
LncRNAs and microRNAs as Essential Regulators of Stemness in Breast Cancer Stem Cells

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. The Mammary Glands: Main Biological and Molecular Characteristics
3. Breast Cancer Stem Cells Origin and Their Main Cell Markers
4. Signaling Pathways Governing the Breast Cancer Stem Cell-Like Phenotype
4.1. The Notch Pathway
4.2. The Hedgehog Pathway
4.3. The Wnt Pathway
4.4. The Hippo Signaling Pathway
4.5. The JAK/STAT Pathway
4.6. NF-κB Signaling Pathway
4.7. PI3K/AKT/mTOR Signaling Pathway
4.8. The TGF-β Pathway
5. The Prominent Role of miRNAs in Breast Cancer Stem Cells
6. The lncRNA: Key Molecules in the Breast Cancer Stem Cells
7. Strategies of Elimination or Resistance in bCSC: microRNA and lncRNAs as Protagonist
8. Clinical Trials
9. Closing Remarks
Authors Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kozłowski, J.; Kozłowska, A.; Kocki, J. Breast cancer metastasis—Insight into selected molecular mechanisms of the phenomenon. Postepy Hig. Med. Dosw. 2015, 69, 447–451. [Google Scholar] [CrossRef]
- Islam, F.; Qiao, B.; Smith, R.A.; Gopalan, V.; Lam, A.K. Cancer stem cell: Fundamental experimental pathological concepts and updates. Exp. Mol. Pathol. 2015, 98, 184–191. [Google Scholar] [CrossRef]
- Lau, E.Y.-T.; Ho, N.P.-Y.; Lee, T.K.-W. Cancer Stem Cells and Their Microenvironment: Biology and Therapeutic Implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, E.; Conti, L.; Cavallo, F. Breast cancer stem cell antigens as targets for immunotherapy. Semin. Immunol. 2020, 47, 10. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Takahashi, A.; Torigoe, T.; Morita, R.; Tamura, Y.; Tsukahara, T.; Kanaseki, T.; Kubo, T.; Watarai, K.; Kondo, T.; et al. Preferential expression of cancer/testis genes in cancer stem-like cells: Proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens 2013, 81, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Siddika, M.A.; Asha, S.Y.; Aktar, S.; Rakib, M.A.; Khanam, J.A.; Pillai, S.; Islam, F. MicroRNAs, a Promising Target for Breast Cancer Stem Cells. Mol. Diagn. Ther. 2020, 24, 69–83. [Google Scholar] [CrossRef]
- Vahidian, F.; Mohammadi, H.; Ali-Hasanzadeh, M.; Derakhshani, A.; Mostaan, M.; Hemmatzadeh, M.; Baradaran, B. MicroRNAs and breast cancer stem cells: Potential role in breast cancer therapy. J. Cell. Physiol. 2019, 234, 3294–3306. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36 (Suppl. 1), 59–72. [Google Scholar] [CrossRef] [PubMed]
- Taurin, S.; Alkhalifa, H. Breast cancers, mammary stem cells, and cancer stem cells, characteristics, and hypotheses. Neoplasia 2020, 22, 663–678. [Google Scholar] [CrossRef]
- Sreekumar, A.; Roarty, K.; Rosen, J.M. The mammary stem cell hierarchy: A looking glass into heterogeneous breast cancer landscapes. Endocr. Relat. Cancer 2015, 22, T161–T176. [Google Scholar] [CrossRef]
- Zeng, Y.A.; Nusse, R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell 2010, 6, 568–577. [Google Scholar] [CrossRef]
- Lindvall, C.; Evans, N.C.; Zylstra, C.R.; Li, Y.; Alexander, C.M.; Williams, B.O. The Wnt signaling receptor Lrp5 is required for mammary ductal stem cell activity and Wnt1-induced tumorigenesis. J. Biol. Chem. 2006, 281, 35081–35087. [Google Scholar] [CrossRef]
- Brisken, C.; Heineman, A.; Chavarria, T.; Elenbaas, B.; Tan, J.; Dey, S.K.; McMahon, J.A.; McMahon, A.P.; Weinberg, R.A. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000, 14, 650–654. [Google Scholar] [PubMed]
- Kouros-Mehr, H.; Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev. Dyn. 2006, 235, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Pal, B.; Vaillant, F.; Harburg, G.; Asselin-Labat, M.-L.; Oakes, S.R.; Lindeman, G.J.; Visvader, J.E. Notch Signaling Regulates Mammary Stem Cell Function and Luminal Cell-Fate Commitment. Cell Stem Cell 2008, 3, 429–441. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Wei, Y.; Romano, R.A.; DeCoste, C.; Kang, Y.; Sinha, S. Elf5 regulates mammary gland stem/progenitor cell fate by influencing notch signaling. Stem Cells 2012, 30, 1496–1508. [Google Scholar] [CrossRef]
- Shi, P.; Feng, J.; Chen, C. Hippo pathway in mammary gland development and breast cancer. Acta Biochim. Biophys. Sin. 2014, 47, 53–59. [Google Scholar] [CrossRef]
- Li, H.; Gumbiner, B.M. Deregulation of the Hippo pathway in mouse mammary stem cells promotes mammary tumorigenesis. Mamm. Genome 2016, 27, 556–564. [Google Scholar] [CrossRef]
- Denson, K.E.; Mussell, A.L.; Shen, H.; Truskinovsky, A.; Yang, N.; Parashurama, N.; Chen, Y.; Frangou, C.; Yang, F.; Zhang, J. The Hippo Signaling Transducer TAZ Regulates Mammary Gland Morphogenesis and Carcinogen-induced Mammary Tumorigenesis. Sci. Rep. 2018, 8, 6449. [Google Scholar] [CrossRef] [PubMed]
