
A Newly Assigned Role of CTCF in Cellular Response to Broken DNAs


Abstract
1. Introduction
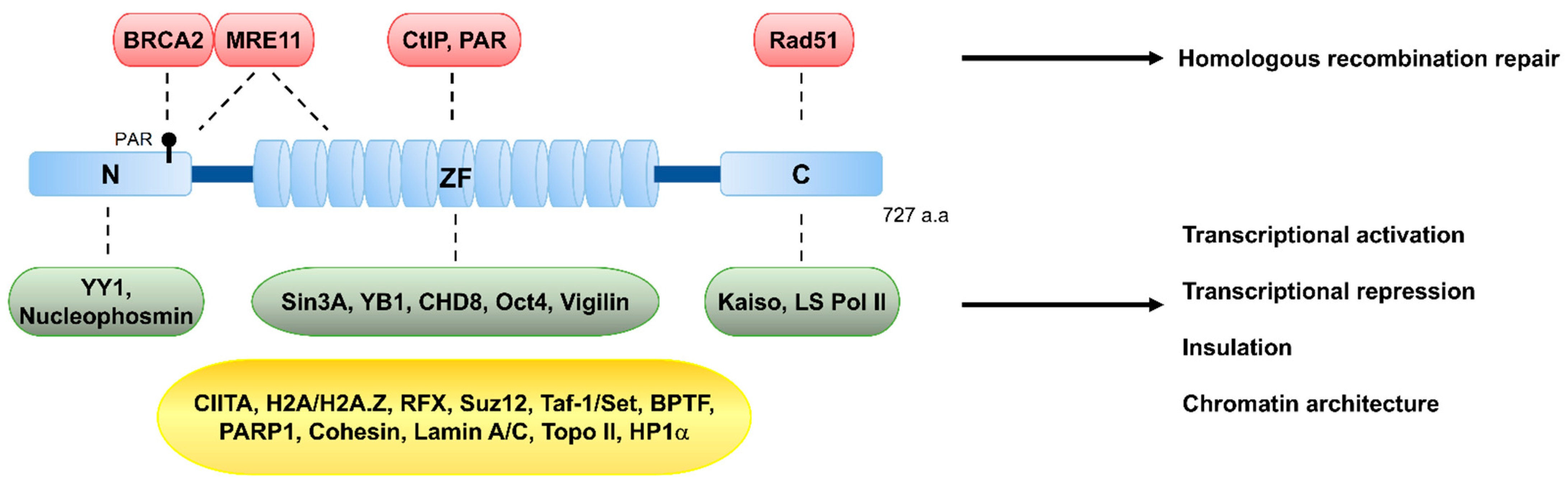
2. The Multifunctional CTCF Protein and Its Classical Roles
2.1. A Transcriptional Repressor or Activator
2.2. An Enhancer Blocker or Helper
2.3. A Regulator of Transcriptional Pausing and Alternative Splicing
2.4. A Regulator of Somatic V(D)J Recombination at Antibody and Antigen Receptor Loci
2.5. A Chromatin Organizer for Global Genome Architecture
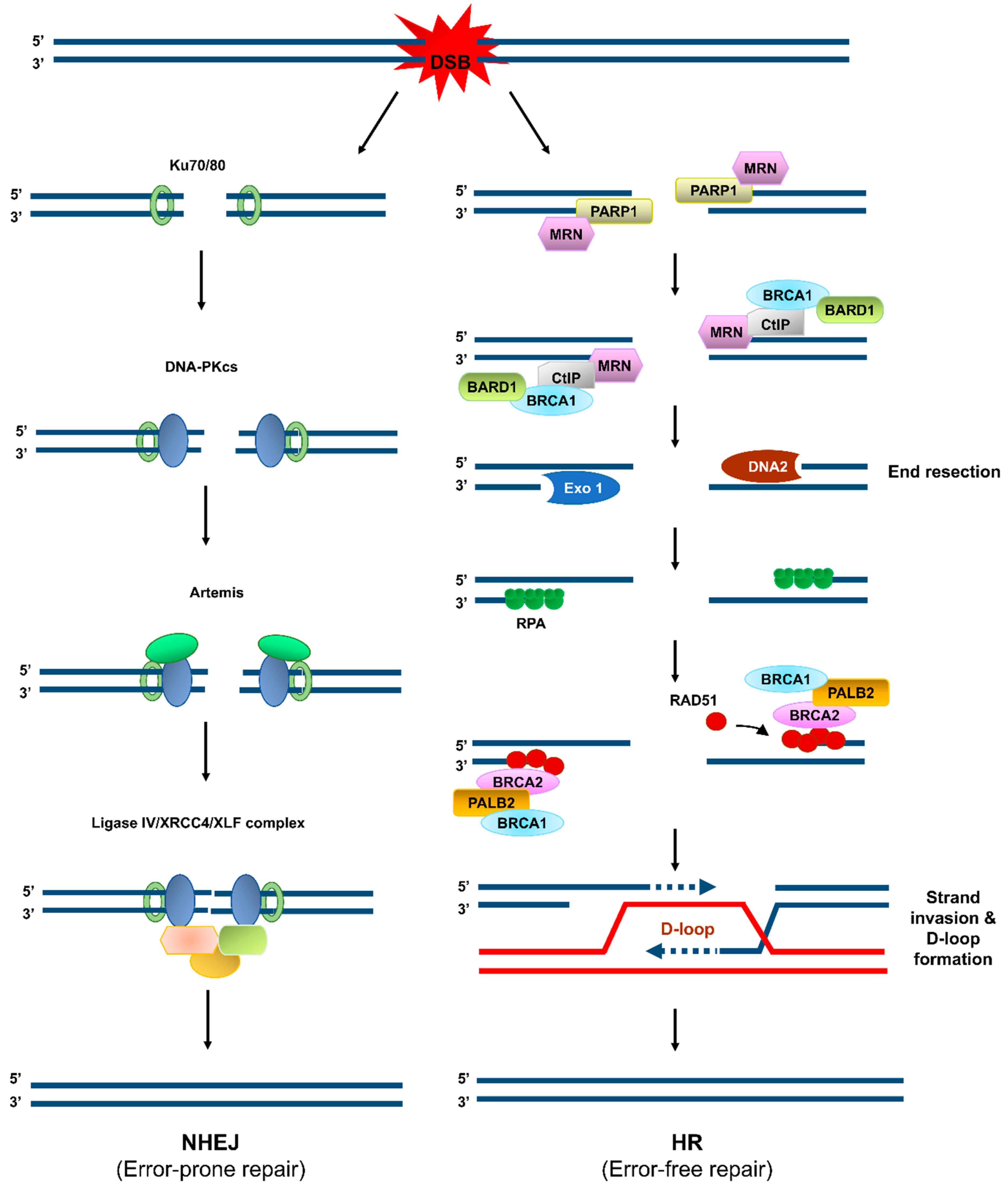
3. Updated Role of CTCF in Genome Integrity
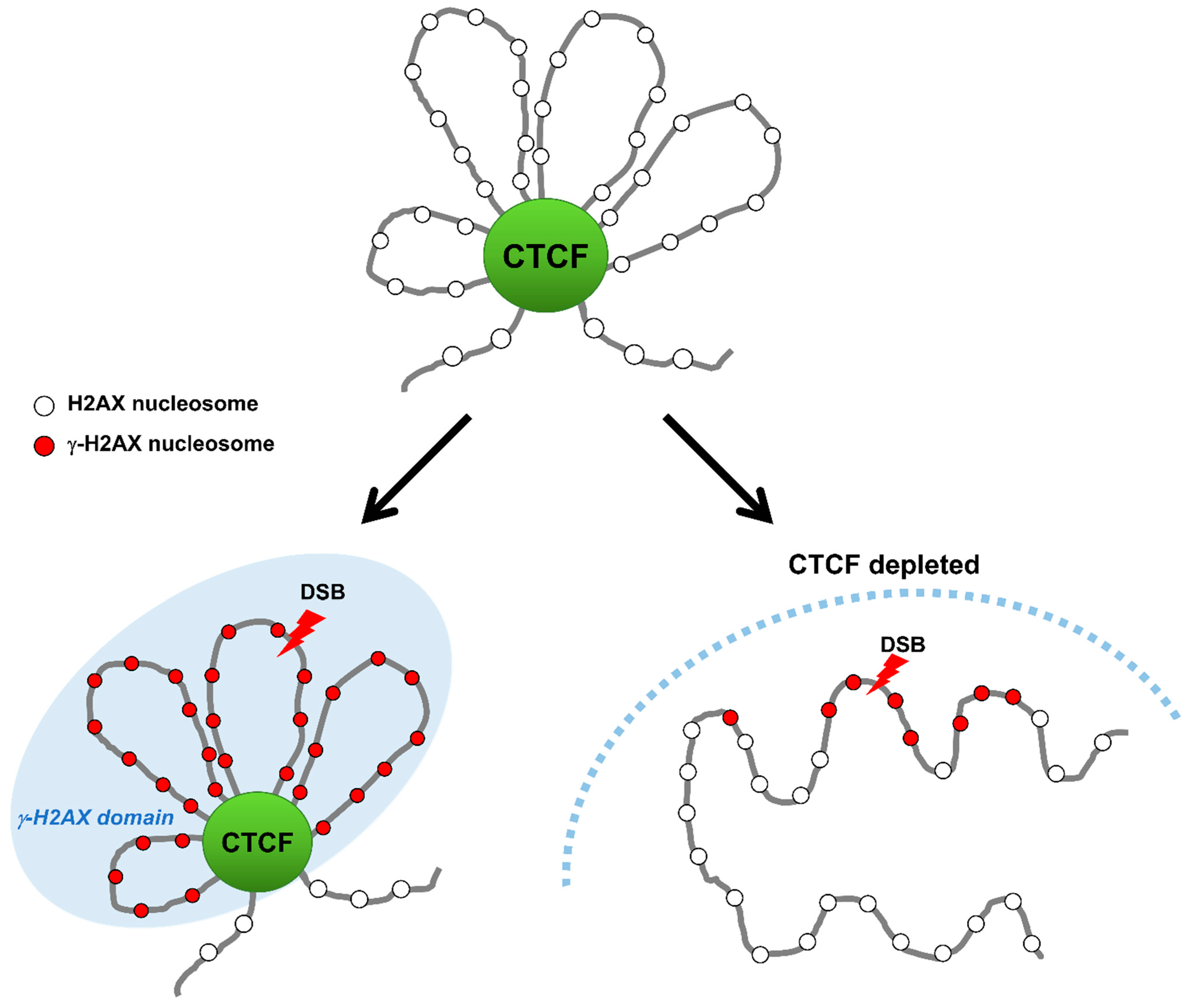
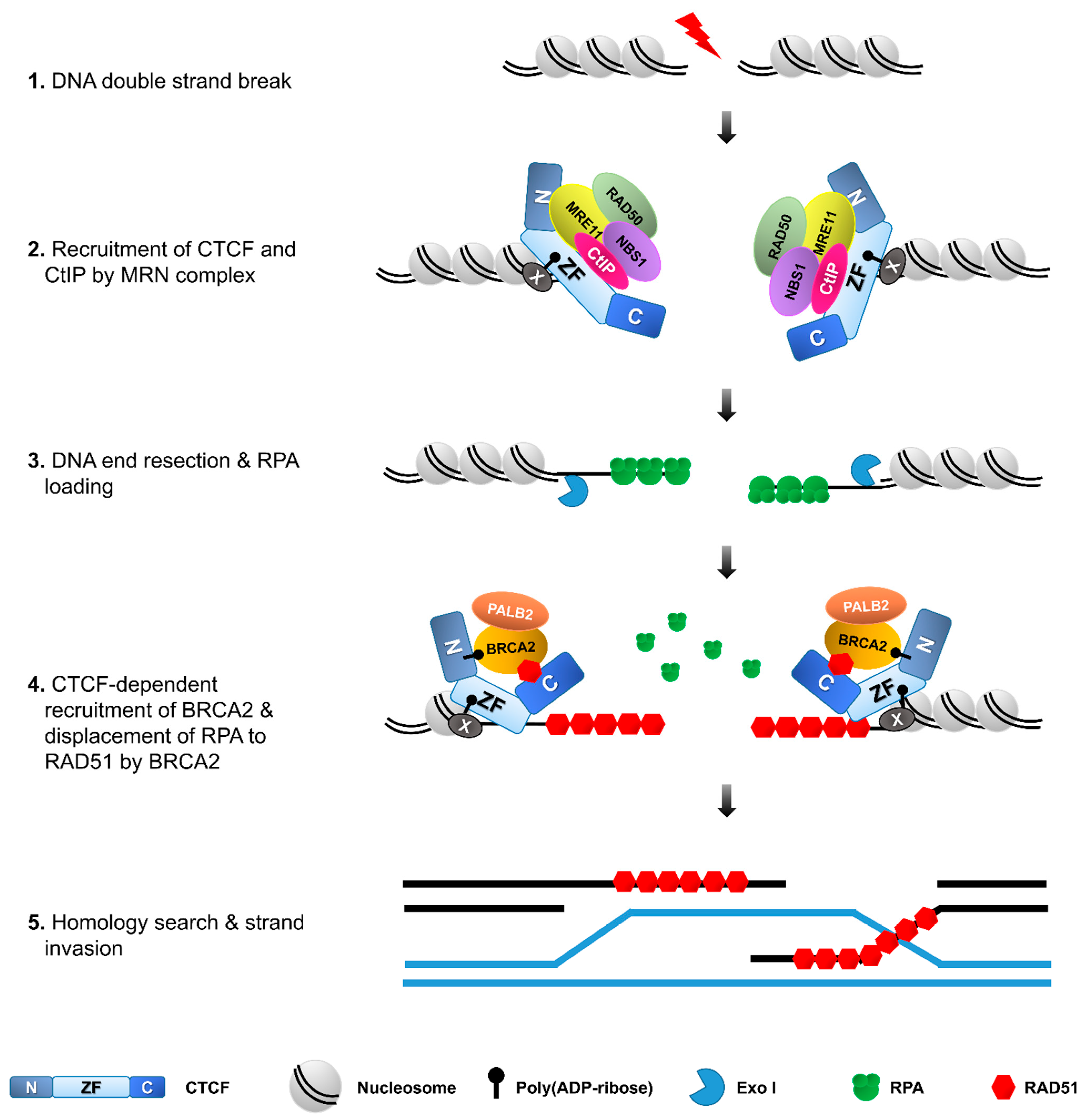
3.1. How and Why Does CTCF Accumulate at DNA Llesions?
3.2. Colocalization of CTCF with γ-H2AX Chromatin to Delimit γ-H2AX Chromatin Domain
3.3. A Promoter of DNA Repair via HR and Beyond
4. Future Questions and Perspectives
4.1. A Genome Repair Tool and Beyond
4.2. A Guardian of Genome
4.3. Other Jobs in DNA Damage Responses?
4.4. Candidate Collaborators of CTCF in HR
4.5. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| CTCF | CCCTC-binding factor |
| DSBs | double-strand breaks |
| NHEJ | non-homologous end joining |
| HR | homologous recombination |
| IR | ionizing radiation |
| UV | ultraviolet |
| DDR | DNA damage response |
| MRN | MRE11-RAD50-NBS1 |
| ssDNA | single-stranded DNA |
| D-loop | displacement loop |
| ChIP | chromatin immunoprecipitation |
| ICR | imprinting control region |
| TCR | T cell receptor |
| CBE | CTCF-binding element |
| TAD | topologically associated domain |
| PAR | poly(ADP-ribose) |
References
- Woodbine, L.; Brunton, H.; Goodarzi, A.A.; Shibata, A.; Jeggo, P.A. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011, 39, 6986–6997. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.P.; Hader, D.P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Bordin, D.L.; Lima, M.; Lenz, G.; Saffi, J.; Meira, L.B.; Mesange, P.; Soares, D.G.; Larsen, A.K.; Escargueil, A.E.; Henriques, J.A.P. DNA alkylation damage and autophagy induction. Mutat. Res. 2013, 753, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Loeb, L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 2001, 447, 7–21. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Kundson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Altieri, F.; Grillo, C.; Maceroni, M.; Chichiarelli, S. DNA damage and repair: From molecular mechanisms to health implications. Antioxid. Redox. Signal 2008, 10, 891–937. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Lamber, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef]
- Betermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef]
- Cruz-Garcia, A.; Lopez-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef]
- Zhao, W.; Vaithiyalingam, S.; San Filippo, J.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol. Cell 2015, 59, 176–187. [Google Scholar] [CrossRef]
- Sonoda, E.; Sasaki, M.S.; Morrison, C.; Yamaguchi-Iwai, Y.; Takata, M.; Takeda, S. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol. Cell Biol. 1999, 19, 5166–5169. [Google Scholar] [CrossRef]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Kang, M.A.; Baik, C.J.; Lee, Y.; Hang, N.T.; Kim, B.G.; Han, J.S.; Jeong, J.H.; Park, D.C.; Myung, K.; et al. CTCF cooperates with CtIP to drive homologous recombination repair of double-strand breaks. Nucleic Acids Res. 2019, 47, 9160–9179. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, K.; Jangal, M.; Marques, M.; Zhao, T.; Saad, A.; Zhang, C.; Luo, V.M.; Syme, A.; Rejon, C.; Yu, Z.; et al. CTCF facilitates DNA double-strand break repair by enhancing homologous recombination repair. Sci. Adv. 2017, 3, e1601898. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Li, X.; Zheng, W.; Li, Z.; Lu, D.; Chen, G.; Gong, D.; Yang, L.; Fu, J.; Shi, P.; et al. CTCF prevents genomic instability by promoting homologous recombination-directed DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2017, 114, 10912–10917. [Google Scholar] [CrossRef]
- Nakahashi, H.; Kwon, K.R.K.; Resch, W.; Vian, L.; Dose, M.; Stavreva, D.; Hakim, O.; Pruett, N.; Nelson, S.; Yamane, A.; et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013, 3, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Renda, M.; Baglivo, I.; Burgess-Beusse, B.; Esposito, S.; Fattorusso, R.; Felsenfeld, G.; Pedone, P.V. Critical DNA binding interactions of the insulator protein CTCF: A small number of zinc fingers mediate strong binding, and a single finger-DNA interaction controls binding at imprinted loci. J. Biol. Chem. 2007, 282, 33336–33345. [Google Scholar] [CrossRef] [PubMed]
