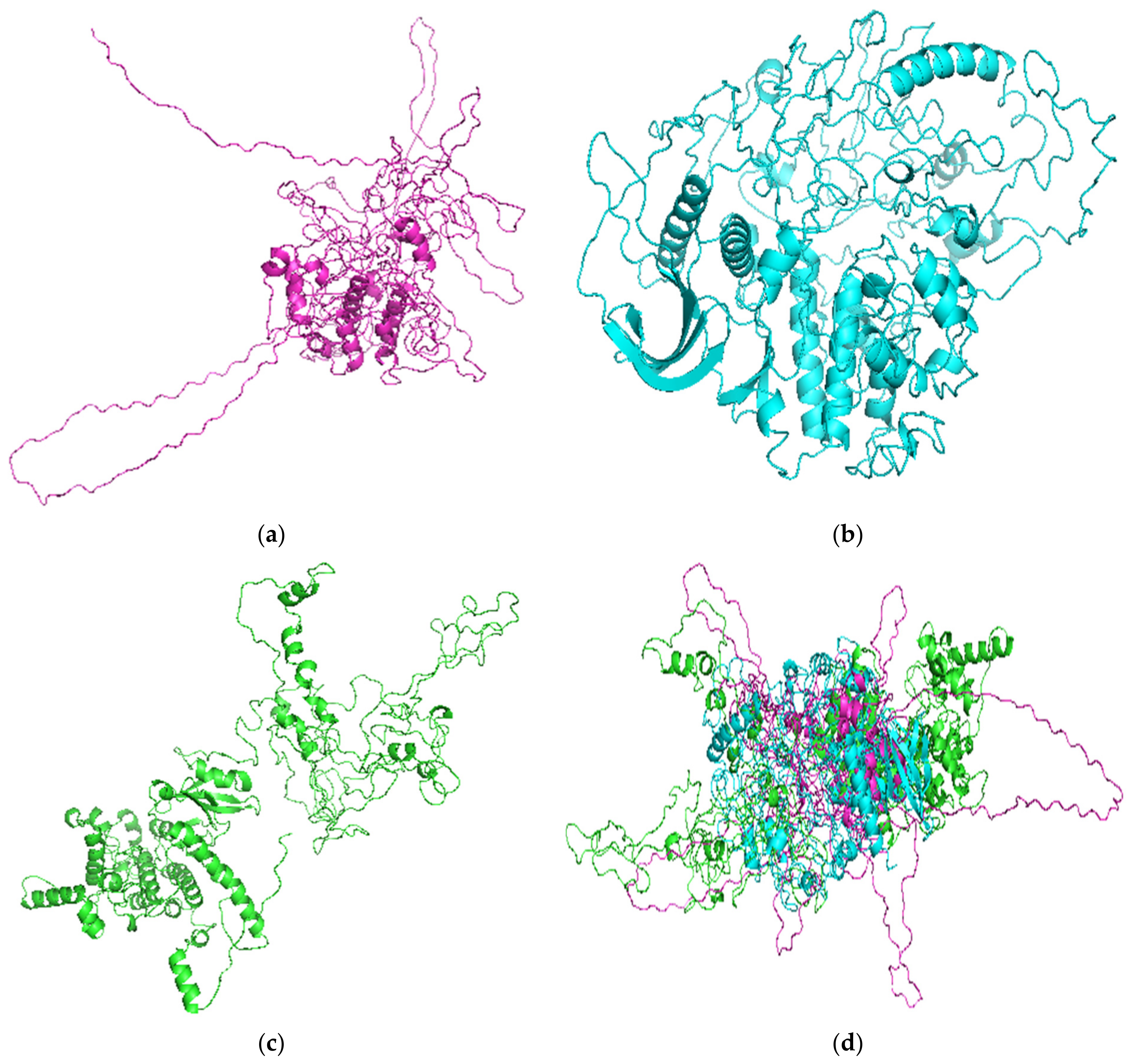
3.1.2. Structure Prediction Using Modeller
3.1.3. Structure Prediction Using I-TASSER
3.1.4. Structure Prediction Using Robetta
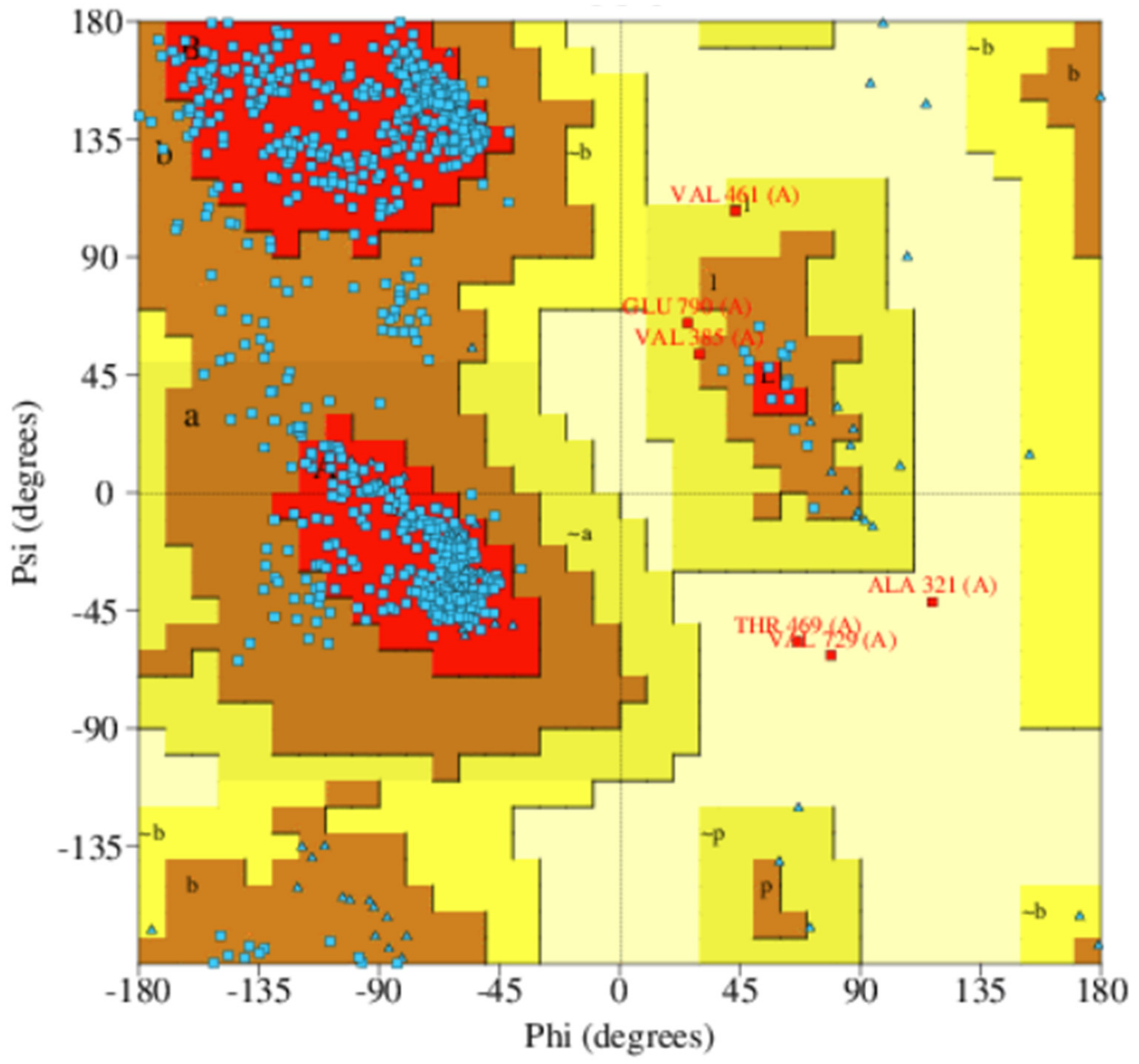
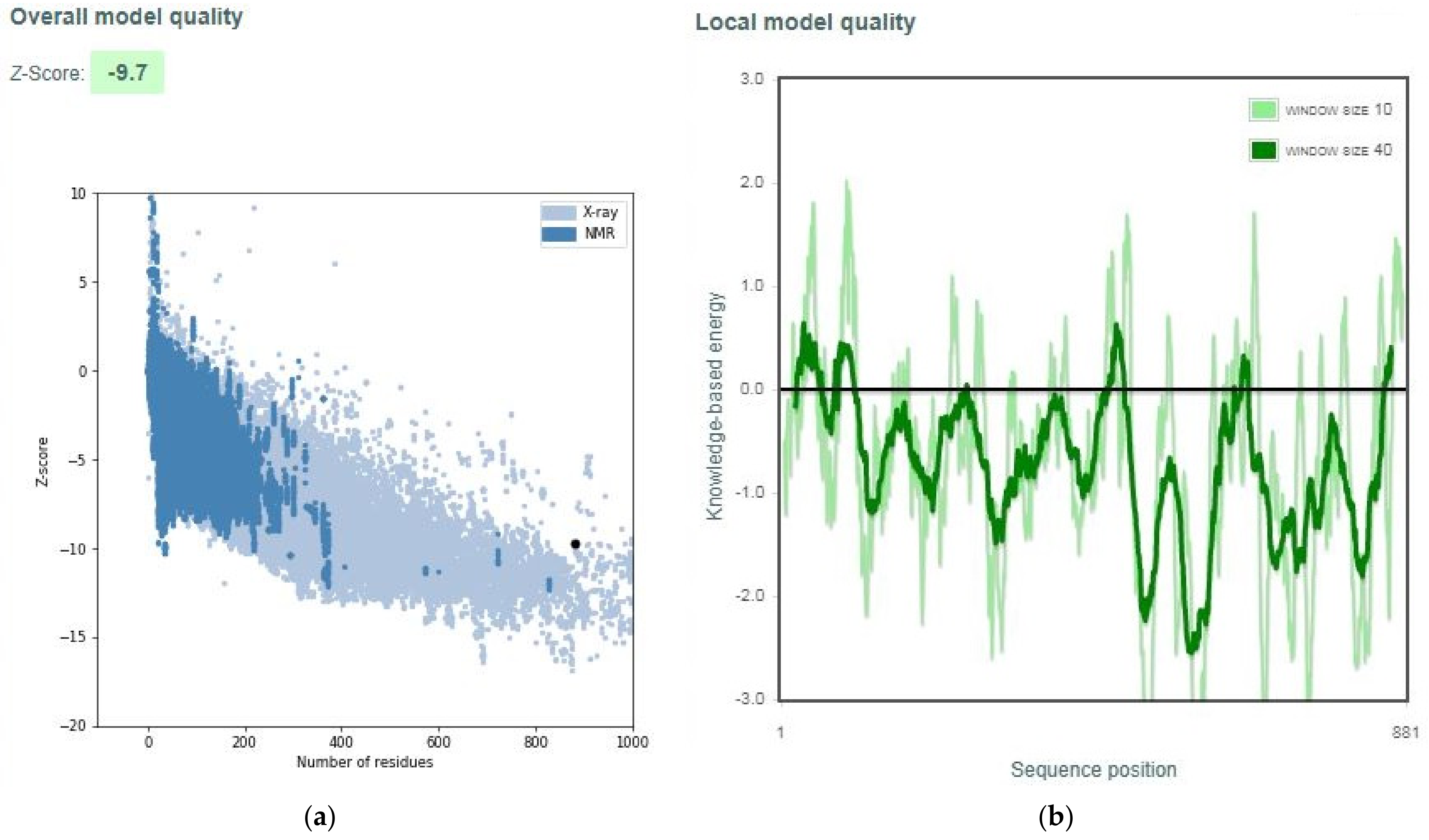
3.2. Quality Assessment of Selected Models
3.3. Binding Site Characterization
3.4. Preparation of Screening Library
3.5. Virtual Screening of Compounds
3.5.1. Screening the Library against LdCRK12
3.5.2. Re-Docking Compounds against the CDK9
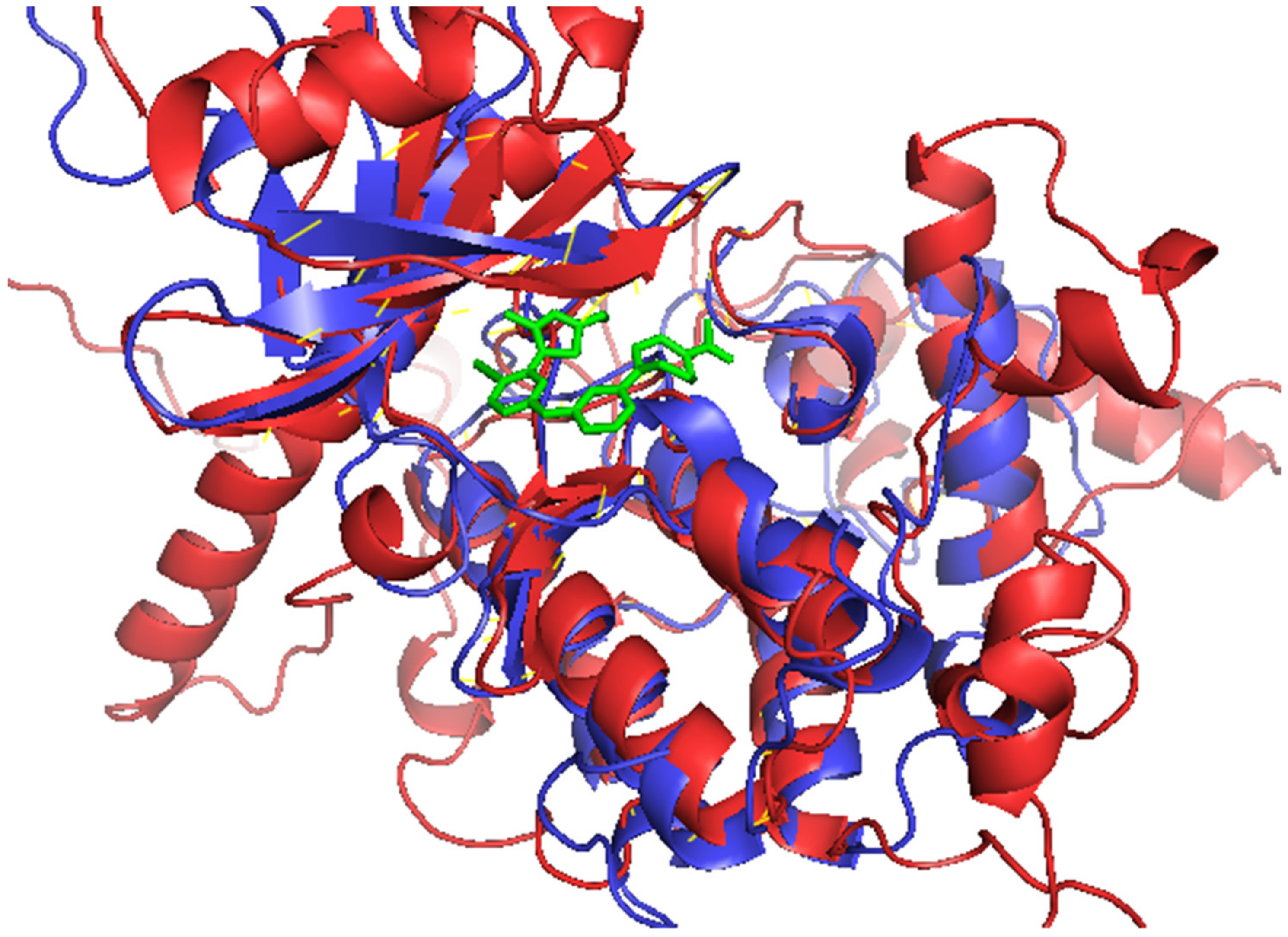
3.6. Characterisation of Mechanisms of Binding
3.6.1. Characterization of LdCRK12–Ligand Interactions
3.6.2. Characterization of Human CDK9–Ligand Interactions
3.7. ADMET Prediction
3.8. Toxicity Prediction with OSIRIS Property Explorer
3.9. Biological Activities of Hits
3.10. Ligand Efficiency-Based Metrics for Selected Compounds
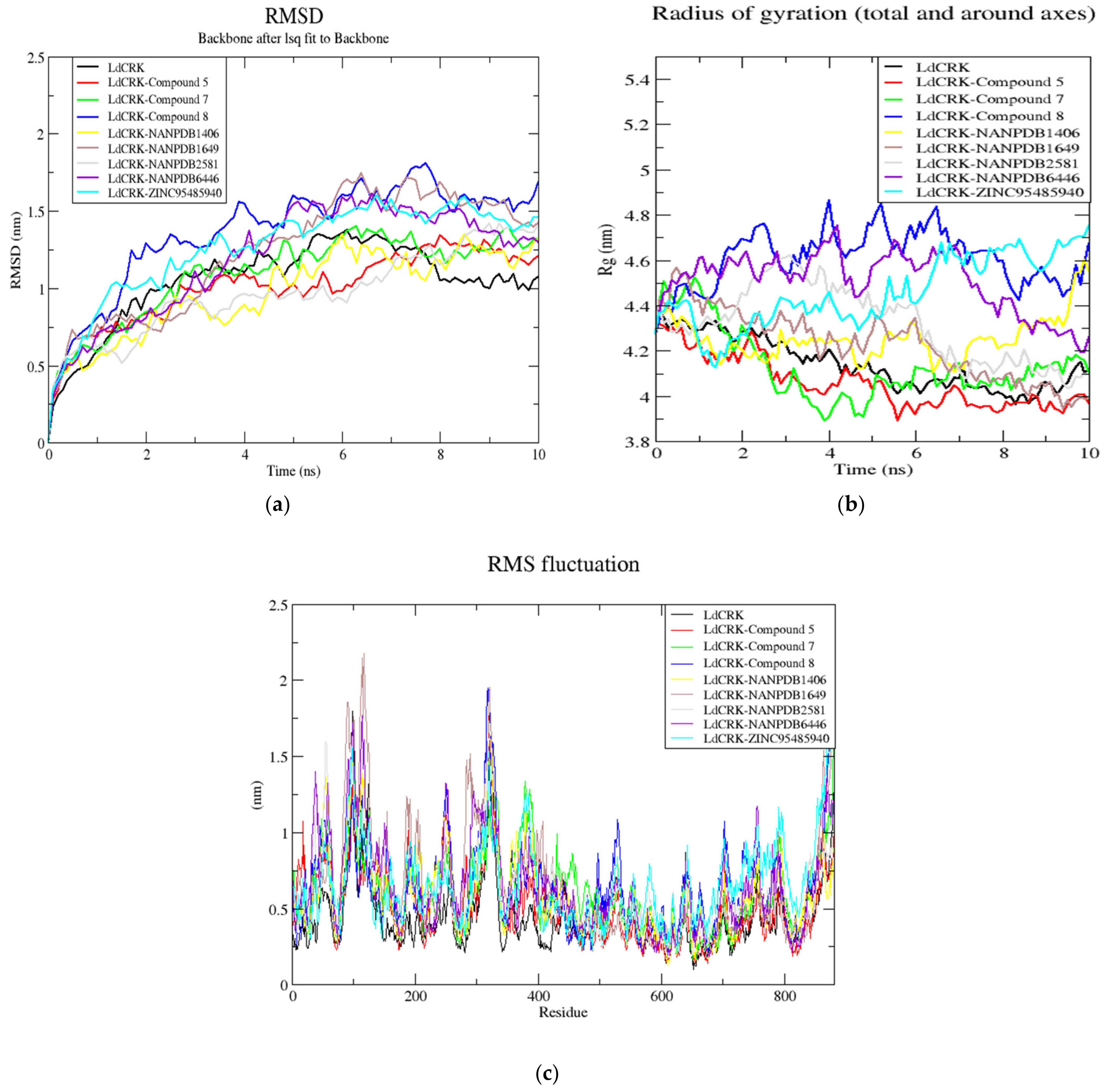
3.11. Molecular Dynamics Simulations
3.11.1. The Root Mean Square Deviation (RMSD) of the Complexes
3.11.2. The Radius of Gyration (Rg) of Complexes
3.11.3. The Root Mean Square Fluctuation (RMSF) of the Complexes
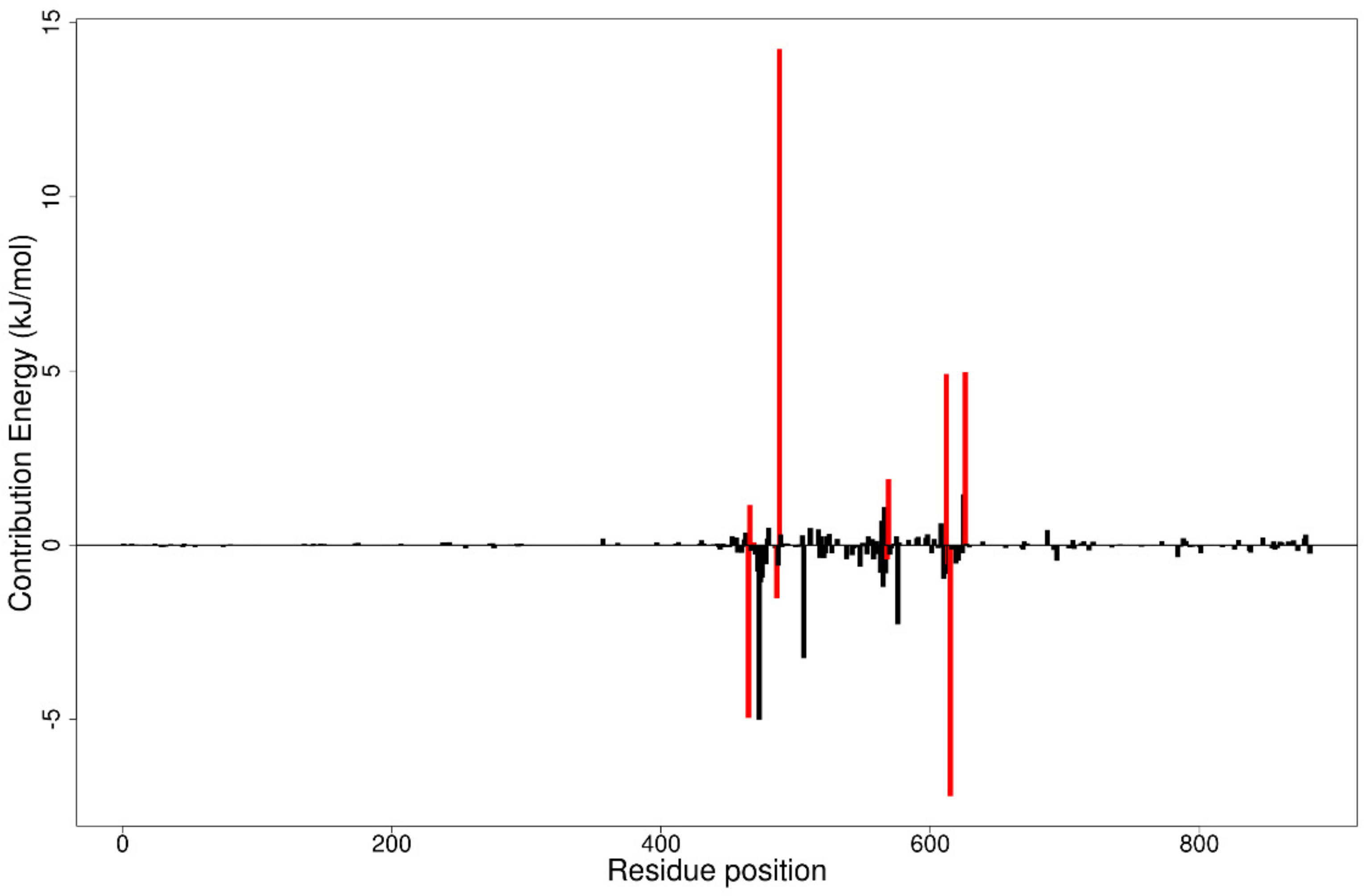
3.12. MM/PBSA Computations
3.12.1. Contributing Energy Terms
3.12.2. Per-Residue Energy Decomposition
3.13. Future Outlook and Implication of the Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Research 2017, 6, 750. [Google Scholar] [CrossRef] [PubMed]
- Gebremichael Tedla, D.; Bariagabr, F.H.; Abreha, H.H. Incidence and Trends of Leishmaniasis and Its Risk Factors in Humera, Western Tigray. J. Parasitol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Corrales, R.M.; Sereno, D.; Mathieu-Daudé, F. Deciphering the Leishmania exoproteome: What we know and what we can learn. FEMS Immunol. Med. Microbiol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.A. Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int. J. Parasitol. 2007, 37, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Kamhawi, S. Phlebotomine sand flies and Leishmania parasites: Friends or foes? Trends Parasitol. 2006, 22, 439–445. [Google Scholar] [CrossRef]
- Ready, P.D. Biology of Phlebotomine Sand Flies as Vectors of Disease Agents. Annu. Rev. Entomol. 2013. [Google Scholar] [CrossRef]
- Dostálová, A.; Volf, P. Leishmania development in sand flies: Parasite-vector interactions overview. Parasites Vectors 2012, 5, 276. [Google Scholar] [CrossRef]
- Babiker, A.M.; Ravagnan, S.; Fusaro, A.; Hassan, M.M.; Bakheit, S.M.; Mukhtar, M.M.; Cattoli, G.; Capelli, G. Concomitant infection with leishmania donovani and L. major in single ulcers of cutaneous leishmaniasis patients from sudan. J. Trop. Med. 2014. [Google Scholar] [CrossRef]
- Showler, A.J.; Boggild, A.K. Protozoan Diseases: Leishmaniasis. In The International Encyclopedia of Public Health, 2nd ed.; Quah, S.R., Cockerham, Eds.; Academic Press: Oxford, UK, 2016; Volume 6, pp. 97–102. ISBN 9780128037089. [Google Scholar]
- Alvar, J.; Aparicio, P.; Aseffa, A.; Den Boer, M.; Cañavate, C.; Dedet, J.P.; Gradoni, L.; Ter Horst, R.; López-Vélez, R.; Moreno, J. The relationship between leishmaniasis and AIDS: The second 10 years. Clin. Microbiol. Rev. 2008, 21, 334–359. [Google Scholar] [CrossRef]
- Manomat, J.; Leelayoova, S.; Bualert, L.; Tan-ariya, P.; Siripattanapipong, S.; Mungthin, M.; Naaglor, T.; Piyaraj, P. Prevalence and risk factors associated with Leishmania infection in Trang Province, southern Thailand. PLoS Negl. Trop. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Badirzadeh, A.; Mohebali, M.; Sabzevari, S.; Ghafoori, M.; Arzamani, K.; Seyyedin, M.; Hashemi, S.A. Case Report: First coinfection report of mixed leishmania infantum/leishmania major and human immunodeficiency virus-acquired immune deficiency syndrome: Report of a case of disseminated cutaneous Leishmaniasis in Iran. Am. J. Trop. Med. Hyg. 2018. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, I.N.; Shuwa, H.A.; Emeribe, A.U.; Olayemi, L.O.; Yunusa, T.; Ahmad, A.E.; Musa, P.O. Serosurvey and factors associated with Leishmania donovani infection in febrile HIV infected individuals attending Abuja Teaching Hospital, Nigeria. Med. J. Zambia 2018, 45, 216–225. [Google Scholar]