- Halaoui, R.; Rejon, C.; Chatterjee, S.J.; Szymborski, J.; Meterissian, S.; Muller, W.J.; Omeroglu, A.; McCaffrey, L. Progressive polarity loss and luminal collapse disrupt tissue organization in carcinoma. Genes Dev. 2017, 31, 1573–1587. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Vlachou, T.; Carminati, M.; Pelicci, P.G.; Mapelli, M. Molecular mechanisms of asymmetric divisions in mammary stem cells. EMBO Rep. 2016, 17, 1700–1720. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef]
- Chiche, A.; Moumen, M.; Petit, V.; Jonkers, J.; Medina, D.; Deugnier, M.A.; Faraldo, M.M.; Glukhova, M.A. Somatic loss of p53 leads to stem/progenitor cell amplification in both mammary epithelial compartments, basal and luminal. Stem Cells 2013, 31, 1857–1867. [Google Scholar] [CrossRef]
- Chiche, A.; Di-Cicco, A.; Sesma-Sanz, L.; Bresson, L.; de la Grange, P.; Glukhova, M.A.; Faraldo, M.M.; Deugnier, M.-A. p53 controls the plasticity of mammary luminal progenitor cells downstream of Met signaling. Breast Cancer Res. BCR 2019, 21, 13. [Google Scholar] [CrossRef] [PubMed]
- Moumen, M.; Chiche, A.; Deugnier, M.A.; Petit, V.; Gandarillas, A.; Glukhova, M.A.; Faraldo, M.M. The proto-oncogene Myc is essential for mammary stem cell function. Stem Cells 2012, 30, 1246–1254. [Google Scholar] [CrossRef]
- Yamanaka, S. Induction of pluripotent stem cells from mouse fibroblasts by four transcription factors. Cell Prolif 2008, 41, 51–56. [Google Scholar] [CrossRef]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012, 31, 1354–1365. [Google Scholar] [CrossRef]
- Al-Dhfyan, A. Embryonic signature in breast cancers; Pluripotency roots of cancer stem cells. Saudi Pharm. J. 2013, 21, 229–232. [Google Scholar] [CrossRef]
- Gwak, J.M.; Kim, M.; Kim, H.J.; Jang, M.H.; Park, S.Y. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget 2017, 8, 36305–36318. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef]
- Sin, W.C.; Lim, C.L. Breast cancer stem cells-from origins to targeted therapy. Stem Cell Investig. 2017, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, J.S.; Miele, L. Breast Cancer Stem Cells. Biomedicines 2018, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Mironchik, Y.; Winnard, P.T., Jr.; Vesuna, F.; Kato, Y.; Wildes, F.; Pathak, A.P.; Kominsky, S.; Artemov, D.; Bhujwalla, Z.; Van Diest, P.; et al. Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res. 2005, 65, 10801–10809. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar] [CrossRef]
- Gooding, A.J.; Schiemann, W.P. Epithelial–Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol. Cancer Res. 2020, 18, 1257–1270. [Google Scholar] [CrossRef]
- Sarrio, D.; Franklin, C.K.; Mackay, A.; Reis-Filho, J.S.; Isacke, C.M. Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells 2012, 30, 292–303. [Google Scholar] [CrossRef]
- Kristiansen, G.; Winzer, K.-J.; Mayordomo, E.; Bellach, J.; Schlüns, K.; Denkert, C.; Dahl, E.; Pilarsky, C.; Altevogt, P.; Guski, H.; et al. CD24 Expression Is a New Prognostic Marker in Breast Cancer. Clin. Cancer Res. 2003, 9, 4906–4913. [Google Scholar] [PubMed]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol 2011, 64, 937–946. [Google Scholar] [CrossRef]
- Honeth, G.; Bendahl, P.O.; Ringnér, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lövgren, K.; Grabau, D.; Fernö, M.; Borg, A.; Hegardt, C. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, H.E.; Li, H.; Shipitsin, M.; Gelman, R.; Polyak, K. Heterogeneity for Stem Cell–Related Markers According to Tumor Subtype and Histologic Stage in Breast Cancer. Clin. Cancer Res. 2010, 16, 876–887. [Google Scholar] [CrossRef]
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R., Jr.; Badve, S.; Nakshatri, H. CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006, 8, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24-and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [PubMed]
- Cantile, M.; Collina, F.; D’Aiuto, M.; Rinaldo, M.; Pirozzi, G.; Borsellino, C.; Franco, R.; Botti, G.; Di Bonito, M. Nuclear Localization of Cancer Stem Cell Marker CD133 in Triple-Negative Breast Cancer: A Case Report. Tumori J. 2013, 99, e245–e250. [Google Scholar] [CrossRef]
- Brugnoli, F.; Grassilli, S.; Piazzi, M.; Palomba, M.; Nika, E.; Bavelloni, A.; Capitani, S.; Bertagnolo, V. In triple negative breast tumor cells, PLC-β2 promotes the conversion of CD133 high to CD133 low phenotype and reduces the CD133-related invasiveness. Mol. Cancer 2013, 12, 165. [Google Scholar] [CrossRef]
- Duru, N.; Gernapudi, R.; Lo, P.-K.; Yao, Y.; Wolfson, B.; Zhang, Y.; Zhou, Q. Characterization of the CD49f+/CD44+/CD24− single-cell derived stem cell population in basal-like DCIS cells. Oncotarget 2016, 7, 47511. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhong, X.; Qiu, Y.; Yang, L.; Wei, B.; Zhang, Z.; Bu, H. CD49f Can Act as a Biomarker for Local or Distant Recurrence in Breast Cancer. J. Breast Cancer 2017, 20, 142–149. [Google Scholar] [CrossRef]