- Quitschke, W.W.; Taheny, M.J.; Fochtmann, L.J.; Vostrov, A.A. Differential effect of zinc finger deletions on the binding of CTCF to the promoter of the amyloid precursor protein gene. Nucleic Acids Res. 2000, 28, 3370–3378. [Google Scholar] [CrossRef][Green Version]
- Ohlsson, R.; Renkawitz, R.; Lobanenkov, V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001, 17, 520–527. [Google Scholar] [CrossRef]
- Ohlsson, R.; Lobanenkov, V.; Klenova, E. Does CTCF mediate between nuclear organization and gene expression? BioEssays 2010, 32, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Zlatanova, J.; Caiafa, P. CTCF and its protein partners: Divide and rule? J. Cell Sci. 2009, 122, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Song, C.; Malakouti, N.; Murray, D.; Hariz, A.; Zimmerman, M.; Gygax, D.; Alhazmi, A.; Landry, J.W. Functional interactions between NURF and CTCF regulate gene expression. Mol. Cell Biol. 2015, 35, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegue, L.; Raurell-Vila, H.; Thackray, J.K.; Gonzalez, J.; Casal, C.; Kane-Goldsmith, N.; Vizoso, M.; Brown, J.P.; Gomez, A.; Ausio, J.; et al. Mammalian HP1 Isoforms Have Specific Roles in Heterochromatin Structure and Organization. Cell Rep. 2017, 21, 2048–2057. [Google Scholar] [CrossRef]
- Yu, X.; Liu, Q.; He, J.; Huang, Y.; Jiang, L.; Xie, X.; Liu, J.; Chen, L.; Wei, L.; Qin, Y. Vigilin interacts with CTCF and is involved in the maintenance of imprinting of IGF2 through a novel RNA-mediated mechanism. Int. J. Biol. Macromol. 2018, 108, 515–522. [Google Scholar] [CrossRef]
- Uuskula-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [CrossRef]
- Li, Y.; Haarhuls, J.H.I.; Cacclatore, A.S.; Oldenkamp, R.; van Rulten, M.S.; Willems, L.; Teunlssen, H.; Mulr, K.W.; de Wlt, E.; Rowland, B.D.; et al. The structural basis for cohesin-CTCF-anchored loops. Nature 2020, 578, 472–476. [Google Scholar] [CrossRef]
- Wutz, G.; Varnai, C.; Nagasaka, K.; Cisneros, D.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topological associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef]
- Baniahmad, A.; Steiner, C.; Kohne, A.C.; Renkawitz, R. Modular structure of a chicken lysozyme silencer: Involvement of an unusual thyroid hormone receptor binding site. Cell 1990, 61, 505–514. [Google Scholar] [CrossRef]
- Klenova, E.M.; Nicolas, R.H.; Paterson, H.F.; Carne, A.F.; Heath, C.M.; Goodwin, G.H.; Neiman, P.E.; Lobanenkov, V.V. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell Biol. 1993, 13, 7612–7624. [Google Scholar] [CrossRef]
