
QSAR-Based Virtual Screening of Natural Products Database for Identification of Potent Antimalarial Hits

 , ,
, , 
Abstract
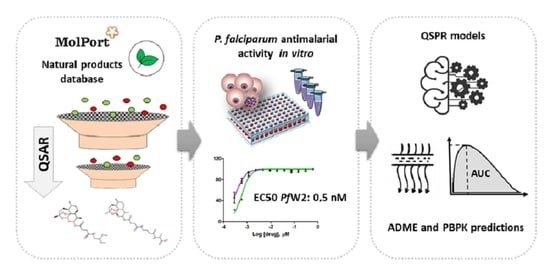
1. Introduction
2. Materials and Methods
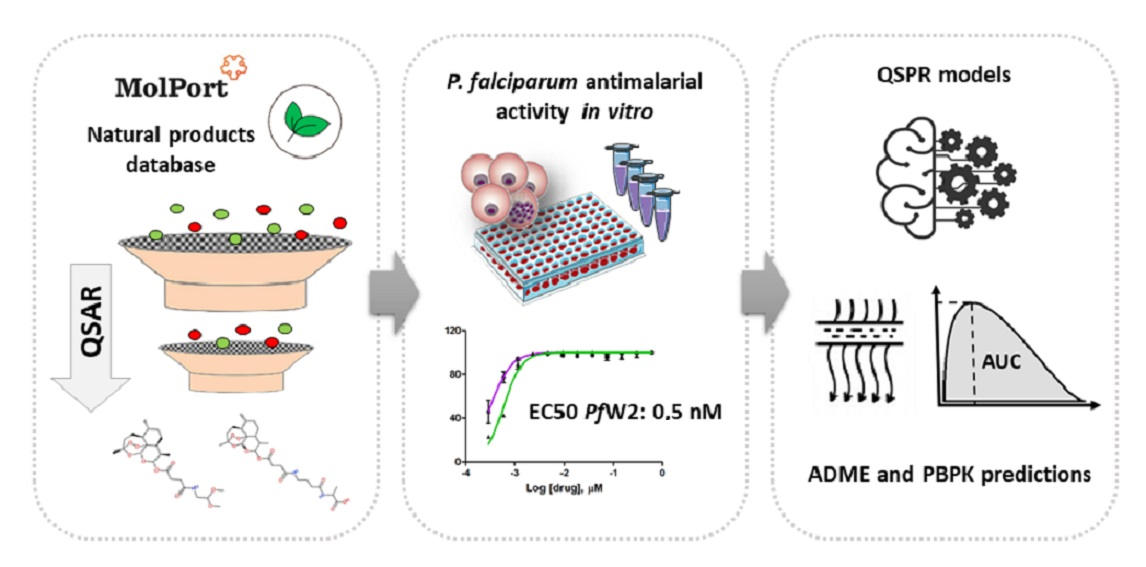
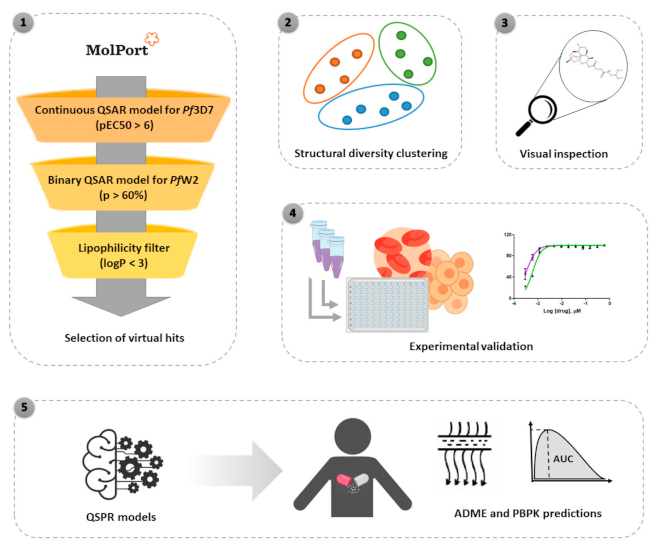
2.1. Virtual Screening and Structural Diversity Clustering
2.2. Compound Preparation
2.3. Plasmodium falciparum In Vitro Culture
2.4. In Vitro Assays for P. falciparum Growth Inhibition
2.5. Citotoxicity Assays
2.6. In Silico Predictions of Metabolism, ADME and PBPK
3. Results
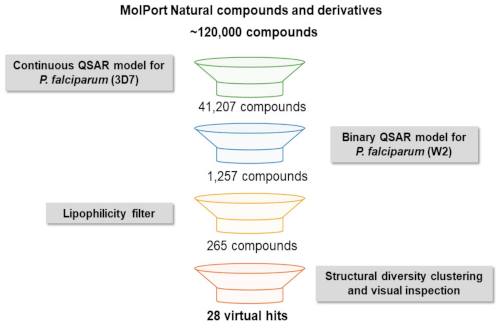
3.1. QSAR-Based Virtual Screening
3.2. Structural Diversity Clustering
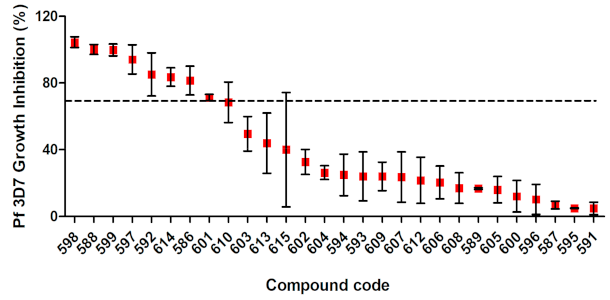
3.3. In Vitro Screening against P. falciparum
3.4. Cytotoxicity against Human Cells
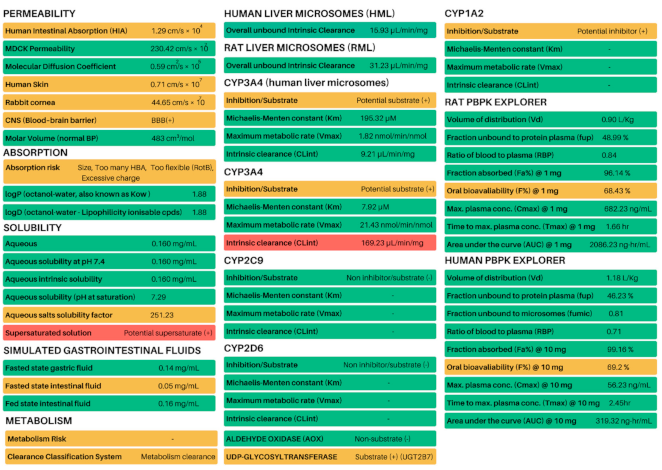
3.5. In Silico Predictions of Metabolism, ADME and PBPK
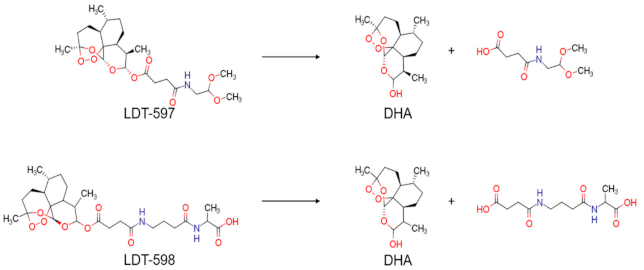
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Rogerson, S.J.; Beeson, J.G.; Laman, M.; Poespoprodjo, J.R.; William, T.; Simpson, J.A.; Price, R.N.; The ACREME Investigators. Identifying and combating the impacts of COVID-19 on malaria. BMC Med. 2020, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sherrard-Smith, E.; Hogan, A.B.; Hamlet, A.; Watson, O.J.; Whittaker, C.; Winskill, P.; Ali, F.; Mohammad, A.B.; Uhomoibhi, P.; Maikore, I.; et al. The potential public health consequences of COVID-19 on malaria in Africa. Nat. Med. 2020, 26, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Haldar, K.; Bhattacharjee, S.; Safeukui, I. Drug resistance in Plasmodium. Nat. Rev. Genet. 2018, 16, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Noedl, H.; Se, Y.; Schaecher, K.; Smith, B.L.; Socheat, D.; Fukuda, M.M. Evidence of Artemisinin-Resistant Malaria in Western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. [Google Scholar] [CrossRef] [PubMed]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.-L.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.-B.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Achan, J.; O Talisuna, A.