
Majeed Syndrome: A Review of the Clinical, Genetic and Immunologic Features


Abstract
1. Introduction
1.1. Autoinflammatory Disorders
1.2. Chronic Recurrent Multifocal Osteomyelitis (CRMO)
1.3. Autoinflammatory Bone Disease Syndromes
2. Majeed Syndrome
Clinical Features of Majeed Syndrome
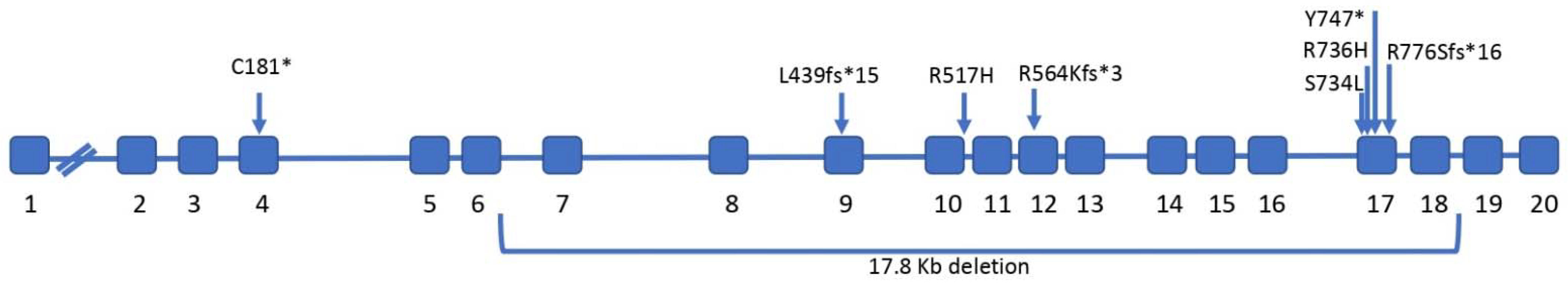
3. Genetics of Majeed Syndrome
3.1. Pathogenesis of Majeed Syndrome
3.2. Majeed Syndrome as an Inflammasomopathy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Masters, S.L.; Simon, A.; Aksentijevich, I.; Kastner, D.L. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease (*). Annu. Rev. Immunol. 2009, 27, 621–668. [Google Scholar] [CrossRef]
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297. [Google Scholar] [CrossRef]
- A candidate gene for familial Mediterranean fever. The French FMF Consortium. Nat. Genet. 1997, 17, 25–31. [CrossRef] [PubMed]
- Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 1997, 90, 797–807. [CrossRef]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef]
- Oda, H.; Kastner, D.L. Genomics, Biology, and Human Illness: Advances in the Monogenic Autoinflammatory Diseases. Rheum. Dis. Clin. N. Am. 2017, 43, 327–345. [Google Scholar] [CrossRef]
- Cox, A.J.; Zhao, Y.; Ferguson, P.J. Chronic Recurrent Multifocal Osteomyelitis and Related Diseases-Update on Pathogenesis. Curr. Rheumatol. Rep. 2017, 19, 18. [Google Scholar] [CrossRef]
- Lenert, A.; Ferguson, P.J. Comparing children and adults with chronic nonbacterial osteomyelitis. Curr. Opin. Rheumatol. 2020, 32, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.R.; Kapplusch, F.; Mabert, K.; Hedrich, C.M. The molecular pathophysiology of chronic non-bacterial osteomyelitis (CNO)-a systematic review. Mol. Cell. Pediatr. 2017, 4, 7. [Google Scholar] [CrossRef]
- Jansson, A.; Renner, E.D.; Ramser, J.; Mayer, A.; Haban, M.; Meindl, A.; Grote, V.; Diebold, J.; Jansson, V.; Schneider, K.; et al. Classification of non-bacterial osteitis: Retrospective study of clinical, immunological and genetic aspects in 89 patients. Rheumatology 2007, 46, 154–160. [Google Scholar] [CrossRef]
- Wipff, J.; Costantino, F.; Lemelle, I.; Pajot, C.; Duquesne, A.; Lorrot, M.; Faye, A.; Bader-Meunier, B.; Brochard, K.; Despert, V.; et al. A large national cohort of French patients with chronic recurrent multifocal osteitis. Arthritis Rheumatol. 2015, 67, 1128–1137. [Google Scholar] [CrossRef]
- Ferguson, P.J.; Sandu, M. Current understanding of the pathogenesis and management of chronic recurrent multifocal osteomyelitis. Curr. Rheumatol. Rep. 2012, 14, 130–141. [Google Scholar] [CrossRef]
- Hofmann, S.R.; Kapplusch, F.; Girschick, H.J.; Morbach, H.; Pablik, J.; Ferguson, P.J.; Hedrich, C.M. Chronic Recurrent Multifocal Osteomyelitis (CRMO): Presentation, Pathogenesis, and Treatment. Curr. Osteoporos. Rep. 2017, 15, 542–554. [Google Scholar] [CrossRef]
- King, S.M.; Laxer, R.M.; Manson, D.; Gold, R. Chronic recurrent multifocal osteomyelitis: A noninfectious inflammatory process. Pediatr. Infect. Dis. J. 1987, 6, 907–911. [Google Scholar] [CrossRef]
- Giedion, A.; Holthusen, W.; Masel, L.F.; Vischer, D. Subacute and chronic "symmetrical" osteomyelitis. Ann. Radiol. 1972, 15, 329–342. [Google Scholar]
- Girschick, H.J.; Raab, P.; Surbaum, S.; Trusen, A.; Kirschner, S.; Schneider, P.; Papadopoulos, T.; Muller-Hermelink, H.K.; Lipsky, P.E. Chronic non-bacterial osteomyelitis in children. Ann. Rheum. Dis. 2005, 64, 279–285. [Google Scholar] [CrossRef]
- Zhao, Y.; Ferguson, P.J. Chronic non-bacterial osteomyelitis and autoinflammatory bone diseases. Clin. Immunol. 2020, 216, 108458. [Google Scholar] [CrossRef]
- Chamot, A.M.; Benhamou, C.L.; Kahn, M.F.; Beraneck, L.; Kaplan, G.; Prost, A. Acne-pustulosis-hyperostosis-osteitis syndrome. Results of a national survey. 85 cases. Rev. Rhum. Mal. Osteoartic 1987, 54, 187–196. [Google Scholar]
- Cianci, F.; Zoli, A.; Gremese, E.; Ferraccioli, G. Clinical heterogeneity of SAPHO syndrome: Challenging diagnose and treatment. Clin. Rheumatol. 2017, 36, 2151–2158. [Google Scholar] [CrossRef]