- Lobba, A.R.; Forni, M.F.; Carreira, A.C.; Sogayar, M.C. Differential expression of CD90 and CD14 stem cell markers in malignant breast cancer cell lines. Cytom. A 2012, 81, 1084–1091. [Google Scholar] [CrossRef]
- Shaikh, M.V.; Kala, M.; Nivsarkar, M. CD90 a potential cancer stem cell marker and a therapeutic target. Cancer Biomark 2016, 16, 301–307. [Google Scholar] [CrossRef]
- Kim, R.-J.; Nam, J.-S. OCT4 Expression Enhances Features of Cancer Stem Cells in a Mouse Model of Breast Cancer. Lab. Anim. Res. 2011, 27, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Ghanei, Z.; Jamshidizad, A.; Joupari, M.D.; Shamsara, M. Isolation and characterization of breast cancer stem cell-like phenotype by Oct4 promoter-mediated activity. J. Cell. Physiol. 2020, 235, 7840–7848. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55. [Google Scholar] [CrossRef]
- Atkinson, R.L.; Yang, W.T.; Rosen, D.G.; Landis, M.D.; Wong, H.; Lewis, M.T.; Creighton, C.J.; Sexton, K.R.; Hilsenbeck, S.G.; Sahin, A.A. Cancer stem cell markers are enriched in normal tissue adjacent to triple negative breast cancer and inversely correlated with DNA repair deficiency. Breast Cancer Res. 2013, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, D.; Sheng, D.; Xu, J.; Chen, W.; Qin, Y.; Du, R.; Yang, X.; He, X.; Xie, N.; et al. NOTCH4 maintains quiescent mesenchymal-like breast cancer stem cells via transcriptionally activating SLUG and GAS1 in triple-negative breast cancer. Theranostics 2020, 10, 2405–2421. [Google Scholar] [CrossRef]
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Shared signaling pathways in normal and breast cancer stem cells. J. Carcinog. 2011, 10, 1477–3163. [Google Scholar]
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, X.; Wang, T.; Guo, Q.; Xi, T.; Zheng, L. Emerging agents that target signaling pathways in cancer stem cells. J. Hematol. Oncol. 2020, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.-S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Valenti, G.; Quinn, H.M.; Heynen, G.; Lan, L.; Holland, J.D.; Vogel, R.; Wulf-Goldenberg, A.; Birchmeier, W. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017, 77, 2134–2147. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef]
- Cai, W.-Y.; Wei, T.-Z.; Luo, Q.-C.; Wu, Q.-W.; Liu, Q.-F.; Yang, M.; Ye, G.-D.; Wu, J.-F.; Chen, Y.-Y.; Sun, G.-B.; et al. The Wnt–β-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J. Cell Sci. 2013, 126, 2877–2889. [Google Scholar] [CrossRef] [PubMed]
- Barnawi, R.; Al-Khaldi, S.; Bakheet, T.; Fallatah, M.; Alaiya, A.; Ghebeh, H.; Al-Alwan, M. Fascin Activates β-Catenin Signaling and Promotes Breast Cancer Stem Cell Function Mainly Through Focal Adhesion Kinase (FAK): Relation With Disease Progression. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, P.; Zhang, H.; Xu, H.; Gao, N.; Li, M.; Liu, C. Nestin positively regulates the Wnt/β-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014, 16, 408. [Google Scholar] [CrossRef]
- Shan, S.; Lv, Q.; Zhao, Y.; Liu, C.; Sun, Y.; Xi, K.; Xiao, J.; Li, C. Wnt/β-catenin pathway is required for epithelial to mesenchymal transition in CXCL12 over expressed breast cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 12357–12367. [Google Scholar]
- Xu, L.; Zhang, L.; Hu, C.; Liang, S.; Fei, X.; Yan, N.; Zhang, Y.; Zhang, F. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int. J. Oncol. 2016, 48, 1175–1186. [Google Scholar] [CrossRef]
- Huang, J.; Tao, C.; Yu, Y.; Yu, F.; Zhang, H.; Gao, J.; Wang, D.; Chen, Y.; Zhang, G.; Zhou, G.; et al. Simultaneous Targeting of Differentiated Breast Cancer Cells and Breast Cancer Stem Cells by Combination of Docetaxel- and Sulforaphane-Loaded Self-Assembled Poly(D, L-lactide-co-glycolide)/Hyaluronic Acid Block Copolymer-Based Nanoparticles. J. Biomed. Nanotechnol. 2016, 12, 1463–1477. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.-K.; Hung, M.-C.; et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, N.; Gray, R.S.; Li, H.; Ewald, A.J.; Zahnow, C.A.; Pan, D. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev. 2014, 28, 432–437. [Google Scholar] [CrossRef]
- Yang, C.E.; Lee, W.Y.; Cheng, H.W.; Chung, C.H.; Mi, F.L.; Lin, C.W. The antipsychotic chlorpromazine suppresses YAP signaling, stemness properties, and drug resistance in breast cancer cells. Chem. Biol. Interact. 2019, 302, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-W.; Shen, H.; Frangou, C.; Yang, N.; Guo, J.; Xu, B.; Bshara, W.; Shepherd, L.; Zhu, Q.; Wang, J.; et al. Characterization of TAZ domains important for the induction of breast cancer stem cell properties and tumorigenesis. Cell Cycle 2015, 14, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Maugeri-Saccà, M.; De Maria, R. Hippo pathway and breast cancer stem cells. Crit. Rev. Oncol. Hematol. 2016, 99, 115–122. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Zhou, B.; Damrauer, J.S.; Bailey, S.T.; Hadzic, T.; Jeong, Y.; Clark, K.; Fan, C.; Murphy, L.; Lee, C.Y.; Troester, M.A.; et al. Erythropoietin promotes breast tumorigenesis through tumor-initiating cell self-renewal. J. Clin. Investig. 2014, 124, 553–563. [Google Scholar] [CrossRef][Green Version]