- Boelaert, M.; Meheus, F.; Sanchez, A.; Singh, S.P.; Vanlerberghe, V.; Picado, A.; Meessen, B.; Sundar, S. The poorest of the poor: A poverty appraisal of households affected by visceral leishmaniasis in Bihar, India. Trop. Med. Int. Health 2009. [Google Scholar] [CrossRef] [PubMed]
- Pigott, D.M.; Bhatt, S.; Golding, N.; Duda, K.A.; Battle, K.E.; Brady, O.J.; Messina, J.P.; Balard, Y.; Bastien, P.; Pratlong, F.; et al. Global distribution maps of the Leishmaniases. Elife 2014. [Google Scholar] [CrossRef]
- Hotez, P.J.; Kamath, A. Neglected tropical diseases in sub-Saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 2009, 3, e412. [Google Scholar] [CrossRef] [PubMed]
- Okwor, I.; Uzonna, J. Social and economic burden of human leishmaniasis. Am. J. Trop. Med. Hyg. 2016, 94, 489–493. [Google Scholar] [CrossRef]
- Van Griensven, J.; Balasegaram, M.; Meheus, F.; Alvar, J.; Lynen, L.; Boelaert, M. Combination therapy for visceral leishmaniasis. Lancet Infect. Dis. 2010, 377, 443–444. [Google Scholar] [CrossRef]
- Catta-Preta, C.M.C.; Mottram, J.C. Drug candidate and target for leishmaniasis. Nature 2018, 560, 171–172. [Google Scholar] [CrossRef]
- Wyllie, S.; Thomas, M.; Patterson, S.; Crouch, S.; De Rycker, M.; Lowe, R.; Gresham, S.; Urbaniak, M.D.; Otto, T.D.; Stojanovski, L.; et al. Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature 2018, 560, 192–197. [Google Scholar] [CrossRef]
- Monnerat, S.; Almeida Costa, C.I.; Forkert, A.C.; Benz, C.; Hamilton, A.; Tetley, L.; Burchmore, R.; Novo, C.; Mottram, J.C.; Hammarton, T.C. Identification and Functional Characterisation of CRK12:CYC9, a Novel Cyclin-Dependent Kinase (CDK)-Cyclin Complex in Trypanosoma brucei. PLoS ONE 2013, 8, e67327. [Google Scholar] [CrossRef]
- Baker, N.; Catta-Preta, C.M.C.; Neish, R.; Sadlova, J.; Powell, B.; Alves-Ferreira, E.V.C.; Geoghegan, V.; Carnielli, J.B.T.; Newling, K.; Hughes, C.; et al. Systematic functional analysis of Leishmania protein kinases identifies regulators of differentiation and survival. Nat Commun. 2020, 12, 1244. [Google Scholar] [CrossRef] [PubMed]
- Badjatia, N.; Park, S.H.; Ambrósio, D.L.; Kirkham, J.K.; Günzl, A. Cyclin-Dependent Kinase CRK9, Required for Spliced Leader trans Splicing of Pre-mRNA in Trypanosomes, Functions in a Complex with a New L-Type Cyclin and a Kinetoplastid-Specific Protein. PLoS Pathog. 2016, 12, e1005498. [Google Scholar] [CrossRef]
- Farombi, E.O. African indigenous plants with chemotherapeutic potentials and biotechnological approach to the production of bioactive prophylactic agents. African J. Biotechnol. 2003, 2, 662–671. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa—A Gromacs tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014. [Google Scholar] [CrossRef] [PubMed]
- Campanera, J.M.; Pouplana, R. MMPBSA decomposition of the binding energy throughout a molecular dynamics simulation of amyloid-beta (Aß10-35) aggregation. Molecules 2010, 15, 2730. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Poroikov, V.V.; Filimonov, D.A.; Ihlenfeldt, W.D.; Gloriozova, T.A.; Lagunin, A.A.; Borodina, Y.V.; Stepanchikova, A.V.; Nicklaus, M.C. PASS biological activity spectrum predictions in the enhanced open NCI Database Browser. J. Chem. Inf. Comput. Sci. 2003, 43, 228–236. [Google Scholar] [CrossRef]