- Wu, N.; Shao, Y.; Huo, J.; Zhang, Y.; Cao, Y.; Jing, H.; Zhang, F.; Chenyang, Y.; Yanying, Y.; Li, C.; et al. Clinical characteristics of pediatric synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome: The first Chinese case series from a single center. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef]
- Hayem, G.; Bouchaud-Chabot, A.; Benali, K.; Roux, S.; Palazzo, E.; Silbermann-Hoffman, O.; Kahn, M.F.; Meyer, O. SAPHO syndrome: A long-term follow-up study of 120 cases. Semin Arthritis Rheum. 1999, 29, 159–171. [Google Scholar] [CrossRef]
- Ferguson, P.J.; El-Shanti, H.I. Autoinflammatory bone disorders. Curr. Opin. Rheumatol. 2007, 19, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, P.J.; Brown, D.R.; Sundel, R.; Killian, J.; El-Shanti, H.I. Chronic inflammatory disorders in first and second degree relatives of children with chronic recurrent multifocal osteomyelitis. Arthritis Rheum. 2005, 52, S301–S302. [Google Scholar]
- Paller, A.S.; Pachman, L.; Rich, K.; Esterly, N.B.; Gonzalez-Crussi, F. Pustulosis palmaris et plantaris: Its association with chronic recurrent multifocal osteomyelitis. J. Am. Acad. Dermatol. 1985, 12, 927–930. [Google Scholar] [CrossRef]
- Laxer, R.M.; Shore, A.D.; Manson, D.; King, S.; Silverman, E.D.; Wilmot, D.M. Chronic recurrent multifocal osteomyelitis and psoriasis--a report of a new association and review of related disorders. Semin Arthritis Rheum. 1988, 17, 260–270. [Google Scholar] [CrossRef]
- Bergdahl, K.; Bjorksten, B.; Gustavson, K.H.; Liden, S.; Probst, F. Pustulosis palmoplantaris and its relation to chronic recurrent multifocal osteomyelitis. Dermatologica 1979, 159, 37–45. [Google Scholar] [CrossRef]
- Bognar, M.; Blake, W.; Agudelo, C. Chronic recurrent multifocal osteomyelitis associated with Crohn’s disease. Am. J. Med. Sci. 1998, 315, 133–135. [Google Scholar]
- Ben Becher, S.; Essaddam, H.; Nahali, N.; Ben Hamadi, F.; Mouelhi, M.H.; Hammou, A.; Hadj Romdhane, L.; Ben Cheikh, M.; Boudhina, T.; Dargouth, M. Recurrent multifocal periostosis in children. Report of a familial form. Ann. Pediatr. 1991, 38, 345–349. [Google Scholar]
- Festen, J.J.; Kuipers, F.C.; Schaars, A.H. Multifocal recurrent periostitis responsive to colchicine. Scand. J. Rheumatol. 1985, 14, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Golla, A.; Jansson, A.; Ramser, J.; Hellebrand, H.; Zahn, R.; Meitinger, T.; Belohradsky, B.H.; Meindl, A. Chronic recurrent multifocal osteomyelitis (CRMO): Evidence for a susceptibility gene located on chromosome 18q21.3–18q22. Eur. J. Hum. Genet. 2002, 10, 217–221. [Google Scholar] [CrossRef]
- Cox, A.J.; Ferguson, P.J. Update on the genetics of nonbacterial osteomyelitis in humans. Curr. Opin. Rheumatol. 2018, 30, 521–525. [Google Scholar] [CrossRef]
- El-Shanti, H.I.; Ferguson, P.J. Chronic recurrent multifocal osteomyelitis: A concise review and genetic update. Clin. Orthop. Relat. Res. 2007, 462, 11–19. [Google Scholar] [CrossRef]
- Majeed, H.A.; Kalaawi, M.; Mohanty, D.; Teebi, A.S.; Tunjekar, M.F.; al-Gharbawy, F.; Majeed, S.A.; al-Gazzar, A.H. Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings. J. Pediatr. 1989, 115, 730–734. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Masters, S.L.; Ferguson, P.J.; Dancey, P.; Frenkel, J.; van Royen-Kerkhoff, A.; Laxer, R.; Tedgard, U.; Cowen, E.W.; Pham, T.-H.; et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 2009, 360, 2426–2437. [Google Scholar] [CrossRef]
- Reddy, S.; Jia, S.; Geoffrey, R.; Lorier, R.; Suchi, M.; Broeckel, U.; Hessner, M.J.; Verbsky, J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 2009, 360, 2438–2444. [Google Scholar] [CrossRef]
- Reiff, A.; Bassuk, A.G.; Church, J.A.; Campbell, E.; Bing, X.; Ferguson, P.J. Exome sequencing reveals RAG1 mutations in a child with autoimmunity and sterile chronic multifocal osteomyelitis evolving into disseminated granulomatous disease. J. Clin. Immunol. 2013, 33, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Cox, A.; Takamatsu, N.; Velez, G.; Laxer, R.M.; Tse, S.M.L.; Mahajan, V.B.; Bassuk, A.G.; Fuchs, H.; Ferguson, P.J.; et al. Gain-of-function mutations in a member of the Src family kinases cause autoinflammatory bone disease in mice and humans. Proc. Natl. Acad. Sci. USA 2019, 116, 11872–11877. [Google Scholar] [CrossRef]