- Kohne, A.C.; Baniahmad, A.; Renkawitz, R. NeP1. A ubiquitous transcription factor synergizes with v-ERBA in transcriptional silencing. J. Mol. Biol. 1993, 232, 747–755. [Google Scholar] [CrossRef]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed]
- Vostrov, A.A.; Quitschke, W.W. The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta-protein precursor promoter. Evidence for a role in transcriptional activation. J. Biol. Chem. 1997, 272, 33353–33359. [Google Scholar] [CrossRef]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef]
- Kanduri, C.; Loukinov, V.P.D.; Pugacheva, E.; Qi, C.F.; Wolffe, A.; Ohlsson, R.; Lobanenkov, V.V. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 2000, 10, 853–856. [Google Scholar] [CrossRef]
- Gaszner, M.; Felsenfeldd, G. Insulators: Exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 2006, 7, 703–713. [Google Scholar] [CrossRef]
- Negre, N.; Brown, C.D.; Shah, P.K.; Kheradpour, P.; Morrison, C.A.; Henikoff, J.G.; Feng, X.; Ahmad, K.; Russell, S.; White, R.A.H.; et al. A comprehensive map of insulator elements for the Drosophila genome. PLoS Genet. 2010, 6, e1000814. [Google Scholar] [CrossRef]
- Kim, S.; Yu, N.K.; Kaang, B.K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef]
- Kyrchanova, O.; Maksimenko, O.; Ibragimov, A.; Sokolov, V.; Postika, N.; Lukyanova, M.; Schedl, P.; Georgiev, P. The insulator functions of the Drosophila polydactyl C2H2 zinc finger protein CTCF: Necessity versus sufficiency. Sci. Adv. 2020, 6, eaaz3152. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef]
- Schoenherr, C.J.; Levorse, J.M.; Tilghman, S.M. CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 2003, 33, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Chen, H.; Bo, X.; Wang, S. Genome-wide analysis of the relationships between DNaseI HS, histone modifications and gene expression reveals distinct modes of chromatin domains. Nucleic Acids Res. 2011, 39, 7428–7443. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, J.; Skok, J.A. The role of CTCF in regulating V(D)J recombination. Curr. Opin. Immunol. 2012, 24, 153–159. [Google Scholar] [CrossRef]
- Schatz, D.G.; Swanson, P.C. V(D)J recombination: Mechanisms of initiation. Ann. Rev. Genet. 2011, 45, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Malu, S.; Malshetty, V.; Francis, D.; Cortes, P. Role of non-homologous end joining in V(D)J recombination. Immunol. Res. 2012, 54, 233–246. [Google Scholar] [CrossRef]