- Fiser, A.; Šali, A. MODELLER: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 5.6.1, 5.6.30–6. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.; Kim, D.; Kellogg, E.; Dimaio, F.; Lange, O.; et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins Struct. Funct. Bioinform. 2009, 77, 89–99. [Google Scholar] [CrossRef]
- Song, Y.; Dimaio, F.; Wang, R.Y.R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32 (Suppl. 2), W526–W531. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M.; Consortium, U.P. UniProt Knowledgebase: A hub of integrated protein data. Database 2011. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Kuntal, B.K.; Aparoy, P.; Reddanna, P. EasyModeller: A graphical interface to MODELLER. BMC Res. Notes 2010, 3, 226. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins Struct. Funct. Genet. 1993, 17, 355–362. [Google Scholar] [CrossRef]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003, 31, 3352–3355. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Rother, K. Introduction to PyMOL. Methods Mol. Biol. Clift. Nj 2005, 635, 1–32. [Google Scholar] [CrossRef]
- DeLano, W. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 700. [Google Scholar]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.A.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A Resource for Natural Products from Northern African Sources. J. Nat. Prod. 2017. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A Select Highly Potent and Diverse Natural Product Library from African Medicinal Plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, EfficientOptimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Lindstrom, W.; Olson, A.J.; Belew, R.K. Analysis of HIV wild-type and mutant structures via in silico docking against diverse ligand libraries. J. Chem. Inf. Model. 2007, 47, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand À Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V.; Brooks, C.L.; Huang, R. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Broni, E.; Teye, J.; Quansah, E.; Issah, I.; Wilson, M.D.; Miller, W.A., III; Tiburu, E.K.; Bonney, J.H.K. Pharmacoinformatics-based identification of potential bioactive compounds against Ebola virus protein VP24. Comput. Biol. Med. 2019, 113, 103414. [Google Scholar] [CrossRef]
- Islam, M.A.; Pillay, T.S. Identification of promising anti-DNA gyrase antibacterial compounds using de novo design, molecular docking and molecular dynamics studies. J. Biomol. Struct. Dyn. 2019, 38, 1798–1809. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE, Version 5.1.19.; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Found. Stat. Comput. 2016. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.A.; Noah, J.W.; Hurt, D. Comparison of common homology modeling algorithms: Application of user-defined alignments. Methods Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Nayeem, A. A comparative study of available software for high-accuracy homology modeling: From sequence alignments to structural models. Protein Sci. 2006, 15, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Thomas, E.C.; Sessions, R.B.; Lawlor, D.A.; Tavaré, J.M.; Day, I.N.M. Structural and Population-Based Evaluations of TBC1D1 p.Arg125Trp. PLoS ONE 2013, 8, e63897. [Google Scholar] [CrossRef]
- Basta, H.A.; Sgro, J.Y.; Palmenberg, A.C. Modeling of the human rhinovirus C capsid suggests a novel topography with insights on receptor preference and immunogenicity. Virology 2014, 448, 176–184. [Google Scholar] [CrossRef]
- Palazzotti, D.; Bissaro, M.; Bolcato, G.; Astolfi, A.; Felicetti, T.; Sabatini, S.; Sturlese, M.; Cecchetti, V.; Barreca, M.L.; Moro, S. Deciphering the molecular recognition mechanism of multidrug resistance staphylococcus aureus nora efflux pump using a supervised molecular dynamics approach. Int. J. Mol. Sci. 2019, 20, 4041. [Google Scholar] [CrossRef]