- Abe, K.; Fuchs, H.; Lisse, T.; Hans, W.; Hrabe de Angelis, M. New ENU-induced semidominant mutation, Ali18, causes inflammatory arthritis, dermatitis, and osteoporosis in the mouse. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2006, 17, 915–926. [Google Scholar] [CrossRef]
- Hofmann, S.R.; Bottger, F.; Range, U.; Luck, C.; Morbach, H.; Girschick, H.J.; Suttorp, M.; Hedrich, C.M. Serum Interleukin-6 and CCL11/Eotaxin May Be Suitable Biomarkers for the Diagnosis of Chronic Nonbacterial Osteomyelitis. Front. Pediatr. 2017, 5, 256–266. [Google Scholar] [CrossRef]
- Hofmann, S.R.; Kubasch, A.S.; Ioannidis, C.; Rosen-Wolff, A.; Girschick, H.J.; Morbach, H.; Hedrich, C. Altered expression of IL-10 family cytokines in monocytes from CRMO patients result in enhanced IL-1beta expression and release. Clin. Immunol. 2015, 161, 300–307. [Google Scholar] [CrossRef]
- Hofmann, S.R.; Kubasch, A.S.; Range, U.; Laass, M.W.; Morbach, H.; Girschick, H.J.; Hedrich, C.M. Serum biomarkers for the diagnosis and monitoring of chronic recurrent multifocal osteomyelitis (CRMO). Rheumatol. Int. 2016, 36, 769–779. [Google Scholar] [CrossRef]
- Hofmann, S.R.; Morbach, H.; Schwarz, T.; Rosen-Wolff, A.; Girschick, H.J.; Hedrich, C.M. Attenuated TLR4/MAPK signaling in monocytes from patients with CRMO results in impaired IL-10 expression. Clin. Immunol. 2012, 145, 69–76. [Google Scholar] [CrossRef]
- Scianaro, R.; Insalaco, A.; Bracci Laudiero, L.; De Vito, R.; Pezzullo, M.; Teti, A.; De Benedetti, F.; Prencipe, G. Deregulation of the IL-1beta axis in chronic recurrent multifocal osteomyelitis. Pediatr. Rheumatol. Online J. 2014, 12, 30. [Google Scholar] [CrossRef]
- Hofmann, S.R.; Schwarz, T.; Moller, J.C.; Morbach, H.; Schnabel, A.; Rosen-Wolff, A.; Girschick, H.; Hedrich, C.M. Chronic non-bacterial osteomyelitis is associated with impaired Sp1 signaling, reduced IL10 promoter phosphorylation, and reduced myeloid IL-10 expression. Clin. Immunol. 2011, 141, 317–327. [Google Scholar] [CrossRef]
- Harrus, S.; Waner, T.; Aizenberg Safra, N.; Mosenco, A.; Radoshitsky, M.; Bark, H. Development of hypertrophic osteodystrophy and antibody response in a litter of vaccinated Weimaraner puppies. J. Small Anim. Pract. 2002, 43, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Safra, N.; Hitchens, P.L.; Maverakis, E.; Mitra, A.; Korff, C.; Johnson, E.; Kol, A.; Bannasch, M.J.; Pedersen, N.C.; Bannasch, D.L. Serum levels of innate immunity cytokines are elevated in dogs with metaphyseal osteopathy (hypertrophic osteodytrophy) during active disease and remission. Vet. Immunol. Immunopathol. 2016, 179, 32–35. [Google Scholar] [CrossRef]
- Safra, N.; Johnson, E.G.; Lit, L.; Foreman, O.; Wolf, Z.T.; Aguilar, M.; Karmi, N.; Finno, C.J.; Bannasch, D.L. Clinical manifestations, response to treatment, and clinical outcome for Weimaraners with hypertrophic osteodystrophy: 53 cases (2009-2011). J. Am. Vet. Med. Assoc. 2013, 242, 1260–1266. [Google Scholar] [CrossRef]
- Backues, K.A.; Hoover, J.P.; Bahr, R.J.; Confer, A.W.; Chalman, J.A.; Larry, M.L. Multifocal pyogranulomatous osteomyelitis resembling chronic recurrent multifocal osteomyelitis in a lemur. J. Am. Vet. Med. Assoc. 2001, 218, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.J.; Chyuan, I.T.; Wu, C.S.; Lin, S.W.; Chen, K.H.; Tsai, H.F.; Hsu, P.-N. Increased neutrophil infiltration, IL-1 production and a SAPHO syndrome-like phenotype in PSTPIP2-deficient mice. Rheumatology 2015, 54, 1317–1326. [Google Scholar] [CrossRef]
- Byrd, L.; Grossmann, M.; Potter, M.; Shen-Ong, G.L. Chronic multifocal osteomyelitis, a new recessive mutation on chromosome 18 of the mouse. Genomics 1991, 11, 794–798. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, X.; Yu, H.; Xu, Q.; Li, X.; Yang, Y.; Meng, X.; Huang, C.; Li, J. PSTPIP2 attenuates joint damage and suppresses inflammation in adjuvant-induced arthritis. Eur. J. Pharmacol. 2019, 859, 172558. [Google Scholar] [CrossRef]
- Ferguson, P.J.; Chen, S.; Tayeh, M.K.; Ochoa, L.; Leal, S.M.; Pelet, A.; Munnich, A.; Lyonnet, S.; Majeed, H.A.; El-Shanti, E. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J. Med. Genet. 2005, 42, 551–557. [Google Scholar] [CrossRef]