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Bosman, A.; Mendis, K.N. A Major Transition in Malaria Treatment: The Adoption and Deployment of Artemisinin-Based Combination Therapies. Am. J. Trop. Med. Hyg. 2007, 77, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.K. Back to the Future: Lessons Learned in Modern Target-based and Whole-Cell Lead Optimization of Antimalarials. Curr. Top. Med. Chem. 2012, 12, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Advances in computational methods to predict the biological activity of compounds. Expert Opin. Drug Discov. 2010, 5, 633–654. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in Drug Design—A Review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Zhang, L.; Fourches, D.; Sedykh, A.; Zhu, H.; Golbraikh, A.; Ekins, S.; Clark, J.; Connelly, M.C.; Sigal, M.; Hodges, D.; et al. Discovery of Novel Antimalarial Compounds Enabled by QSAR-Based Virtual Screening. J. Chem. Inf. Model. 2013, 53, 475–492. [Google Scholar] [CrossRef]
- Neves, B.J.; Dantas, R.F.; Senger, M.R.; Melo-Filho, C.C.; Valente, W.C.G.; De Almeida, A.C.M.; Rezende-Neto, J.M.; Lima, E.F.C.; Paveley, R.; Furnham, N.; et al. Discovery of New Anti-Schistosomal Hits by Integration of QSAR-Based Virtual Screening and High Content Screening. J. Med. Chem. 2016, 59, 7075–7088. [Google Scholar] [CrossRef]
- Lima, M.N.N.; Melo-Filho, C.C.; Cassiano, G.C.; Neves, B.J.; Alves, V.M.; Braga, R.C.; Cravo, P.V.L.; Muratov, E.N.; Calit, J.; Bargieri, D.Y.; et al. QSAR-Driven Design and Discovery of Novel Compounds With Antiplasmodial and Transmission Blocking Activities. Front. Pharmacol. 2018, 9, 146. [Google Scholar] [CrossRef]
- Gomes, M.N.; Alcântara, L.M.; Neves, B.J.; Melo-Filho, C.C.; Freitas-Junior, L.H.; Moraes, C.B.; Ma, R.; Franzblau, S.G.; Muratov, E.N.; Andrade, C.H. Computer-aided discovery of two novel chalcone-like compounds active and selective against Leishmania infantum. Bioorg. Med. Chem. Lett. 2017, 27, 2459–2464. [Google Scholar] [CrossRef]
- Melo-Filho, C.C.; Dantas, R.F.; Braga, R.C.; Neves, B.J.; Senger, M.R.; Valente, W.C.G.; Rezende-Neto, J.M.; Chaves, W.T.; Muratov, E.N.; Paveley, R.A.; et al. QSAR-Driven Discovery of Novel Chemical Scaffolds Active against Schistosoma mansoni. J. Chem. Inf. Model. 2016, 56, 1357–1372. [Google Scholar] [CrossRef]
- Xie, N.; Wang, C.; Lian, Y.; Wu, C.; Zhang, H.; Zhang, Q. Inhibition of mitochondrial fission attenuates Aβ-induced microglia apoptosis. Neuroscience 2014, 256, 36–42. [Google Scholar] [CrossRef]