- Ba, Z.; Lou, J.; Ye, A.Y.; Dai, H.Q.; Dring, E.W.; Lin, S.G.; Jain, S.; Kyritsis, N.; Kieffer-Kwon, K.R.; Casellas, R.; et al. CTCF orchestrates long-range cohesin-deriven V(D)J recombination scanning. Nature 2020, 586, 305–310. [Google Scholar] [CrossRef]
- Dai, H.Q.; Hu, H.; Lou, J.; Ye, A.Y.; Ba, Z.; Zhang, X.; Zhang, Y.; Zhao, L.; Yoon, H.S.; Chapdelaine-Williams, A.M.; et al. Loop extrusion mediates physiological Igh locus contraction for RAG scanning. Nature 2021, 590, 338–343. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 2017, 169, 930–944. [Google Scholar] [CrossRef]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.J.A.; Ye, Z.; Kolovos, P.; Brouwer, R.W.W.; van de Corput, M.P.C.; Werken, H.J.G.; Knoch, T.A.; IJcken, W.F.J.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef]
- Han, D.; Chen, Q.; Shi, J.; Zhang, F.; Yu, X. CTCF participates in DNA damage response via poly (ADP-ribosyl) lation. Sci. Rep. 2017, 7, 43530. [Google Scholar] [CrossRef]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Horl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef]
- Caron, P.; Aymard, F.; Iacovoni, J.S.; Briois, S.; Canitrot, Y.; Bugler, B.; Massip, L.; Losada, A.; Legube, G. Cohesin protects gene against γH2AX induced by DNA double-strand breaks. PLoS Genet. 2012, 8, e1002460. [Google Scholar] [CrossRef] [PubMed]
- Arnould, C.; Legube, G. The secret life of chromosome loops upon DNA double-strand break. J. Mol. Biol. 2020, 432, 724–736. [Google Scholar] [CrossRef]
- Collins, P.L.; Purman, C.; Porter, S.I.; Nganga, V.; Saini, A.; Hayer, K.E.; Gurewitz, G.L.; Sleckman, B.P.; Bednarski, J.J.; Bassing, C.H.; et al. DNA double-strand breaks induce H2AX phosphorylation domains in contact-dependent manner. Nat. Commun. 2020, 11, 3158. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, J. N terminus of CtIP is critical for homologous recombination-mediated double-strand break repair. J. Biol. Chem. 2009, 284, 31746–31752. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.K.; Morimatsu, M.; Albercht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichelee, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef]
- O’Donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar] [CrossRef]