- Majeed, H.A.; Al-Tarawna, M.; El-Shanti, H.; Kamel, B.; Al-Khalaileh, F. The syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia. Report of a new family and a review. Eur. J. Pediatr. 2001, 160, 705–710. [Google Scholar] [CrossRef]
- Majeed, H.A.; El-Shanti, H.; Al-Rimawi, H.; Al-Masri, N. On mice and men: An autosomal recessive syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia. J. Pediatr. 2000, 137, 441–442. [Google Scholar] [CrossRef]
- Ferguson, P.J.; Bing, X.; Vasef, M.A.; Ochoa, L.A.; Mahgoub, A.; Waldschmidt, T.J.; Tygrett, L.T.; Schlueter, A.J.; El-shanti, E. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone 2006, 38, 41–47. [Google Scholar] [CrossRef]
- Grosse, J.; Chitu, V.; Marquardt, A.; Hanke, P.; Schmittwolf, C.; Zeitlmann, L.; Schropp, P.; Barth, B.; Yu, P.; Paffenholz, R.; et al. Mutation of mouse Mayp/Pstpip2 causes a macrophage autoinflammatory disease. Blood 2006, 107, 3350–3358. [Google Scholar] [CrossRef]
- Abe, K.; Klaften, M.; Narita, A.; Kimura, T.; Imai, K.; Kimura, M.; Rubio-Aliaga, I.; Wagner, S.; Jakob, T.; Hrabe de Angelis, M. Genome-wide search for genes that modulate inflammatory arthritis caused by Ali18 mutation in mice. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2009, 20, 152–161. [Google Scholar] [CrossRef]
- Abe, K.; Wechs, S.; Kalaydjiev, S.; Franz, T.J.; Busch, D.H.; Fuchs, H.; Soewarto, D.; Behrendt, H.; Wagner, S.; Jakob, T.; et al. Novel lymphocyte-independent mechanisms to initiate inflammatory arthritis via bone marrow-derived cells of Ali18 mutant mice. Rheumatology 2008, 47, 292–300. [Google Scholar] [CrossRef][Green Version]
- Cox, A.J.; Darbro, B.W.; Laxer, R.M.; Velez, G.; Bing, X.; Finer, A.L.; Erives, A.; Mahajan, V.B.; Bassuk, A.G.; Ferguson, P.J. Recessive coding and regulatory mutations in FBLIM1 underlie the pathogenesis of chronic recurrent multifocal osteomyelitis (CRMO). PLoS ONE 2017, 12, e0169687. [Google Scholar] [CrossRef]
- Cassel, S.L.; Janczy, J.R.; Bing, X.; Wilson, S.P.; Olivier, A.K.; Otero, J.E.; Iwakura, Y.; Shayakhmetov, D.M.; Bassuk, A.G.; Abu-Amer, Y.; et al. Inflammasome-independent IL-1beta mediates autoinflammatory disease in Pstpip2-deficient mice. Proc. Natl. Acad. Sci. USA 2014, 111, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Gross, J.M.; Calabrese, C.; Iwakura, Y.; Lamkanfi, M.; Vogel, P.; Kanneganti, T.-D. Critical role for inflammasome-independent IL-1beta production in osteomyelitis. Proc. Natl. Acad. Sci. USA 2014, 111, 1066–1071. [Google Scholar] [CrossRef]
- Young, S.; Sharma, N.; Lee, J.H.; Chitu, V.; Neumeister, V.; Sohr, E.; Stanley, E.R.; Hedrich, C.M.; Craig, A.W.B. Mast cells enhance sterile inflammation in chronic nonbacterial osteomyelitis. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Drobek, A.; Kralova, J.; Skopcova, T.; Kucova, M.; Novak, P.; Angelisova, P.; Otahal, P.; Alberich-Jorda, M.; Brdicka, T. PSTPIP2, a Protein Associated with Autoinflammatory Disease, Interacts with Inhibitory Enzymes SHIP1 and Csk. J. Immunol. 2015, 195, 3416–3426. [Google Scholar] [CrossRef]
- Herlin, T.; Fiirgaard, B.; Bjerre, M.; Kerndrup, G.; Hasle, H.; Bing, X.; Ferguson, P.J. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann. Rheum. Dis. 2013, 72, 410–413. [Google Scholar] [CrossRef]
- Lorden, G.; Sanjuan-Garcia, I.; de Pablo, N.; Meana, C.; Alvarez-Miguel, I.; Perez-Garcia, M.T.; Pelegrin, P.; Balsinde, J.; Balboa, M.A. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J. Exp. Med. 2017, 214, 511–528. [Google Scholar] [CrossRef]
- Valdearcos, M.; Esquinas, E.; Meana, C.; Peña, L.; Gil-de-Gómez, L.; Balsinde, J.; Balboa, M.A. Lipin-2 reduces proinflammatory signaling induced by saturated fatty acids in macrophages. J. Biol. Chem. 2012, 287, 10894–10904. [Google Scholar] [CrossRef]
- Jesus, A.A.; Osman, M.; Silva, C.A.; Kim, P.W.; Pham, T.H.; Gadina, M.; Yang, B.; Bertola, D.R.; Carneiro-Sampaio, M.; Ferguson, P.J.; et al. A novel mutation of IL1RN in the deficiency of interleukin-1 receptor antagonist syndrome: Description of two unrelated cases from Brazil. Arthritis Rheum. 2011, 63, 4007–4017. [Google Scholar] [CrossRef] [PubMed]