- Song, I.H.; Kim, Y.-A.; Heo, S.-H.; Park, I.A.; Lee, M.; Bang, W.S.; Park, H.S.; Gong, G.; Lee, H.J. ADAR1 expression is associated with tumour-infiltrating lymphocytes in triple-negative breast cancer. Tumor Biol. 2017, 39, 1010428317734816. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, D.; Gacquer, D.; Rothé, F.; Lefort, A.; Libert, F.; Brown, D.; Kheddoumi, N.; Shlien, A.; Konopka, T.; Salgado, R.; et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015, 13, 277–289. [Google Scholar] [CrossRef]
- Xu, L.D.; Öhman, M. ADAR1 Editing and its Role in Cancer. Genes 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Cottrell, K.A.; Ryu, S.; Bramel, E.R.; Kladney, R.D.; Bross, E.A.; Maggi, L.; Weber, J.D. ADAR1 editing dependency in triple-negative breast cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kung, C.P.; Cottrell, K.A.; Ryu, S.; Bramel, E.R.; Kladney, R.D.; Bao, E.A.; Freeman, E.C.; Sabloak, T.; Maggi, L., Jr.; Weber, J.D. Evaluating the therapeutic potential of ADAR1 inhibition for triple-negative breast cancer. Oncogene 2021, 40, 189–202. [Google Scholar] [CrossRef] [PubMed]
- de Santiago, P.R.; Blanco, A.; Morales, F.; Marcelain, K.; Harismendy, O.; Sjöberg Herrera, M.; Armisén, R. Immune-related IncRNA LINC00944 responds to variations in ADAR1 levels and it is associated with breast cancer prognosis. Life Sci. 2020, 268, 118956. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, E.A.; Blanco, A.; Sagredo, A.I.; Pérez, P.; Sepúlveda-Hermosilla, G.; Morales, F.; Müller, B.; Verdugo, R.; Marcelain, K.; Harismendy, O.; et al. ADAR1-mediated RNA-editing of 3’UTRs in breast cancer. Biol. Res. 2018, 51, 36. [Google Scholar] [CrossRef]
- Vazquez-Santillan, K.; Melendez-Zajgla, J.; Jimenez-Hernandez, L.E.; Gaytan-Cervantes, J.; Muñoz-Galindo, L.; Piña-Sanchez, P.; Martinez-Ruiz, G.; Torres, J.; Garcia-Lopez, P.; Gonzalez-Torres, C.; et al. NF-kappaΒ-inducing kinase regulates stem cell phenotype in breast cancer. Sci. Rep. 2016, 6, 37340. [Google Scholar] [CrossRef]
- Kong, L.; Guo, S.; Liu, C.; Zhao, Y.; Feng, C.; Liu, Y.; Wang, T.; Li, C. Overexpression of SDF-1 activates the NF-κB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells. Int. J. Oncol. 2016, 48, 1085–1094. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Futakuchi, M.; Lami, K.; Tachibana, Y.; Yamamoto, Y.; Furukawa, M.; Fukuoka, J. The Effects of TGF-β Signaling on Cancer Cells and Cancer Stem Cells in the Bone Microenvironment. Int. J. Mol. Sci. 2019, 20, 5117. [Google Scholar] [CrossRef] [PubMed]
- Futakuchi, M.; Nannuru, K.C.; Varney, M.L.; Sadanandam, A.; Nakao, K.; Asai, K.; Shirai, T.; Sato, S.Y.; Singh, R.K. Transforming growth factor-beta signaling at the tumor-bone interface promotes mammary tumor growth and osteoclast activation. Cancer Sci. 2009, 100, 71–81. [Google Scholar] [CrossRef]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef]
- Woosley, A.N.; Dalton, A.C.; Hussey, G.S.; Howley, B.V.; Mohanty, B.K.; Grelet, S.; Dincman, T.; Bloos, S.; Olsen, S.K.; Howe, P.H. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene 2019, 38, 3794–3811. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Wang, C.; Zhou, Z.; Chen, C.; Liu, R.; Wang, D. Transforming growth factor-beta increases breast cancer stem cell population partially through upregulating PMEPA1 expression. Acta Biochim. Biophys. Sin. 2016, 48, 194–201. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Y.; Tsuyada, A.; Ren, X.; Wu, X.; Stubblefield, K.; Rankin-Gee, E.K.; Wang, S.E. Transforming growth factor-β regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene 2011, 30, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Detassis, S.; Grasso, M.; Del Vescovo, V.; Denti, M.A. microRNAs Make the Call in Cancer Personalized Medicine. Front. Cell Dev. Biol. 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Yu, F.; Jiao, Y.; Zhu, Y.; Wang, Y.; Zhu, J.; Cui, X.; Liu, Y.; He, Y.; Park, E.Y.; Zhang, H.; et al. MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J. Biol. Chem. 2012, 287, 465–473. [Google Scholar] [CrossRef]
- Kang, L.; Mao, J.; Tao, Y.; Song, B.; Ma, W.; Lu, Y.; Zhao, L.; Li, J.; Yang, B.; Li, L. MicroRNA-34a suppresses the breast cancer stem cell-like characteristics by downregulating Notch1 pathway. Cancer Sci. 2015, 106, 700–708. [Google Scholar] [CrossRef]
- Park, E.Y.; Chang, E.; Lee, E.J.; Lee, H.W.; Kang, H.G.; Chun, K.H.; Woo, Y.M.; Kong, H.K.; Ko, J.Y.; Suzuki, H.; et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res. 2014, 74, 7573–7582. [Google Scholar] [CrossRef]