- Neves, B.; Moreira-Filho, J.; Silva, A.; Borba, J.; Mottin, M.; Alves, V.; Braga, R.; Muratov, E.; Andrade, C. Automated Framework for Developing Predictive Machine Learning Models for Data-Driven Drug Discovery. J. Braz. Chem. Soc. 2021, 1–13. [Google Scholar] [CrossRef]
- Lima, M.N.N.; Cassiano, G.C.; Tomaz, K.C.P.; Silva, A.C.; Sousa, B.K.P.; Ferreira, L.T.; Tavella, T.A.; Calit, J.; Bargieri, D.Y.; Neves, B.J.; et al. Integrative Multi-Kinase Approach for the Identification of Potent Antiplasmodial Hits. Front. Chem. 2019, 7, 773. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Curation of chemogenomics data. Nat. Chem. Biol. 2015, 11, 535. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fu, A.Y.; Lai, L. A New Atom-Additive Method for Calculating Partition Coefficients. J. Chem. Inf. Comput. Sci. 1997, 37, 615–621. [Google Scholar] [CrossRef]
- Butina, D. Unsupervised Data Base Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way to Cluster Small and Large Data Sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Sydow, D.; Morger, A.; Driller, M.; Volkamer, A. TeachOpenCADD: A teaching platform for computer-aided drug design using open source packages and data. J. Cheminform. 2019, 11, 29. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Trager, J.J.W. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Radfar, A.; Méndez, D.; Moneriz, C.; Linares, M.; Marín-García, P.; Puyet, A.; Diez, A.; Bautista, J.M. Synchronous culture of Plasmodium falciparum at high parasitemia levels. Nat. Protoc. 2009, 4, 1899–1915. [Google Scholar] [CrossRef]
- Hartwig, C.L.; Ahmed, A.O.A.; Cooper, R.A.; Stedman, T.T. SYBR Green I®-Based Parasite Growth Inhibition Assay for Measurement of Antimalarial Drug Susceptibility in Plasmodium Falciparum. In Methods in Malaria Research; Moll, K., Kaneko, A., Scherf, A., Wahlgren, M., Eds.; EVIMalaR: Manassas, Virginia, 2013; pp. 122–129. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-De-La-Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. BioTransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11, 1–25. [Google Scholar] [CrossRef]
- Cui, L.; Su, X.-Z. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti-Infect. Ther. 2009, 7, 999–1013. [Google Scholar] [CrossRef]
- Karunajeewa, H.A. Artemisinins: Artemisinin, Dihydroartemisinin, Artemether and Artesunate. In Treatment and Prevention of Malaria; Metzler, J.B., Ed.; Springer Basel: Basel, Switzerland, 2011; pp. 157–190. [Google Scholar]