- Eddy, S.R. Where did the BLOSUM62 alignment score matrix come from? Nat. Biotechnol. 2004, 22, 1035–1036. [Google Scholar] [CrossRef] [PubMed]
- Melo, F.; Sánchez, R.; Sali, A. Statistical potentials for fold assessment. Protein Sci. 2009, 11, 430–448. [Google Scholar] [CrossRef]
- Hoda, S.; Gupta, L.; Shankar, J.; Gupta, A.K.; Vijayaraghavan, P. cis-9-Hexadecenal, a Natural Compound Targeting Cell Wall Organization, Critical Growth Factor, and Virulence of Aspergillus fumigatus. ACS Omega 2020, 5, 10077–10088. [Google Scholar] [CrossRef]
- Bolsover, S.R.; Hyams, J.S.; Shephard, E.A.; White, H.A.; Wiedemann, C.G. How Proteins Work. In Cell Biology; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Purich, D.L. Active Sites and their Chemical Properties. In Enzyme Kinetics: Catalysis & Control; Elsevier BV: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Edelsbrunner, H.; Facello, M.; Fu, P.; Liang, J. Measuring proteins and voids in proteins. In Proceedings of the Annual Hawaii International Conference on System Sciences, Wailea, HI, USA, 3–6 January 1995. [Google Scholar]
- Kwofie, S.K.; Broni, E.; Asiedu, S.O.; Kwarko, G.B.; Dankwa, B.; Enninful, K.S.; Tiburu, E.K.; Wilson, M.D. Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules 2021, 26, 406. [Google Scholar] [CrossRef]
- Wen, C.C.; Kuo, Y.H.; Jan, J.T.; Liang, P.H.; Wang, S.Y.; Liu, H.G.; Lee, C.K.; Chang, S.T.; Kuo, C.J.; Lee, S.S.; et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef]
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef]
- Lee, S.; Lee, K.H.; Song, M.; Lee, D. Building the process-drug–side effect network to discover the relationship between biological Processes and side effects. BMC Bioinform. 2011, 12, S2. [Google Scholar] [CrossRef]
- Hole, A.J.; Baumli, S.; Shao, H.; Shi, S.; Huang, S.; Pepper, C.; Fischer, P.M.; Wang, S.; Endicott, J.A.; Noble, M.E. Comparative structural and functional studies of 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile CDK9 inhibitors suggest the basis for isotype selectivity. J. Med. Chem. 2013, 56, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Shi, S.; Huang, S.; Hole, A.J.; Abbas, A.Y.; Baumli, S.; Liu, X.; Lam, F.; Foley, D.W.; Fischer, P.M.; et al. Substituted 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines are highly active CDK9 inhibitors: Synthesis, X-ray crystal structures, structure-activity relationship, and anticancer activities. J. Med. Chem. 2013, 56, 640–659. [Google Scholar] [CrossRef]
- Lücking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 2017, 12, 1776. [Google Scholar] [CrossRef]
- Baumli, S.; Hole, A.J.; Wang, L.Z.; Noble, M.E.M.; Endicott, J.A. The CDK9 tail determines the reaction pathway of positive transcription elongation factor b. Structure 2012, 20, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Mirza, M.U.; Ikram, N. Integrated computational approach for virtual hit identification against ebola viral proteins VP35 and VP40. Int. J. Mol. Sci. 2016, 17, 1748. [Google Scholar] [CrossRef]
- Suenderhauf, C.; Hammann, F.; Huwyler, J. Computational prediction of blood-brain barrier permeability using decision tree induction. Molecules 2012, 17, 10429–10445. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Perez, J.; Fuertes, M.; Nguewa, P.; Castilla, J.; Alonso, C. Anticancer Compounds as Leishmanicidal Drugs: Challenges in Chemotherapy and Future Perspectives. Curr. Med. Chem. 2008, 15, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, L.; Yardley, V.; Croft, S.L. Activity of anti-cancer protein kinase inhibitors against Leishmania spp. J. Antimicrob. Chemother. 2014, 69, 1888–1891. [Google Scholar] [CrossRef]