- Li, C.Y.; Miao, K.L.; Chen, Y.; Liu, L.Y.; Zhao, G.B.; Lin, M.H.; Jiang, C. Jagged2 promotes cancer stem cell properties of triple negative breast cancer cells and paclitaxel resistance via regulating microRNA-200. Eur. Rev. Med. Pharm. Sci. 2018, 22, 6008–6014. [Google Scholar]
- Sun, X.; Xu, C.; Tang, S.C.; Wang, J.; Wang, H.; Wang, P.; Du, N.; Qin, S.; Li, G.; Xu, S.; et al. Let-7c blocks estrogen-activated Wnt signaling in induction of self-renewal of breast cancer stem cells. Cancer Gene Ther. 2016, 23, 83–89. [Google Scholar] [CrossRef]
- Sun, X.; Jiang, S.; Liu, J.; Wang, H.; Zhang, Y.; Tang, S.-C.; Wang, J.; Du, N.; Xu, C.; Wang, C.; et al. MiR-208a stimulates the cocktail of SOX2 and β-catenin to inhibit the let-7 induction of self-renewal repression of breast cancer stem cells and formed miR208a/let-7 feedback loop via LIN28 and DICER1. Oncotarget 2015, 6, 32944–32954. [Google Scholar] [CrossRef]
- Liang, R.; Li, Y.; Wang, M.; Tang, S.C.; Xiao, G.; Sun, X.; Li, G.; Du, N.; Liu, D.; Ren, H. MiR-146a promotes the asymmetric division and inhibits the self-renewal ability of breast cancer stem-like cells via indirect upregulation of Let-7. Cell Cycle 2018, 17, 1445–1456. [Google Scholar] [CrossRef]
- El Helou, R.; Pinna, G.; Cabaud, O.; Wicinski, J.; Bhajun, R.; Guyon, L.; Rioualen, C.; Finetti, P.; Gros, A.; Mari, B.; et al. miR-600 Acts as a Bimodal Switch that Regulates Breast Cancer Stem Cell Fate through WNT Signaling. Cell Rep. 2017, 18, 2256–2268. [Google Scholar] [CrossRef]
- Isobe, T.; Hisamori, S.; Hogan, D.J.; Zabala, M.; Hendrickson, D.G.; Dalerba, P.; Cai, S.; Scheeren, F.; Kuo, A.H.; Sikandar, S.S.; et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife 2014, 18, 01977. [Google Scholar]
- Wang, L.; Tian, H.; Yuan, J.; Wu, H.; Wu, J.; Zhu, X. CONSORT: Sam68 Is Directly Regulated by MiR-204 and Promotes the Self-Renewal Potential of Breast Cancer Cells by Activating the Wnt/Beta-Catenin Signaling Pathway. Medicine 2015, 94, e2228. [Google Scholar] [CrossRef]
- Liu, T.; Hu, K.; Zhao, Z.; Chen, G.; Ou, X.; Zhang, H.; Zhang, X.; Wei, X.; Wang, D.; Cui, M.; et al. MicroRNA-1 down-regulates proliferation and migration of breast cancer stem cells by inhibiting the Wnt/β-catenin pathway. Oncotarget 2015, 6, 41638–41649. [Google Scholar] [CrossRef]
- Cheng, S.; Huang, Y.; Lou, C.; He, Y.; Zhang, Y.; Zhang, Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin β3/Wnt signaling under miR-137 regulation. Cancer Biol. Ther. 2019, 20, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Li, F.; Li, X.; Tian, Y.; Zhang, Y.; Sheng, X.; Song, Y.; Meng, Q.; Yuan, S.; Luan, L.; et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 2017, 8, 1036. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, T.Y.; Zou, D.L.; Zhou, L.; Lou, M.; Liu, J.S.; Li, Y.Z.; Ding, D.Y.; Li, Y.C.; Zhang, N.; et al. miR-520b Promotes Breast Cancer Stemness Through Hippo/YAP Signaling Pathway. Oncol. Targets Ther. 2019, 12, 11691–11700. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Patel, S.H.; Ginestier, C.; Ibarra, I.; Martin-Trevino, R.; Bai, S.; McDermott, S.P.; Shang, L.; Ke, J.; Ou, S.J.; et al. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet. 2012, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Finlay-Schultz, J.; Cittelly, D.M.; Hendricks, P.; Patel, P.; Kabos, P.; Jacobsen, B.M.; Richer, J.K.; Sartorius, C.A. Progesterone downregulation of miR-141 contributes to expansion of stem-like breast cancer cells through maintenance of progesterone receptor and Stat5a. Oncogene 2015, 34, 3676–3687. [Google Scholar] [CrossRef]
- Li, B.; Lu, Y.; Yu, L.; Han, X.; Wang, H.; Mao, J.; Shen, J.; Wang, B.; Tang, J.; Li, C.; et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-κB/COX-2 activation. Chem. Biol. Interact. 2017, 277, 33–42. [Google Scholar] [CrossRef]
- Li, B.; Lu, Y.; Wang, H.; Han, X.; Mao, J.; Li, J.; Yu, L.; Wang, B.; Fan, S.; Yu, X.; et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed. Pharm. 2016, 79, 93–101. [Google Scholar] [CrossRef]
- Han, M.; Liu, M.; Wang, Y.; Chen, X.; Xu, J.; Sun, Y.; Zhao, L.; Qu, H.; Fan, Y.; Wu, C. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS ONE 2012, 7, e39520. [Google Scholar] [CrossRef] [PubMed]
- Bahena-Ocampo, I.; Espinosa, M.; Ceballos-Cancino, G.; Lizarraga, F.; Campos-Arroyo, D.; Schwarz, A.; Garcia-Lopez, P.; Maldonado, V.; Melendez-Zajgla, J. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016, 17, 201642700. [Google Scholar] [CrossRef]
- Gao, L.; Guo, Q.; Li, X.; Yang, X.; Ni, H.; Wang, T.; Zhao, Q.; Liu, H.; Xing, Y.; Xi, T.; et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine 2019, 41, 395–407. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, F.; Chen, L.; Yang, Y.; Cao, S.; Ye, Y.; Wang, X.; Mu, J.; Li, Z.; Li, L. Blockage of TGFβ-SMAD2 by demethylation-activated miR-148a is involved in caffeic acid-induced inhibition of cancer stem cell-like properties in vitro and in vivo. FEBS Open Biol. 2015, 5, 466–475. [Google Scholar] [CrossRef]