- Sung, P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 1994, 265, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Shi, J.; Bian, C.; Yu, X. Poly (ADP-ribose) mediates the BRCA2-dependent early DNA damage response. Cell Rep. 2015, 13, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Caiafa, P.; Zlatanova, J. CCCTC-binding factor meets poly (ADP-ribose) polymerase-1. J. Cell. Physiol. 2009, 219, 265–270. [Google Scholar] [CrossRef]
- Katainen, R.; Dave, K.; Pitkanen, E.; Palin, K.; Kivioja, T.; Valimaki, N.; Gylfe, A.E.; Ristolainen, H.; Hanninen, U.A.; Cajuso, T.; et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 2015, 47, 818–821. [Google Scholar] [CrossRef]
- Guo, Y.A.; Chang, M.M.; Huang, W.; Ooi, W.F.; Xing, M.; Tan, P.; Skanderup, A.J. Mutation hotspots at CTCF binding sites coupled to chromosomal instability in gastrointestinal cancers. Nat. Commun. 2018, 9, 1520. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome organization drives chromosome fragility. Cell 2017, 170, 507–521. [Google Scholar] [CrossRef]
- Gothe, H.J.; Bouwman, B.A.M.; Gusmao, E.G.; Piccinno, R.; Petrosino, G.; Sayols, S.; Drechsel, O.; Minneker, V.; Josipovic, N.; Mizi, A.; et al. Spatial chromosome folding and active transcription drive DNA fragility and formation of oncogenic MLL translocations. Mol. Cell 2019, 75, 267–283. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Huang, S.Y.; Wutz, G.; Tang, W.; Zagnoli-Vieira, G.; Callen, E.; Wong, N.; Day, A.; Peters, J.M.; et al. Topoisomerase II-induced chromosome breakage and translocation is determined by chromosome architecture and transcriptional activity. Mol. Cell 2019, 75, 252–266. [Google Scholar] [CrossRef]
- Sanders, J.; Freeman, T.F.; Xu, Y.; Golloshi, R.; Stallard, M.A.; Hill, A.M.; San Martin, R.; Balajee, A.S.; McCord, R.P. Radiation-induced DNA damage and repair effects on 3D genome organization. Nat. Commun. 2020, 11, 6178. [Google Scholar] [CrossRef]
- Ochs, F.; Karemore, G.; Miron, E.; Brown, J.; Sedlackova, H.; Rask, M.B.; Lampe, M.; Buckle, V.; Schermelleh, L.; Lukas, J.; et al. Stabilization of chromatin topology safeguards genome integrity. Nature 2019, 574, 571–574. [Google Scholar] [CrossRef]