- Pulivarthi, D.; Steinberg, K.M.; Monzote, L.; Piñón, A.; Setzer, W.N. Antileishmanial activity of compounds isolated from sassafras albidum. Nat. Prod. Commun. 2015, 10, 1934578X1501000723. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Delmas, F.; Akhmedjanova, V.; Ollivier, E.; Bessonova, I.; Riad, E.; Timon-David, P. In vitro antileishmanial activity of diphyllin isolated from Haplophyllum bucharicum. Planta Med. 2005, 71, 366–369. [Google Scholar] [CrossRef]
- Gertsch, J.; Thöni Tobler, R.; Brun, R.; Sticher, O.; Heilmann, J. Antifungal, antiprotozoal, cytotoxic and piscicidal properties of justicidin B and a new arylnaphthalide lignan from Phyllanthus piscatorum. Planta Med. 2003, 69, 420–424. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Reynolds, R.C. Group Additivity in Ligand Binding Affinity: An Alternative Approach to Ligand Efficiency. J. Chem. Inf. Model. 2017, 57, 3086–3093. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Schultes, S.; De Graaf, C.; Haaksma, E.E.J.; De Esch, I.J.P.; Leurs, R.; Krämer, O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discov. Today Technol. 2010, 7, e157–e162. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.Y.; Coumar, M.S.; Shiao, H.Y.; Wang, W.C.; Chen, C.W.; Song, J.S.; Chen, C.H.; Lin, W.H.; Wu, S.H.; Hsu, J.T.A.; et al. Ligand efficiency based approach for efficient virtual screening of compound libraries. Eur. J. Med. Chem. 2014, 83, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.H.; Tounge, B.A.; Bembenek, S.D. Ligand binding efficiency: Trends, physical basis, and implications. J. Med. Chem. 2008, 51, 2432–2438. [Google Scholar] [CrossRef]
- Keserü, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef]
- Cheng, X.; Ivanov, I. Molecular dynamics. Methods Mol. Biol. 2012, 929, 243–285. [Google Scholar]
- Liao, K.H.; Chen, K.B.; Lee, W.Y.; Sun, M.F.; Lee, C.C.; Chen, C.Y.C. Ligand-based and structure-based investigation for Alzheimer’s disease from traditional Chinese medicine. Evid. Based Complement. Altern. Med. 2014. [Google Scholar] [CrossRef]
- Sinha, S.; Wang, S.M. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput. Struct. Biotechnol. J. 2020, 18, 723–736. [Google Scholar] [CrossRef]
- Dong, Y.W.; Liao, M.L.; Meng, X.L.; Somero, G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. USA 2018, 115, 1274–1279. [Google Scholar] [CrossRef]
- Perez, A.; Morrone, J.A.; Simmerling, C.; Dill, K.A. Advances in free-energy-based simulations of protein folding and ligand binding. Curr. Opin. Struct. Biol. 2016, 36, 25–31. [Google Scholar] [CrossRef]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Chaudhary, N.; Aparoy, P. MM-PBSA and per-residue decomposition energy studies on 7-Phenyl-imidazoquinolin-4(5H)-one derivatives: Identification of crucial site points at microsomal prostaglandin E synthase-1 (mPGES-1) active site. Int. J. Biol. Macromol. 2018, 119, 352–359. [Google Scholar] [CrossRef]
- Deng, N.J.; Zhang, P.; Cieplak, P.; Lai, L. Elucidating the energetics of entropically driven protein-ligand association: Calculations of absolute binding free energy and entropy. J. Phys. Chem. B 2011, 115, 11902–11910. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, S.K.; Dankwa, B.; Enninful, K.S.; Adobor, C.; Broni, E.; Ntiamoah, A.; Wilson, M.D. Molecular docking and dynamics simulation studies predict munc18b as a target of mycolactone: A plausible mechanism for granule exocytosis impairment in Buruli Ulcer Pathogenesis. Toxins 2019, 11, 181. [Google Scholar] [CrossRef]
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef]
- Hassan, P.; Fergusson, D.; Grant, K.M.; Mottram, J.C. The CRK3 protein kinase is essential for cell cycle progression of Leishmania mexicana. Mol. Biochem. Parasitol. 2001, 113, 189–198. [Google Scholar] [CrossRef]