- Kutukculer, N.; Puel, A.; Eren Akarcan, S.; Moriya, K.; Edeer Karaca, N.; Migaud, M.; Cassanova, J.-L.; Aksu, G. Deficiency of Interleukin-1 Receptor Antagonist: A Case with Late Onset Severe Inflammatory Arthritis, Nail Psoriasis with Onychomycosis and Well Responsive to Adalimumab Therapy. Case Rep. Immunol. 2019, 2019, 1902817. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, L.O.; Grossi, A.; Caroli, F.; de Oliveira, R.A.; Kalil, J.; Castro, F.F.M.; Pontillo, A.; Ceccherini, I.; Barros, M.A.M.T.; Gattorno, M. A case report of a novel compound heterozygous mutation in a Brazilian patient with deficiency of Interleukin-1 receptor antagonist (DIRA). Pediatr. Rheumatol. Online J. 2020, 18, 67. [Google Scholar] [CrossRef]
- Mendonca, L.O.; Malle, L.; Donovan, F.X.; Chandrasekharappa, S.C.; Montealegre Sanchez, G.A.; Garg, M.; Tedgard, U.; Castells, M.; Saini, S.S.; Dutta, S.; et al. Deficiency of Interleukin-1 Receptor Antagonist (DIRA): Report of the First Indian Patient and a Novel Deletion Affecting IL1RN. J. Clin. Immunol. 2017, 37, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Minkis, K.; Aksentijevich, I.; Goldbach-Mansky, R.; Magro, C.; Scott, R.; Davis, J.G.; Sardana, N.; Herzog, R. Interleukin 1 receptor antagonist deficiency presenting as infantile pustulosis mimicking infantile pustular psoriasis. Arch. Dermatol. 2012, 148, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sakran, W.; Shalev, S.A.; Sakran, W.; Shalev, S.A.; El-Shanti, H.; Uziel, Y.; Uziel, Y. Chronic recurrent multifocal osteomyelitis and deficiency of interleukin-1-receptor antagonist. Pediatr. Infect. Dis. J. 2013, 32, 94. [Google Scholar] [CrossRef]
- Schnellbacher, C.; Ciocca, G.; Menendez, R.; Aksentijevich, I.; Goldbach-Mansky, R.; Duarte, A.M.; Rivas-Chacon, R. Deficiency of interleukin-1 receptor antagonist responsive to anakinra. Pediatr. Dermatol. 2013, 30, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Stenerson, M.; Dufendach, K.; Aksentijevich, I.; Brady, J.; Austin, J.; Reed, A.M. The first reported case of compound heterozygous IL1RN mutations causing deficiency of the interleukin-1 receptor antagonist. Arthritis Rheum. 2011, 63, 4018–4022. [Google Scholar] [CrossRef]
- Ulusoy, E.; Karaca, N.E.; El-Shanti, H.; Kilicoglu, E.; Aksu, G.; Kutukculer, N. Interleukin-1 receptor antagonist deficiency with a novel mutation; late onset and successful treatment with canakinumab: A case report. J. Med. Case Rep. 2015, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Ziaee, V.; Youssefian, L.; Faghankhani, M.; Jazayeri, A.; Saeidian, A.H.; Vahidnezhad, H.; Uitto, J. Homozygous IL1RN Mutation in Siblings with Deficiency of Interleukin-1 Receptor Antagonist (DIRA). J. Clin. Immunol. 2020, 40, 637–642. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B.; Welzel, T.; Hoertnagel, K.; Tsiflikas, I.; Hospach, A.; Liu, X.; Schlipf, S.; HAnsmann, S.; Samba, S.D.; Griesinger, A. New variant in the IL1RN-gene (DIRA) associated with late-onset, CRMO-like presentation. Rheumatology 2020, 59, 3259–3263. [Google Scholar] [CrossRef]
- Sozeri, B.; Gerceker-Turk, B.; Yildiz-Atikan, B.; Mir, S.; Berdeli, A. A novel mutation of interleukin-1 receptor antagonist (IL1RN) in a DIRA patient from Turkey: Diagnosis and treatment. Turk. J. Pediatr. 2018, 60, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Al Mosawi, Z.; Madan, W.; Al Moosawi, B.; Al-Wadaei, S.; Naser, H.; Ali, F. Dramatic Response of Familial Majeed Syndrome to Interleukin-1 Antagonist Therapy: Case report. Arch. Rheumatol. 2019, 34, 352–356. [Google Scholar] [CrossRef]
- Al-Mosawi, Z.S.; Al-Saad, K.K.; Ijadi-Maghsoodi, R.; El-Shanti, H.I.; Ferguson, P.J. A splice site mutation confirms the role of LPIN2 in Majeed syndrome. Arthritis Rheum. 2007, 56, 960–964. [Google Scholar] [CrossRef]
- Bhuyan, F.; de Jesus, A.A.; Mitchell, J.; Leikina, E.; VanTries, R.; Herzog, R.; Onel, K.B.; Oler, A.; Montealegre Sanchez, G.A.; Johnson, K.A.; et al. Novel Majeed syndrome causing LPIN2 mutations link bone inflammation to inflammatory M2 macrophages and accelerated osteoclastogenesis. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Hu, X.Y.; Zhao, Z.P.; Guo, R.L.; Guo, J.; Li, W.; Hao, C.-J.; Xu, B.-P. Compound heterozygous LPIN2 pathogenic variants in a patient with Majeed syndrome with recurrent fever and severe neutropenia: Case report. BMC Med. Genet. 2019, 20, 182. [Google Scholar] [CrossRef]