- Xu, H.; Yan, Y.; Deb, S.; Rangasamy, D.; Germann, M.; Malaterre, J.; Eder, N.C.; Ward, R.L.; Hawkins, N.J.; Tothill, R.W.; et al. Cohesin Rad21 mediates loss of heterozygosity and is upregulated via Wnt promoting transcriptional dysregulation in gastrointestinal tumors. Cell Rep. 2014, 9, 1781–1797. [Google Scholar] [CrossRef]
- Sonoda, E.; Matsusaka, T.; Morrison, C.; Vagnarelli, P.; Hoshi, O.; Ushiki, T.; Nojima, K.; Fukagawa, T.; Waizenegger, I.C.; Peters, J.M.; et al. Scc1/Rad21, Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev. Cell 2001, 1, 759–770. [Google Scholar] [CrossRef]
- Countryman, P.; Fan, Y.; Gorthi, A.; Pan, H.; Strickland, J.; Kaur, P.; Wang, X.; Lin, J.; Lei, X.; White, C.; et al. Cohesin SA2 is a sequence-independent DNA-binding protein that recognizes DNA replication and repair intermediates. J. Biol. Chem. 2018, 293, 1054–1069. [Google Scholar] [CrossRef]
- Unal, E.; Arbel-Eden, A.; Sattler, U.; Shroff, R.; Lichten, M.; Haber, J.E.; Koshland, D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell 2004, 16, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Strom, L.; Lindroos, H.B.; Shirahige, K.; Sjogren, C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 2004, 16, 1003–1015. [Google Scholar] [CrossRef]
- Bauerschmidt, C.; Woodcock, M.; Stevens, D.L.; Hill, M.A.; Rothkamm, K.; Helleday, T. Cohesin phosphorylation and mobility of SMC1 at ionizing radiation-induced DNA double-strand breaks in human cells. Exp. Cell Res. 2011, 317, 330–337. [Google Scholar] [CrossRef]
- Kim, J.S.; Krasieva, T.B.; LaMorte, V.; Tayor, M.R.; Yokomori, K. Specific recruitment of human cohesin to laser-induced DNA damage. J. Biol. Chem. 2002, 277, 45149–45153. [Google Scholar] [CrossRef]
- Piazza, A.; Bordelet, H.; Dumont, A.; Thierry, A.; Savocco, J.; Girard, F.; Koszul, R. Cohesin regulates homology search during recombinational DNA repair. BioRxiv 2020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Partner | Functions of CTCF–Protein Interaction | Refs | |
|---|---|---|---|
| Transcription enzyme | RNA polymerase II large subunit (LS pol II) | Regulation of transcription and insulator functions | [23] |
| Transcription regulatory factor | CIITA | HLA-DRB1 and HLA-DQA1 gene transcription | [23,24] |
| Regulatory factor X (RFX) | HLA-DRB1 and HLA-DQA1 gene transcription | [23,24] | |
| Kaiso | Regulation of insulator functions Transcriptional repression of RB gene following CTCF-binding sites methylation | [23,24] | |