- Naula, C.; Parsons, M.; Mottram, J.C. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim. Biophys. Acta Proteins Proteom. 2005, 1754, 151–159. [Google Scholar] [CrossRef]
- Tu, X.; Wang, C.C. Pairwise knockdowns of cdc2-related kinases (CRKs) in Trypanosoma brucei identified the CRKs for G1/S and G2/M transitions and demonstrated distinctive cytokinetic regulations between two developmental stages of the organism. Eukaryot. Cell 2005, 4, 755–764. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Protein Name | E-Value | BLAST Score | Identity (%) |
|---|---|---|---|---|
| O14098 | C-terminal heptapeptide repeat domain CTD kinase subunit alpha (Schizosaccharomyces pombe (strain 972/ATCC 24843) (Fission yeast)) | 4.2 × 10−34 | 356 | 36 |
| Q9TVL3-2 | Isoform a, of Probable cyclin-dependent kinase 9 (Caenorhabditis elegans) | 9.7 × 10−34 | 348 | 35 |
| Q641Z4 | Cyclin-dependent kinase 9 (Rattus norvegicus) | 7.4 × 10−34 | 345 | 31.3 |
| Q99J95 | Cyclin-dependent kinase 9 (Mus musculus) | 7.4 × 10−34 | 345 | 31.3 |
| P50750 | Cyclin-dependent kinase 9 (Homo sapiens) | 7.4 × 10−34 | 345 | 31.3 |
| Q5EAB2 | Cyclin-dependent kinase 9 (Bos taurus) | 1.4 × 10−33 | 343 | 31 |
| Models | Dope Score | GA341 Score |
|---|---|---|
| MOD1 | −49,545.96484 | 0.36807 |
| MOD2 | −49,137.34766 | 0.20576 |
| MOD3 | −47,466.54688 | 0.10907 |
| MOD4 | −49,459.57422 | 0.28138 |
| MOD5 | −50,486.88281 | 0.21007 |
| Models | ITAS1 | ITAS2 | ITAS3 | ITAS4 | ITAS5 |
|---|---|---|---|---|---|
| C-Score | −3.68 | −3.85 | −2.87 | −2.77 | −2.66 |
| Models | Model Score | |||
|---|---|---|---|---|
| Verify (%) | Errat | Prove (%) | Procheck | |
| ROB1 | 82.97 | 88.0579 | 0.0 (Pass) | 4E, 3W and 2P |
| ROB2 | 66.06 | 87.4259 | 0.0 (Pass) | 5E, 2W and 2P |
| ROB3 | 65.83 | 84.7073 | 6.1 (Error) | 5E, 1W and 3P |
| ROB4 | 67.54 | 83.6538 | 6.8 (Error) | 5E, 2W and 2P |
| ROB5 | 78.55 | 87.822 | 5.8 (Error) | 5E, 0W and 4P |
| Tool | Model Score | ||
|---|---|---|---|
| ROB1 | ITAS5 | MOD5 | |
| Verify (%) | 82.97 | 85.36 | 41.20 |
| Errat (Quality Factor) | 88.0579 | 80.2158 | 10.0536 |
| Prove (%) | 0.0 (Pass) | 9.5 (Error) | 16.1 (Error) |
| Procheck | 4E, 3W and 2P | 6E, 2W and 1P | 5E, 2W and 1P |
| Model | MOD5 | ITAS5 | ROB1 | |||
|---|---|---|---|---|---|---|
| No. of Residues | Percentage | No. of Residues | Percentage | No. of Residues | Percentage | |
| Most favored regions [A, B, L] | 581 | 79.7 | 445 | 61.0 | 598 | 82.0 |
| Additionally allowed regions [a, b, l, p] | 113 | 15.5 | 217 | 29.8 | 125 | 17.1 |
| Generously allowed regions [~a, ~b, ~l, ~p] | 22 | 3.0 | 43 | 5.9 | 3 | 0.4 |
| Disallowed regions | 13 | 1.8 | 24 | 3.3 | 3 | 0.4 |
| Non-glycine and non-proline residues | 729 | 100.0 | 729 | 100.0 | 729 | 100.0 |
| Area (SA)/Å2 | Volume (SA)/Å3 | Residues Lining the Pocket | |
|---|---|---|---|
| 1 | 566.585 | 712.561 | Leu438, Pro439, Ala441, Pro442, Pro443, Pro444, Ser445, Glu463, Lys464, Leu465, Ser466, Glu467, Gly468, Thr469, Tyr470, Val473, Lys475, Ala486, Leu487, Lys488, Glu506, Leu510, Ser544, Phe563, Ala564, Tyr565, Ala566, Thr567, Ala568, Ser569, Ala571, Gly572, Arg575, Arg576, His606, Asp608, Lys610, Asp612, Asn613, Leu615, Thr625, Asp626, Phe627, Leu629, Cys630, Val650, Thr652 |
| 2 | 312.963 | 420.314 | Met492, Thr495, His496, Gly498, Phe499, Pro500, Gln501, Thr502, Arg505, Arg607, Gly628, Leu629, Cys630, Ser631, Arg639, Cys640, Val644, Thr647, Pro648, Ser649, Val650, Ile651, Arg656, Met660, Thr665, Tyr667, Ser708, Ala709, Glu712 |
| 3 | 443.095 | 377.107 | Ile536, Arg597, Lys598, His600, Glu601, Arg603, Pro635, Asp668, Glu669, Lys670, Thr823, Ala825, Glu826, Leu828, Arg829, Leu836, Asp837, Asp838, Ala839, Pro840, Leu841, Leu842, Tyr845, Gln846, Arg847, Val848, Leu849 |