- Pinto-Fernandez, C.; Seoane-Reula, M.E. Efficacy of treatment with IL-1RA in Majeed syndrome. Allergol. Immunopathol. 2017, 45, 99–101. [Google Scholar] [CrossRef]
- Rao, A.P.; Gopalakrishna, D.B.; Bing, X.; Ferguson, P.J. Phenotypic Variability in Majeed Syndrome. J. Rheumatol. 2016, 43, 1258–1259. [Google Scholar] [CrossRef]
- Roy, N.B.A.; Zaal, A.I.; Hall, G.; Wilkinson, N.; Proven, M.; McGowan, S.; Hipkiss, R.; Buckle, V.; Kavirayani, A.; Babbs, C. Majeed syndrome: Description of a novel mutation and therapeutic response to bisphosphonates and IL-1 blockade with anakinra. Rheumatology 2019, 59, 448–451. [Google Scholar] [CrossRef]
- Moussa, T.; Bhat, V.; Kini, V.; Fathalla, B.M. Clinical and genetic association, radiological findings and response to biological therapy in seven children from Qatar with non-bacterial osteomyelitis. Int. J. Rheum. Dis. 2016, 20, 1286–1296. [Google Scholar] [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Congenital dyserythropoietic anemias. Blood 2020, 136, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.C.; Stapleton, F.B.; Bond, M.J.; Barrett, F.F. Sweet’s syndrome with multifocal sterile osteomyelitis. Am. J. Dis. Child. 1986, 140, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Bozgeyik, E.; Mercan, R.; Arslan, A.; Tozkir, H. Next-generation screening of a panel of genes associated with periodic fever syndromes in patients with Familial Mediterranean Fever and their clinical characteristics. Genomics 2020, 112, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- Milledge, J.; Shaw, P.J.; Mansour, A.; Williamson, S.; Bennetts, B.; Roscioli, T.; Curtin, J.; Christodoulou, J. Allogeneic bone marrow transplantation: Cure for familial Mediterranean fever. Blood 2002, 100, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Nurre, L.D.; Rabalais, G.P.; Callen, J.P. Neutrophilic dermatosis-associated sterile chronic multifocal osteomyelitis in pediatric patients: Case report and review. Pediatr. Dermatol. 1999, 16, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Reue, K. Lipin proteins and glycerolipid metabolism: Roles at the ER membrane and beyond. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Reue, K. The lipin family: Mutations and metabolism. Curr. Opin. Lipidol. 2009, 20, 165–170. [Google Scholar] [CrossRef]
- Reue, K.; Wang, H. Mammalian lipin phosphatidic acid phosphatases in lipid synthesis and beyond: Metabolic and inflammatory disorders. J. Lipid. Res. 2019, 60, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, C., Jr.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef]
- Csaki, L.S.; Dwyer, J.R.; Fong, L.G.; Tontonoz, P.; Young, S.G.; Reue, K. Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog. Lipid. Res. 2013, 52, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Verity, M.A.; Reue, K. Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab. 2014, 20, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Reue, K. Lipin-1 flexes its muscle in autophagy. Cell Cycle 2014, 13, 3789–3790. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Z.; Gropler, M.C.; Mitra, M.S.; Finck, B.N. Complex interplay between the lipin 1 and the hepatocyte nuclear factor 4 alpha (HNF4alpha) pathways to regulate liver lipid metabolism. PLoS ONE 2012, 7, e51320. [Google Scholar]
- Chen, Z.; Gropler, M.C.; Norris, J.; Lawrence, J.C.; Harris, T.E., Jr.; Finck, B.N. Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion. Arter. Thromb. Vasc. Biol. 2008, 28, 1738–1744. [Google Scholar] [CrossRef]
- Donkor, J.; Sariahmetoglu, M.; Dewald, J.; Brindley, D.N.; Reue, K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 2007, 282, 3450–3457. [Google Scholar] [CrossRef]