- Smit, F.J.; van Biljon, R.A.; Birkholtz, L.-M.; N’Da, D.D. Synthesis and in vitro biological evaluation of dihydroartemisinyl-chalcone esters. Eur. J. Med. Chem. 2015, 90, 33–44. [Google Scholar] [CrossRef]
- Pedersen, M.M.; Chukwujekwu, J.C.; Lategan, C.A.; Van Staden, J.; Smith, P.J.; Staerk, D. Antimalarial sesquiterpene lactones from Distephanus angulifolius. Phytochemistry 2009, 70, 601–607. [Google Scholar] [CrossRef]
- Francois, G.; Passreiter, C.M. Pseudoguaianolide sesquiterpene lactones with high activities against the human malaria parasitePlasmodium falciparum. Phytother. Res. 2004, 18, 184–186. [Google Scholar] [CrossRef]
- Bordignon, A.; Frédérich, M.; LeDoux, A.; Campos, P.-E.; Clerc, P.; Hermann, T.; Quetin-Leclercq, J.; Cieckiewicz, E. In vitro antiplasmodial and cytotoxic activities of sesquiterpene lactones from Vernonia fimbrillifera Less. (Asteraceae). Nat. Prod. Res. 2017, 32, 1463–1466. [Google Scholar] [CrossRef]
- Avery, M.A.; Alvim-Gaston, M.; Rodrigues, C.R.; Barreiro, E.J.; Cohen, F.E.; Sabnis, Y.A.; Woolfrey, J.R. Structure−Activity Relationships of the Antimalarial Agent Artemisinin. The Development of Predictive In Vitro Potency Models Using CoMFA and HQSAR Methodologies. J. Med. Chem. 2002, 45, 292–303. [Google Scholar] [CrossRef]
- Arey, R.; Reisfeld, B. Predicting the Disposition of the Antimalarial Drug Artesunate and Its Active Metabolite Dihydroartemisinin Using Physiologically Based Pharmacokinetic Modeling. Antimicrob. Agents Chemother. 2020, 65. [Google Scholar] [CrossRef]
- Gordi, T.; Xie, R.; Huong, N.V.; Huong, D.X.; Karlsson, M.O.; Ashton, M. A semiphysiological pharmacokinetic model for artemisinin in healthy subjects incorporating autoinduction of metabolism and saturable first-pass hepatic extraction. Br. J. Clin. Pharmacol. 2005, 59, 189–198. [Google Scholar] [CrossRef]
- Olafuyi, O.; Coleman, M.; Badhan, R.K. Development of a paediatric physiologically based pharmacokinetic model to assess the impact of drug-drug interactions in tuberculosis co-infected malaria subjects: A case study with artemether-lumefantrine and the CYP3A4-inducer rifampicin. Eur. J. Pharm. Sci. 2017, 106, 20–33. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code (MolPort ID) | 2D Structure (Biological Source of NP Precursor) | EC50 a (µM) | CC50 b (µM) | In vitro Therapeutic Index c | |