| 4 | 207.834 | 203.064 | Arg692, His693, Ala695, Gln696, Gln699, Gln700, Arg703, Pro705, Thr711, Glu714, Gln715, Ser717, Thr720, Glu721, Gln749, Ala758, Ala759, Gln760, Ala762 |
| 5 | 110.211 | 105.944 | Thr720, Pro725, Leu726, Pro727, Pro728, Val731, Leu743, Leu746, Glu747, Gln749, Gly750, Arg751, Glu754, Pro761, Ala762, Asn763 |
| 6 | 141.403 | 95.351 | Ala571, Arg575, Lys610, Ser611, Asp612, Thr652, Ala654, Tyr655, Gln682, Leu686, Glu687, Pro688, Tyr691, Arg694, Phe780 |
| 7 | 50.346 | 89.822 | Pro635, Gly636, Ser637, Leu849, Pro850, Thr852 |
| 8 | 159.056 | 87.650 | Leu685, Glu687, Pro688, Pro689, Tyr691, Arg692, Arg694, Ala695, Gln698, Gln699, Arg718, Glu721, Ser774, Phe775, Leu776, Gln778, Gln779, Phe780 |
| 9 | 101.847 | 84.378 | Ala342, Val402, Ala403, Met404, Gly405, Leu412, Arg413, Leu415, Pro417, Tyr420, Arg429 |
| 10 | 60.970 | 41.895 | Phe580, Glu584, Leu587, Leu588, Lys591, Glu619, Gly620, Val622 |
| 11 | 150.732 | 38.172 | Cys574, Phe578, Ala579, Phe580, Thr581, Pro582, Met585, Gln682, Met683, Phe684, Leu686, Ile770, Phe771, Gly785, Trp786, Glu788, Glu790, His799, Arg801, Pro802 |
| 12 | 68.900 | 28.853 | Thr642, His643, Val644, Pro658, Glu659, Leu662, Gly663, Ser664, Leu726, Ser736, His739, Met740, Leu816, Pro818, Arg821 |
| 13 | 69.295 | 22.359 | Pro427, Arg429, Arg430, Val434, Gly435, Phe448, Gln452, Lys456 |
| 14 | 71.553 | 21.095 | Pro658, Leu662, Leu713, Leu716, Ser717, Ile719, Thr720, Gly724, Pro725, Leu726, Val742, Gln745, Leu746, Gln749, Leu816 |
| Compound | Binding Energy (kcal/mol) | Hydrogen Bonds (Bond Length (Å)) | Hydrophobic Bonds | |||
|---|---|---|---|---|---|---|
| LdCRK12 | CDK9 | LdCRK12 | CDK9 | LdCRK12 | CDK9 | |
| ZINC000095485940 | −10.1 | −7.7 | Gly468 (2.93), Ser569 (2.95), Asp626 (2.70) | Arg195 (3.06, 3.22), Glu234 (2.9), Arg343 (3.1) | Leu465, Ser466, Thr469, Val473, Ala486, Lys488, Ser544, Phe563, Asp612, Asn613, Leu615, Thr625 | Arg188, Leu192, Arg195, Thr233, Glu234, Tyr338, Ala340, Arg343 |
| NANPDB1406 | −9.5 | −7.3 | Lys488 (3.26), Ala566 (2.89, 2.97, 3.07), Ser569 (3.01) | Asn232 (2.8), Phe336 (3.08), Ala340 (2.69, 3.15), Arg343 (2.88, 3.1, 3.17, 3.25) | Leu465, Ser466, Gly468, Val473, Ala486, Tyr565, Thr567, Ala568, Asp612, Leu615, Asp626 | Asn232, Thr233, Met335, Phe336, Tyr338, Ala340, Arg343 |
| NANPDB2581 | −9.2 | −7.5 | Lys610 (3.08) | Arg195 (3.32), Arg343 (3.09, 3.35) | Leu465, Ser466, Thr469, Tyr470, Ala568, Ser569, Asp612, Asn613, Leu615, Asp626 | Leu192, Arg195, Thr233, Glu234, Pro341, Pro342, Arg343 |
| NANPDB6446 | −9.1 | −7.3 | Ser569 (2.77, 3.02), Arg575 (2.87, 3.15) | Asn179 (3.04), Tyr259 (2.89) | Leu465, Ser466, Ala568, Gly572, Asp612, Asp626 | Asn179, Pro182, Glu203, Asp205, Trp253, Asn258, Tyr259, Pro300 |
| Compound 8 | −9.1 | −9.0 | Leu723 (2.83) | Lys48 (2.91), Asp149 (2.99, 3.08) | Gly724, Pro725, Leu726, Pro727, Pro728, Val731, Leu743, Glu747, Asn763, Trp764, Gln815, Leu816 | Thr29, Phe30, Leu51, Pro60, Thr62, Asp149, Leu170, Arg188, Val190, Thr191, Leu192, Met335 |
| T6Q | −9.1 | −8.6 | Thr469 (3.06) | - | Leu465, Ser466, Gly468, Thr469, Val473, Lys488, Ala568, Ser569, Arg575, Lys610, Asp612, Asn613, Leu615, Asp626 | Ile25, Phe30, Val33, Ly48, Asp109, Gly112, Ala153, Leu156, Ala166, Asp167, His331, Leu332, Thr333 |
| DDD853651/GSK3186899/Compound 7 | −8.5 | −8.8 | Ser466 (2.96), Gly468 (3.19), Lys488 (3.03), Ser544 (3.27), Thr625 (3.12), Asp626 (3.31, 3.3), Tyr691 (2.98) | Glu107 (3.07, 2.98) | Gly468, Thr469, Tyr470, Val473, Ala486, Lys488, Phe563, Lys610, Asp612, Leu615, Asp626, Tyr691 | Ile25, Val33, Lys35, Lys48, Phe103, Glu107, His108, Asp109, Ala166, Asp167 |