- Peterfy, M.; Phan, J.; Xu, P.; Reue, K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 2001, 27, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Zeharia, A.; Shaag, A.; Houtkooper, R.H.; Hindi, T.; de Lonlay, P.; Erez, G.; Hubert, L.; Saada, A.; de Keyzer, Y.; Eshel, G.; et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am. J. Hum. Genet. 2008, 83, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.B.; Sweet, H.O.; Gordon, J.I.; Birkenmeier, E.H. The fld mutation maps near to but distinct from the Apob locus on mouse chromosome 12. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 1996, 7, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Langner, C.A.; Birkenmeier, E.H.; Ben-Zeev, O.; Schotz, M.C.; Sweet, H.O.; Davisson, M.T.; Gordon, J.I. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J. Biol. Chem. 1989, 264, 7994–8003. [Google Scholar] [CrossRef]
- Nadra, K.; de Preux Charles, A.S.; Medard, J.J.; Hendriks, W.T.; Han, G.S.; Gres, S.; Carman, G.M.; Saulnier-Blache, J.-S.; Verheijen, M.H.G.; Chrast, R. Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev. 2008, 22, 1647–1661. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhu, J.; Zhuang, X.; Zhang, X.; Luo, T.; Esser, K.A.; Hongmei, R. Lipin1 Regulates Skeletal Muscle Differentiation through Extracellular Signal-regulated Kinase (ERK) Activation and Cyclin D Complex-regulated Cell Cycle Withdrawal. J. Biol. Chem. 2015, 290, 23646–23655. [Google Scholar] [CrossRef]
- Farmer, T.A., Jr.; Hammack, W.J.; Frommeyer, W.B., Jr. Idiopathic recurrent rhabdomyolysis associated with myoglobinuria: Report of a case. N. Engl. J. Med. 1961, 264, 60–66. [Google Scholar] [CrossRef]
- Bowden, D.H.; Fraser, D.; Jackson, S.H.; Walker, N.F. Acute recurrent rhabdomyolysis (paroxysmal myohaemoglobinuria); a report of three cases and a review of the literature. Medicine 1956, 35, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T.E.; Saxtrup, O.; Hansen, T.I.; Kristensen, B.H.; Beck, B.L.; Plesner, T.; Krogh, I.M.; Andersen, V.; Standgaard, S. amilial myoglobinuria. A study of muscle and kidney pathophysiology in three brothers. Dan. Med. Bull. 1983, 30, 112–115. [Google Scholar] [PubMed]
- Ramesh, V.; Gardner-Medwin, D. Familial paroxysmal rhabdomyolysis: Management of two cases of the non-exertional type. Dev. Med. Child Neurol. 1992, 34, 73–79. [Google Scholar] [CrossRef]
- Temprano, A.; Sembongi, H.; Han, G.S.; Sebastian, D.; Capellades, J.; Moreno, C.; Guardiola, J.; Wabitsch, M.; Richart, C.; Yanes, O.; et al. edundant roles of the phosphatidate phosphatase family in triacylglycerol synthesis in human adipocytes. Diabetologia 2016, 59, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.; Peterfy, M.; Reue, K. Lipin expression preceding peroxisome proliferator-activated receptor-gamma is critical for adipogenesis in vivo and in vitro. J. Biol. Chem. 2004, 279, 29558–29564. [Google Scholar] [CrossRef]
- Mitra, M.S.; Chen, Z.; Ren, H.; Harris, T.E.; Chambers, K.T.; Hall, A.M.; Nadra, K.; Klein, S.; Chrast, R.; Su, X.; et al. Mice with an adipocyte-specific lipin 1 separation-of-function allele reveal unexpected roles for phosphatidic acid in metabolic regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 642–647. [Google Scholar] [CrossRef]
- Schweitzer, G.G.; Collier, S.L.; Chen, Z.; Eaton, J.M.; Connolly, A.M.; Bucelli, R.C.; Pestronk, A.; Harris, T.E.; Finck, B.N. Rhabdomyolysis-Associated Mutations in Human LPIN1 Lead to Loss of Phosphatidic Acid Phosphohydrolase Activity. JIMD Rep. 2015, 23, 113–122. [Google Scholar]
- Dwyer, J.R.; Donkor, J.; Zhang, P.; Csaki, L.S.; Vergnes, L.; Lee, J.M.; Dewald, J.; Brindley, D.N.; Atti, E.; Tetradis, S.; et al. Mouse lipin-1 and lipin-2 cooperate to maintain glycerolipid homeostasis in liver and aging cerebellum. Proc. Natl. Acad. Sci. USA 2012, 109, E2486–E2495. [Google Scholar] [CrossRef]
- Donkor, J.; Zhang, P.; Wong, S.; O’Loughlin, L.; Dewald, J.; Kok, B.P.; Brindley, D.N.; Reue, K. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J. Biol. Chem. 2009, 284, 29968–29978. [Google Scholar] [CrossRef] [PubMed]
- Jesus, A.A.; Goldbach-Mansky, R. IL-1 blockade in autoinflammatory syndromes. Ann. Rev. Med. 2014, 65, 223–244. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| A1 | A2 | A3 | A4 | B1 | B2 | C1 | D1 | D2 | E1 | E2 | F1 | F2 | G1 | H1 | H2 | I1 | I2 | I3 | I4 | I5 | I6 | J1 | K1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recurrent fever | + | + | + | + | +++ | +++ | + | – | + | – | – | NR | NR | – | + | – | + | NR | NR | – | – | – | + | – |