- Jiang, F.; Li, Y.; Mu, J.; Hu, C.; Zhou, M.; Wang, X.; Si, L.; Ning, S.; Li, Z. Glabridin inhibits cancer stem cell-like properties of human breast cancer cells: An epigenetic regulation of miR-148a/SMAd2 signaling. Mol. Carcinog. 2016, 55, 929–940. [Google Scholar] [CrossRef]
- Hu, X.; Guo, J.; Zheng, L.; Li, C.; Zheng, T.M.; Tanyi, J.L.; Liang, S.; Benedetto , C.; Mitidieri, M.; Katsaros, D.; et al. The Heterochronic microRNA let-7 Inhibits Cell Motility by Regulating the Genes in the Actin Cytoskeleton Pathway in Breast Cancer. Mol. Cancer Res. 2013, 11, 240–250. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef]
- Song, L.; Wang, L.; Li, Y.; Xiong, H.; Wu, J.; Li, J.; Li, M. Sam68 up-regulation correlates with, and its down-regulation inhibits, proliferation and tumourigenicity of breast cancer cells. J. Pathol. 2010, 222, 227–237. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, N.; Liu, L.; Dong, H.; Liu, X. microRNA-128-3p overexpression inhibits breast cancer stem cell characteristics through suppression of Wnt signalling pathway by down-regulating NEK2. J. Cell Mol. Med. 2020, 24, 7353–7369. [Google Scholar] [CrossRef]
- Han, M.; Liu, M.; Wang, Y.; Mo, Z.; Bi, X.; Liu, Z.; Fan, Y.; Chen, X.; Wu, C. Re-expression of miR-21 contributes to migration and invasion by inducing epithelial-mesenchymal transition consistent with cancer stem cell characteristics in MCF-7 cells. Mol. Cell. Biochem. 2012, 363, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J. Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef]
- Lei, X.; Bandyopadhyay, A.; Le, T.; Sun, L. Autocrine TGFbeta supports growth and survival of human breast cancer MDA-MB-231 cells. Oncogene 2002, 21, 7514–7523. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Yang, J.; Nichols, R.W.; Sun, L.Z. Abrogation of TGFbeta signaling induces apoptosis through the modulation of MAP kinase pathways in breast cancer cells. Exp. Cell Res. 2007, 313, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bandyopadhyay, A.; Nichols, R.W.; Wang, L.; Hinck, A.P.; Wang, S.; Sun, L.Z. Blockade of Autocrine TGF-β Signaling Inhibits Stem Cell Phenotype, Survival, and Metastasis of Murine Breast Cancer Cells. J. Stem Cell Res. Ther. 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Soudyab, M.; Iranpour, M.; Ghafouri-Fard, S. The Role of Long Non-Coding RNAs in Breast Cancer. Arch. Iran. Med. 2016, 19, 508–517. [Google Scholar] [PubMed]
- Chen, S.; Zhu, J.; Wang, F.; Guan, Z.; Ge, Y.; Yang, X.; Cai, J. LncRNAs and their role in cancer stem cells. Oncotarget 2017, 8, 110685–110692. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, J.; Koirala, P.; Ding, X.; Chen, B.; Wang, Y.; Wang, Z.; Wang, C.; Zhang, X.; Mo, Y.-Y. Long non-coding RNAs as prognostic markers in human breast cancer. Oncotarget 2016, 7, 20584–20596. [Google Scholar] [CrossRef]
- Huang, Q.Y.; Liu, G.F.; Qian, X.L.; Tang, L.B.; Xiong, L.X. Long Non-Coding RNA: Dual Effects on Breast Cancer Metastasis and Clinical Applications. Cancers 2019, 11, 1802. [Google Scholar] [CrossRef] [PubMed]
- García-Venzor, A.; Mandujano-Tinoco, E.A.; Lizarraga, F.; Zampedri, C.; Krötzsch, E.; Salgado, R.M.; Dávila-Borja, V.M.; Encarnación-Guevara, S.; Melendez-Zajgla, J.; Maldonado, V. Microenvironment-regulated lncRNA-HAL is able to promote stemness in breast cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 12, 8. [Google Scholar] [CrossRef]
- Tu, Z.; Schmöllerl, J.; Cuiffo, B.G.; Karnoub, A.E. Microenvironmental Regulation of Long Noncoding RNA LINC01133 Promotes Cancer Stem Cell-Like Phenotypic Traits in Triple-Negative Breast Cancers. Stem Cells 2019, 37, 1281–1292. [Google Scholar] [CrossRef]
- Zhou, M.; Hou, Y.; Yang, G.; Zhang, H.; Tu, G.; Du, Y.-E.; Wen, S.; Xu, L.; Tang, X.; Tang, S.; et al. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells 2016, 34, 55–66. [Google Scholar] [CrossRef]
- Lu, Z.-D.; Jiao, D.-C.; Qiao, J.-H.; Yang, S.; Liu, Z.-Z. Long non-coding RNA SOX21-AS1 modulates breast cancer stem cells properties and carcinogenesis via targeting SOX2. Oncotarget 2017, 5, 1–11. [Google Scholar] [CrossRef]
- Li, L.; Meng, D.; Wang, R. Long non-coding RNA SOX21-AS1 enhances the stemness of breast cancer cells via the Hippo pathway. FEBS Open Biol. 2020, 26, 2211–5463. [Google Scholar]
- Wang, B.; Ye, Q.; Zou, C. Long Non-Coding RNA THOR Enhances the Stem Cell-Like Traits of Triple-Negative Breast Cancer Cells Through Activating β-Catenin Signaling. Med. Sci. Monit. 2020, 14, 923507. [Google Scholar]