|---|---|---|---|---|---|
| Pf3D7 | PfW2 | HepG2 | |||
| LDT-586 (MolPort-001-745-423) |  (No data available) | 4.52 ± 0.91 | 3.44 ± 1.30 | 98.59 ± 0 | 21.81 |
| LDT-588 (MolPort-000-651-065) |  (No data available) | 3.84 ± 1.45 | 2.09 ± 1.17 | 67.04 ± 2.28 | 17.46 |
| LDT-592 (MolPort-002-323-504) |  (No data available) | 9.09 ± 3.74 | 3.11 ± 0.54 | 17 ± 0 | 1.87 |
| LDT-597 (MolPort-001-732-360) |  (Artemesia annua) | 0.0005 ± 0.00 | 0.0005 ± 0.00 | 18.29 ± 3.51 | 33,870.37 |
| LDT-598 (MolPort-001-732-370) |  (Artemesia annua) | 0.0007 ± 0.00 | 0.0006 ± 0.00 | 25.94 ± 1.13 | 33,299.10 |
| LDT-599 (MolPort-001-737-485) |  (Aspergillus fumigatus) | 3.68 ± 1.92 | 2.74 ± 0.78 | 20.96 ± 2.51 | 5.70 |
| LDT-601 (MolPort-002-506-405) |  (Pachycereus weberi, Pachycereus pringlei, Pachycereus pecten -aboriginum, Backebergia militaris and Carnegiea gigantea) | 6.61 ± 3.20 | 0.65 ± 0.47 | 21.79 ± 4.16 | 3.30 |
| LDT-614 (MolPort-044-180-513) |  (Penicillium patulum) | 5.26 ± 0.52 | 5.65 ± 3.11 | 23.55 ± 1.82 | 4.48 |
| Chloroquine | 0.0079 ± 0.00 | 0.147 ± 0.04 | ND | - | |
| Artesunate | 0.0016 ± 0.00 | ND | ND | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, L.T.; Borba, J.V.B.; Moreira-Filho, J.T.; Rimoldi, A.; Andrade, C.H.; Costa, F.T.M. QSAR-Based Virtual Screening of Natural Products Database for Identification of Potent Antimalarial Hits. Biomolecules 2021, 11, 459. https://doi.org/10.3390/biom11030459
Ferreira LT, Borba JVB, Moreira-Filho JT, Rimoldi A, Andrade CH, Costa FTM. QSAR-Based Virtual Screening of Natural Products Database for Identification of Potent Antimalarial Hits. Biomolecules. 2021; 11(3):459. https://doi.org/10.3390/biom11030459
Chicago/Turabian StyleFerreira, Letícia Tiburcio, Joyce V. B. Borba, José Teófilo Moreira-Filho, Aline Rimoldi, Carolina Horta Andrade, and Fabio Trindade Maranhão Costa. 2021. "QSAR-Based Virtual Screening of Natural Products Database for Identification of Potent Antimalarial Hits" Biomolecules 11, no. 3: 459. https://doi.org/10.3390/biom11030459
APA StyleFerreira, L. T., Borba, J. V. B., Moreira-Filho, J. T., Rimoldi, A., Andrade, C. H., & Costa, F. T. M. (2021). QSAR-Based Virtual Screening of Natural Products Database for Identification of Potent Antimalarial Hits. Biomolecules, 11(3), 459. https://doi.org/10.3390/biom11030459