| Compound 5 | −7.2 | −8.6 | Leu723 (2.98, 3.07) | Cys106 (3.2, 3.0) | Gly724, Pro725, Leu726, Pro727, Pro728, Val731, Tyr732, Leu743, Asn763, Trp764, Gln815, Leu816, Asp817, Gln820 | Ile25, Val33, Ala46, Lys48, Phe103, Phe105, Glu107, His108, Asp109, Gly112, Leu113, Ala153, Asn154, Leu156, Ala166, Asp167 |
| Compound | Binding Energy | NHA | cLogP | Ki (µM) | LE | LE_Scale | FQ | LELP |
|---|---|---|---|---|---|---|---|---|
| ZINC000095485940 | −10.1 | 29 | −0.1814 | 0.039 | 0.348 | 0.347 | 1.003 | 0.521 |
| NANPDB1406 | −9.5 | 23 | 1.5531 | 0.108 | 0.413 | 0.416 | 0.993 | 3.761 |
| NANPDB2581 | −9.2 | 24 | 3.3633 | 0.180 | 0.383 | 0.404 | 0.948 | 8.781 |
| NANPDB6446 | −9.1 | 26 | −0.8296 | 0.213 | 0.35 | 0.380 | 0.921 | 2.370 |
| NANPDB1649 | −8.5 | 26 | 3.2246 | 0.587 | 0.327 | 0.380 | 0.861 | 9.861 |
| Compound ID | Common/IUPAC Name | 2D Structure |
|---|---|---|
| ZINC000095485940 | (1R,2R,4R,7S,8R,10R,11R,12R,13R,16S)-7-(furan-3-yl)-10,13-dihydroxy-8,13-dimethyl-3,6,14-trioxapentacyclo[9.7.0.02,4.02,8.012,16]octadecane-5,18-dione |  |
| NANPDB1406 | methyl ellagic acid |  |
| NANPDB2581 | stylopine |  |
| NANPDB6446 | sennecicannabine |  |
| NANPDB1649 | sesamin |  |
| Compound 5 | 2-methyl-N-[(1r,4r)-4-{[3-(2-methoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-6-yl]amino}cyclohexyl]propane-1-sulfonamide |  |
| GSK3186899 | DDD853651/Compound 7 |  |
| Compound 8 | 2-(2,4-difluorophenyl)-1-(4-{[3-(2-methoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-6-yl]amino}piperidin-1-yl)ethan-1-one |  |
| van der Waal Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy (kJ/mol) | |
|---|---|---|---|---|---|
| Compound 5 | −98.909 ± 22.552 | −9.113 ± 13.340 | 67.471 ± 24.307 | −13.471 ± 2.359 | −54.023 ± 17.067 |
| DDD853651/GSK3186899/Compound 7 | −107.423 ± 24.517 | −43.202 ± 23.069 | 140.188 ± 36.187 | −16.945 ± 2.444 | −27.382 ± 20.792 |
| Compound 8 | −138.191 ± 15.201 | −17.732 ± 9.037 | 103.997 ± 24.258 | −16.683 ± 1.392 | −68.609 ± 13.327 |
| NANPDB1406 | −125.840 ± 9.460 | −40.995 ± 10.072 | 157.502 ± 22.889 | −15.186 ± 0.971 | −24.518 ± 14.412 |
| NANPDB1649 | −111.638 ± 18.534 | −4.178 ± 8.634 | 80.033 ± 25.137 | −14.651 ± 1.960 | −50.434 ± 13.538 |
| NANPDB2581 | −110.229 ± 10.366 | −7.999 ± 7.595 | 83.680 ± 20.147 | −14.826 ± 1.181 | −49.374 ± 14.169 |
| NANPDB6446 | −84.419 ± 19.455 | −64.626 ± 32.749 | 125.008 ± 32.850 | −13.141 ± 2.305 | −37.179 ± 17.980 |
| ZINC000095485940 | −91.882 ± 13.394 | −29.485 ± 12.960 | 136.331 ± 31.350 | −14.372 ± 1.704 | 0.593 ± 16.180 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broni, E.; Kwofie, S.K.; Asiedu, S.O.; Miller, W.A., III; Wilson, M.D. A Molecular Modeling Approach to Identify Potential Antileishmanial Compounds Against the Cell Division Cycle (cdc)-2-Related Kinase 12 (CRK12) Receptor of Leishmania donovani. Biomolecules 2021, 11, 458. https://doi.org/10.3390/biom11030458
Broni E, Kwofie SK, Asiedu SO, Miller WA III, Wilson MD. A Molecular Modeling Approach to Identify Potential Antileishmanial Compounds Against the Cell Division Cycle (cdc)-2-Related Kinase 12 (CRK12) Receptor of Leishmania donovani. Biomolecules. 2021; 11(3):458. https://doi.org/10.3390/biom11030458
Chicago/Turabian StyleBroni, Emmanuel, Samuel K. Kwofie, Seth O. Asiedu, Whelton A. Miller, III, and Michael D. Wilson. 2021. "A Molecular Modeling Approach to Identify Potential Antileishmanial Compounds Against the Cell Division Cycle (cdc)-2-Related Kinase 12 (CRK12) Receptor of Leishmania donovani" Biomolecules 11, no. 3: 458. https://doi.org/10.3390/biom11030458
APA StyleBroni, E., Kwofie, S. K., Asiedu, S. O., Miller, W. A., III, & Wilson, M. D. (2021). A Molecular Modeling Approach to Identify Potential Antileishmanial Compounds Against the Cell Division Cycle (cdc)-2-Related Kinase 12 (CRK12) Receptor of Leishmania donovani. Biomolecules, 11(3), 458. https://doi.org/10.3390/biom11030458