- Tang, T.; Guo, C.; Xia, T.; Zhang, R.; Zen, K.; Pan, Y.; Jin, L. LncCCAT1 Promotes Breast Cancer Stem Cell Function through Activating WNT/β-catenin Signaling. Theranostics 2019, 9, 7384–7402. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Song, X.; Zhang, L.; Zhao, L.; Mao, X.; Wei, M.; Jin, F. Long non-coding RNA LUCAT1/miR-5582-3p/TCF7L2 axis regulates breast cancer stemness via Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 305. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Li, T.-T.; Wang, K.-L.; Xiao, G.-Q.; Wang, J.-H.; Zhao, H.-D.; Kang, Z.-J.; Fan, W.-J.; Zhu, L.-L.; Li, M.; et al. H19/let-7/LIN28 reciprocal negative regulatory circuit promotes breast cancer stem cell maintenance. Cell Death Dis. 2017, 8, e2569. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Kida, K.; Adachi, S.; Yamada, A.; Sugae, S.; Narui, K.; Miyagi, Y.; Nishi, M.; Ryo, A.; Murata, S.; et al. Lnc RNA H19 is associated with poor prognosis in breast cancer patients and promotes cancer stemness. Breast Cancer Res. Treat. 2018, 170, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, Y.; Xiao, G.D.; Zheng, X.Q.; Wang, J.C.; Xu, C.W.; Qin, S.; Ren, H.; Tang, S.C.; Sun, X. H19 regulation of oestrogen induction of symmetric division is achieved by antagonizing Let-7c in breast cancer stem-like cells. Cell Prolif. 2019, 52, 18. [Google Scholar] [CrossRef]
- Deng, J.; Yang, M.; Jiang, R.; An, N.; Wang, X.; Liu, B. Long Non-Coding RNA HOTAIR Regulates the Proliferation, Self-Renewal Capacity, Tumor Formation and Migration of the Cancer Stem-Like Cell (CSC) Subpopulation Enriched from Breast Cancer Cells. PLoS ONE 2017, 12, e0170860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef]
- Zeng, L.; Cen, Y.; Chen, J. Long non-coding RNA MALAT-1 contributes to maintenance of stem cell-like phenotypes in breast cancer cells. Oncol. Lett. 2018, 15, 2117–2122. [Google Scholar] [CrossRef]
- Ma, F.; Liu, X.; Zhou, S.; Li, W.; Liu, C.; Chadwick, M.; Qian, C. Long non-coding RNA FGF13-AS1 inhibits glycolysis and stemness properties of breast cancer cells through FGF13-AS1/IGF2BPs/Myc feedback loop. Cancer Lett. 2019, 450, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, L.; Zhang, Y.; Lu, G.; Li, Y.; Wei, Z. Long non-coding RNA FEZF1-AS1 promotes breast cancer stemness and tumorigenesis via targeting miR-30a/Nanog axis. J. Cell Physiol. 2018, 233, 8630–8638. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Li, Y.; Ma, Y.; Lu, J.; Chen, Y.; Jiang, Q.; Qin, Q.; Zhao, L.; Huang, Q.; Luo, Z.; et al. Long noncoding RNA LINC00511 contributes to breast cancer tumourigenesis and stemness by inducing the miR-185-3p/E2F1/Nanog axis. J. Exp. Clin. Cancer Res. 2018, 37, 289. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, L.; Xu, L.; Qin, K.; Liu, C.; Yu, Y.; Su, D.; Wu, K.; Sheng, Y. Long noncoding RNA linc00617 exhibits oncogenic activity in breast cancer. Mol. Carcinog. 2017, 56, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.-C.; Choi, H.S.; Liu, R.; Kim, J.-H.; Kim, S.-L.; Yun, B.-S.; Lee, D.-S. Inhibitory Effects of Tangeretin, A Citrus Peel-Derived Flavonoid, on Breast Cancer Stem Cell Formation through Suppression of Stat3 Signaling. Molecules 2020, 25, 2599. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xu, C.; Xiao, G.; Meng, J.; Wang, J.; Tang, S.-C.; Qin, S.; Du, N.; Li, G.; Ren, H.; et al. Breast cancer stem-like cells are sensitized to tamoxifen induction of self-renewal inhibition with enforced Let-7c dependent on Wnt blocking. Int. J. Mol. Med. 2018, 41, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Shin, D.H.; Kim, J.S. Let-7 miRNA and CDK4 siRNA co-encapsulated in Herceptin-conjugated liposome for breast cancer stem cells. Asian J. Pharm. Sci. 2020, 15, 472–481. [Google Scholar] [CrossRef]
- Liu, R.; Choi, H.S.; Kim, S.L.; Kim, J.H.; Yun, B.S.; Lee, D.S. 6-Methoxymellein Isolated from Carrot (Daucus carota L.) Targets Breast Cancer Stem Cells by Regulating NF-κB Signaling. Molecules 2020, 25, 4374. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chen, W.; Wei, F.; Xie, X. TV-circRGPD6 Nanoparticle Suppresses Breast Cancer Stem Cell-Mediated Metastasis via the miR-26b/YAF2 Axis. Mol. Ther. 2020, 5, 30462–30467. [Google Scholar]
- Sakunrangsit, N.; Ketchart, W. Plumbagin inhibits cancer stem-like cells, angiogenesis and suppresses cell proliferation and invasion by targeting Wnt/β-catenin pathway in endocrine resistant breast cancer. Pharm. Res. 2019, 150, 3. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liu, Y.; Wei, H.Y.; Lv, K.Z.; Fu, P. Linc-ROR induces epithelial-mesenchymal transition and contributes to drug resistance and invasion of breast cancer cells. Tumour Biol. 2016, 37, 10861–10870. [Google Scholar] [CrossRef]
- Xiu, D.H.; Liu, G.F.; Yu, S.N.; Li, L.Y.; Zhao, G.Q.; Liu, L.; Li, X.F. Long non-coding RNA LINC00968 attenuates drug resistance of breast cancer cells through inhibiting the Wnt2/β-catenin signaling pathway by regulating WNT2. J. Exp. Clin. Cancer Res. 2019, 38, 94. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Marker | Protein | Reference |