| Oct4 | X chromosome pairing and counting | [23] | |
| CTCF | Interaction between distant DNA regions | [23] | |
| YB1 | Transcriptional repression of c-myc and serotonin transporter (5-HTT) gene | [23,24] | |
| YY1 | Tsix gene transactivation | [23,24] | |
| BPTF | Transcriptional suppression of H2-K1 gene via inhibition of Klf4 binding | [25] | |
| Chromatin constituent | CHD8 | Regulation of insulator functions | [23,24] |
| Suz12 | Regulation of insulator function (H19 ICR)Transcriptional repression of Sox2 | [23,24] | |
| Sin3A | Transcriptional repression via recruitment of HDAC | [23,24] | |
| Taf-1/Set | Unknown | [23,24] | |
| H2A/H2A.Z | Co-localize genome-wide | [23,24] | |
| HP1α | Interact in pericentric heterochromatin (PCH) and restricts H4K20me3 and H3K27me3 distribution | [26] | |
| Genome integrity | PARP1 | Regulation of crosstalk between poly(ADP-ribosyl)ation and DNA methylation | [23,24] |
| RNA binding protein | Vigilin | Interacts with CTCF via H19 lncRNA and keep the imprinting of IGF2 | [27] |
| Nuclear architecture | Nucelophosmin | Regulation of insulator function | [23] |
| Cohesin | Co-localize genome-wide Regulation of insulator function (c-myc, H19/Igf2) | [23,24,29,30] | |
| Lamin A/C | Unknown | [23] | |
| Topoisomerase II | Unknown | [23,28] | |
| DNA damage repair | MRE11 | CTCF and CtIP recruitment on DNA double strand break (DSB) sites | [16] |
| CtIP | Induction of DNA end resection | [16] | |
| RAD51 | Homologous pairing and strand invasion | [18] | |
| BRCA2 | Assembly of RAD51 on single stranded DNA | [17] | |
| PARP1 | Chromatin PARylation and early recruitment of CTCF to DSB sites | [17] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.A.; Lee, J.-S. A Newly Assigned Role of CTCF in Cellular Response to Broken DNAs. Biomolecules 2021, 11, 363. https://doi.org/10.3390/biom11030363
Kang MA, Lee J-S. A Newly Assigned Role of CTCF in Cellular Response to Broken DNAs. Biomolecules. 2021; 11(3):363. https://doi.org/10.3390/biom11030363
Chicago/Turabian StyleKang, Mi Ae, and Jong-Soo Lee. 2021. "A Newly Assigned Role of CTCF in Cellular Response to Broken DNAs" Biomolecules 11, no. 3: 363. https://doi.org/10.3390/biom11030363
APA StyleKang, M. A., & Lee, J.-S. (2021). A Newly Assigned Role of CTCF in Cellular Response to Broken DNAs. Biomolecules, 11(3), 363. https://doi.org/10.3390/biom11030363