| Failure to thrive | + | + | + | + | + | + | – | NR | NR | + | – | NR | NR | + | – | – | + | NR | NR | NR | NR | NR | NR | – |
| CNO | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | Joint and bone pain | Joint and bone pain | Knee pains | – | + |
| Age at onset (months) Δ | 12 | 19 | 9 | 1 | 0.75 | 9 | 15 | 6 | 3 | 24 | 96 | 6 | 48 | 72 | 13 | 15 | infant | ? | ? | ? | ? | ? | 6 | 12 |
| LE long bones | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||
| UE long bones | + | + | + | + | + | + | + | + | + | + | + | – | – | NR | NR | ND | NR | NR | ||||||
| Feet | – | – | – | – | – | + | + | – | – | + | – | + | – | + | – | ND | NR | NR | ||||||
| Spine # | – | – | – | – | – | – | – | – | – | – | – | – | – | – | ND | ND | NR | NR | ||||||
| Other | hands | hands | ribs | hands | Limb pain | Limb pain | Knee pain | |||||||||||||||||
| Bone biopsy | osteo | osteo | osteo | NR | NR | NR | ND | Osteo Φ | ND | ND | ND | ND | necrosis | osteo | c/w osteo | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Objective joint swelling | + | + | + | NR | + | + | + | – | + | + | + | + | + | + | + | – | NR | NR | NR | NR | NR | NR | – | – |
| Hepatosplenomegaly | + | + | + | + | + | NR | + | – | – | + | – | NR | NR | NR | – | – | NR | NR | NR | NR | NR | NR | – | NR |
| ↑ ESR/CRP | +++ | +++ | +++ | +++ | ++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | ++ | +++ | ++ | ++ | +++ | +++ | ++ | NR | NR | NR | +++ | +++ |
| Microcytic anemia | + | + | + | + | + 8.8 | + 4.0 | + | +° | +° | + | +* | + | + | NR | 9.3 MCV | 7.7@ | 6.9 | + | + | anemia | anemia | – | 8.5 | 10.1 |
| Transfusion Rx | ++ | +++ | ++ | ++ | ++ | +++ | – | – | – | – | – | – | – | NR | – | – | – | – | – | – | – | – | – | – |
| Dyserythropoiesis on BM | + | + | + | + | + | + | + | + | + | + | ND | ND | + | NR | ND | + | + | ND | ND | ND | ND | ND | – | + |
| Neutropenia (ANC) | + (750) | leuko | – | + (588) | NR | NR | NR | NR | NR | NR | NR | + (400) | – | |||||||||||
| Neutrophilic dermatosis | + | + | – | – | – | – | – | – | – | – | – | – | – | NR | – | – | – | – | – | – | – | – | – | – |
| Treatments | Pred, NSAID | Failed colchicine. Pred, NSAID | Failed colchicine. Pred, NSAID | Pred, NSAID | NSAID | NSAID | NSAID + prednisolone | Steroids, Failed etanercept, improved with anakinra, canakin | Steroids, Failed etanercept, improved with anakinra, canakin | NSAId, MTX, partial pam | MTX | NSAID | NSAID, canak | Anakinra | NSAID, Etanercept (failed), did well on anakinra | Failed etanercept, improved with anakinra | NSAID, steroid, partial resonse to bisphos, anakinra | Anakinra | Anakinra | NSAID | NSAID | none | none | NSAID, canakin |
| Mutation | S734L | S734L | S734L | S734L | C181* | C181* | R776Sfs*66 | L439fs*15 | L439fs*15 | Y747* | Y747* | S734L | S734L | R776Sfs*66 | R776Sfs*66 | R776Sfs*66 | R736H | R736H | R736H | R736H | R736H | R736H | R776Sfs*66 And R564Kfs*3 | R517H and 17.8 kb del |
| Clinical Feature | % |
|---|---|
| Recurrent fever | 46 |
| Failure to thrive | 38 |
| Hepatosplenomegaly | 30 |
| Objective limb swelling | 54 |
| Neutrophilic dermatosis | 8 |
| ↑inflammatory marker | 88 |
| Microcytic anemia/CDA | 92 |
| Neutropenia | 13 |
| Radiographic CNO++ | 83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferguson, P.J.; El-Shanti, H. Majeed Syndrome: A Review of the Clinical, Genetic and Immunologic Features. Biomolecules 2021, 11, 367. https://doi.org/10.3390/biom11030367
Ferguson PJ, El-Shanti H. Majeed Syndrome: A Review of the Clinical, Genetic and Immunologic Features. Biomolecules. 2021; 11(3):367. https://doi.org/10.3390/biom11030367
Chicago/Turabian StyleFerguson, Polly J., and Hatem El-Shanti. 2021. "Majeed Syndrome: A Review of the Clinical, Genetic and Immunologic Features" Biomolecules 11, no. 3: 367. https://doi.org/10.3390/biom11030367
APA StyleFerguson, P. J., & El-Shanti, H. (2021). Majeed Syndrome: A Review of the Clinical, Genetic and Immunologic Features. Biomolecules, 11(3), 367. https://doi.org/10.3390/biom11030367