|---|---|---|
| CD44 | Cell adhesion molecule | [46,47] |
| CD24 | Cell adhesion molecule | [45,46,47] |
| ALDH1 | Aldehyde dehydrogenase 1 | [46,47,51] |
| CD133 | Prominin 1 | [52,53,54] |
| CD49f | α6-integrin | [55,56] |
| CD90 | GPI-anchored glycoprotein | [57,58] |
| miRNA | Target | Hallmarks | Reference |
|---|---|---|---|
| miR-34c | Not apply | Self-renewal, EMT | [108] |
| miR-34a | Not apply | Stemness and chemoresistance | [109,110] |
| miR-200 | Not apply | Stemness and Chemoresistance | [111] |
| Let-7c | Not apply | Self-renewal and tumorigenicity | [112] |
| miR-208a-Let-7 | Lin28, Sox2 and β-catenin | Self-renewal | [113] |
| Let-7 | Lin28 | Expansion and self-renewal | [73] |
| miR-146a-Let-7c | Lin28 | Asymmetric division and Expansion | [114] |
| miR-600 | SCD1 | Self-renewal and Expansion | [115] |
| miR-142 | APC | Proliferation and apoptosis | [116] |
| miR-204 | Sam68 | Self-renewal | [117] |
| miR-1 | Frizzled 7 and TNKS2 | Proliferation and migration | [118] |
| miR-137 | FSTL1 | Chemoresistance, proliferation, and stemness | [119] |
| miR-31 | Xin1, Gsk3β, Dkk1, Smad3 and Smad4 | Self-renewal, maintenance and metastasis | [120] |
| miR-520b | LATS2 | Stemness maintenance | [121] |
| miR-93 | Not apply | Proliferation and differentiation | [122] |
| miR-141 | STAT5A | Proliferation and expansion | [123] |
| miR-221/222 | PTEN | Self-renewal Stemness properties and tumor growth | [124,125] |
| miR-21 | PTEN | EMT and Stemness properties | [126] |
| miR-10b | PTEN | EMT, self-renewal metastasis and migration | [127] |
| miR-873 | PD-L1 | Stemness and chemoresistance | [128] |
| miR-181 | ATM | Mammospheres Formation | [104] |
| miR-148a | SMAD2 | Proliferation, migration, invasion processes and mammospheres Formation | [129,130] |
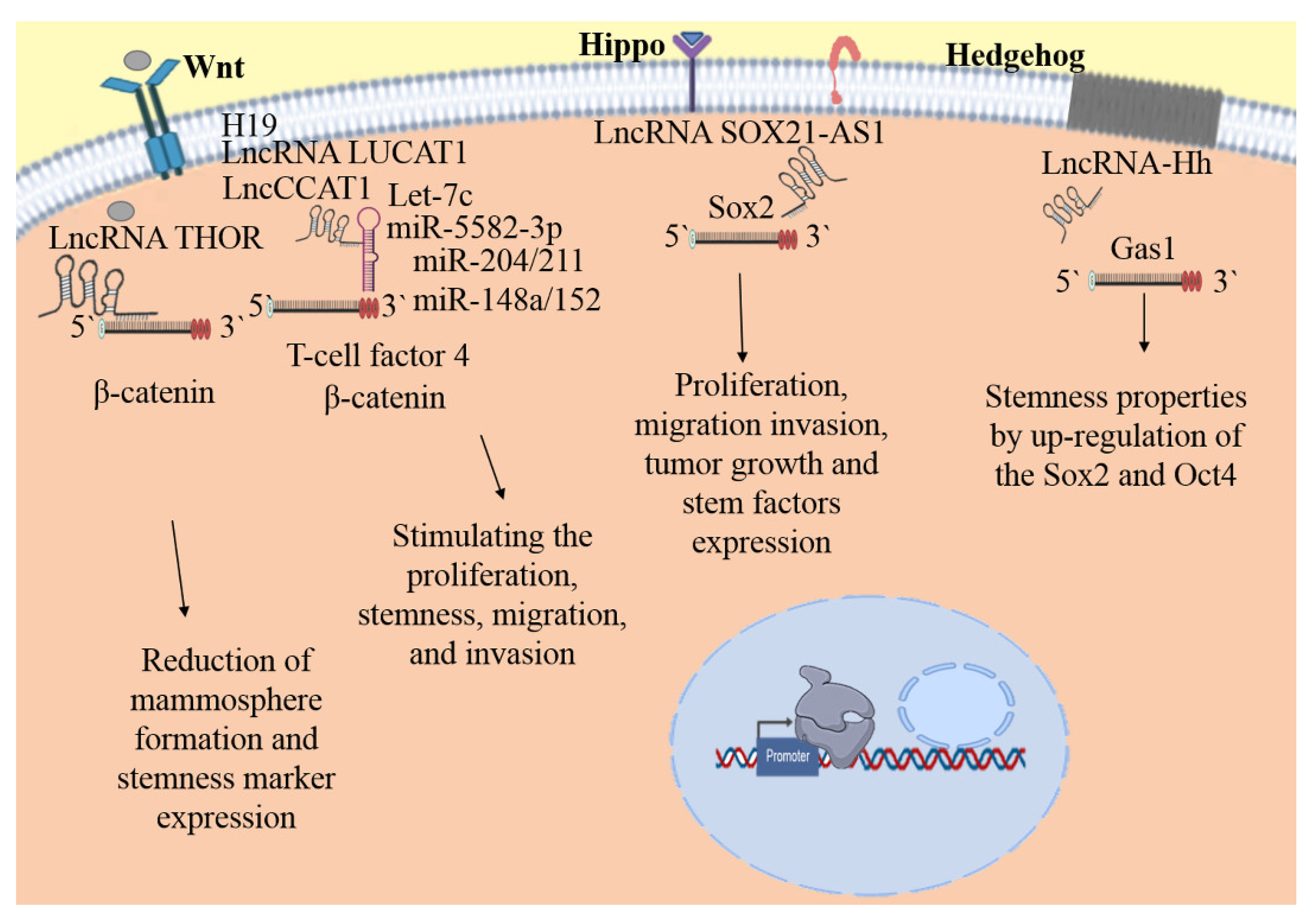
| LncRNA | miRNA | Target (mRNA) | Hallmarks | Reference |
|---|---|---|---|---|
| HAL | Not apply | Not apply | Stemness, proliferation, migration, and cell survival | [145] |
| LINC01133 | miR-199a | KLF4 and FOXP2 | Stemness and growth | [146] |
| HH | Not apply | GAS1 | Stemness maintenance, mammospheres formation, EMT and self-renewal | [147] |
| SOX21-AS1 | miR-429 | SOX2 | Proliferation, invasion, tumor growth, stem factors expression | [148] |
| SOX21-AS1 | Not apply | Not apply | Stemness, proliferation, migration and invasion abilities | [149] |
| THOR | Not apply | β-Catenin | Mammospheres formation and stemness marker expression | [150] |
| CCAT1 | miR-204/211, 148a/152 | TCF4 | Proliferation, stemness, migration, and invasion | [151] |
| LUCAT1 | miR-5582-3p | TCF7L2 | Stemness features, proliferation and tumor growth | [152] |
| H19 | Let-7 | Lin28 | Stemness maintenance, clonogenicity, migration, mammospheres formation, and tumor growth | [153] |
| H19 | miR-103, 107, let-7, and 29b-1, | Not apply | Stemness, mammospheres formation and tumor size | [154] |
| H19 | Let-7c | Not apply | Symmetric division and self-renewal | [155] |
| HOTAIR | miR-34a | Sox2 | Proliferation, invasion, Self-Renewal, Tumor Formation and Migration | [156] |
| HOTAIR | miR-7 | SETDB1 | Invasion, metastasis and EMT | [157] |
| MALAT-1 | Not apply | Sox2 | Stemness maintenance, mammospheres formation, proliferation, migration, and invasion | [158] |
| FGF13-AS1 | Not apply | c-Myc and IGF2BP | proliferation, migration, invasion, glycolysis and stemness properties | [159] |
| FEZF1-AS1 | miR-30a | Nanog | Stemness, mammospheres, growth, proliferation, migration, and invasion | [160] |
| LINC00511 | miR-185-3p | E2F1 | Stemness, proliferation, mammospheres formation and tumor growth | [161] |
| LINC00617 | Not apply | Sox2 | Motility, invasion, EMT and metastasis | [162] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Huerta, N.; Silva-Cázares, M.B.; Arriaga-Pizano, L.A.; Prieto-Chávez, J.L.; López-Camarillo, C. LncRNAs and microRNAs as Essential Regulators of Stemness in Breast Cancer Stem Cells. Biomolecules 2021, 11, 380. https://doi.org/10.3390/biom11030380
Flores-Huerta N, Silva-Cázares MB, Arriaga-Pizano LA, Prieto-Chávez JL, López-Camarillo C. LncRNAs and microRNAs as Essential Regulators of Stemness in Breast Cancer Stem Cells. Biomolecules. 2021; 11(3):380. https://doi.org/10.3390/biom11030380
Chicago/Turabian StyleFlores-Huerta, Nadia, Macrina B. Silva-Cázares, Lourdes A. Arriaga-Pizano, Jessica L. Prieto-Chávez, and César López-Camarillo. 2021. "LncRNAs and microRNAs as Essential Regulators of Stemness in Breast Cancer Stem Cells" Biomolecules 11, no. 3: 380. https://doi.org/10.3390/biom11030380
APA StyleFlores-Huerta, N., Silva-Cázares, M. B., Arriaga-Pizano, L. A., Prieto-Chávez, J. L., & López-Camarillo, C. (2021). LncRNAs and microRNAs as Essential Regulators of Stemness in Breast Cancer Stem Cells. Biomolecules, 11(3), 380. https://doi.org/10.3390/biom11030380