
Circadian Rhythm of NER and ATR Pathways


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
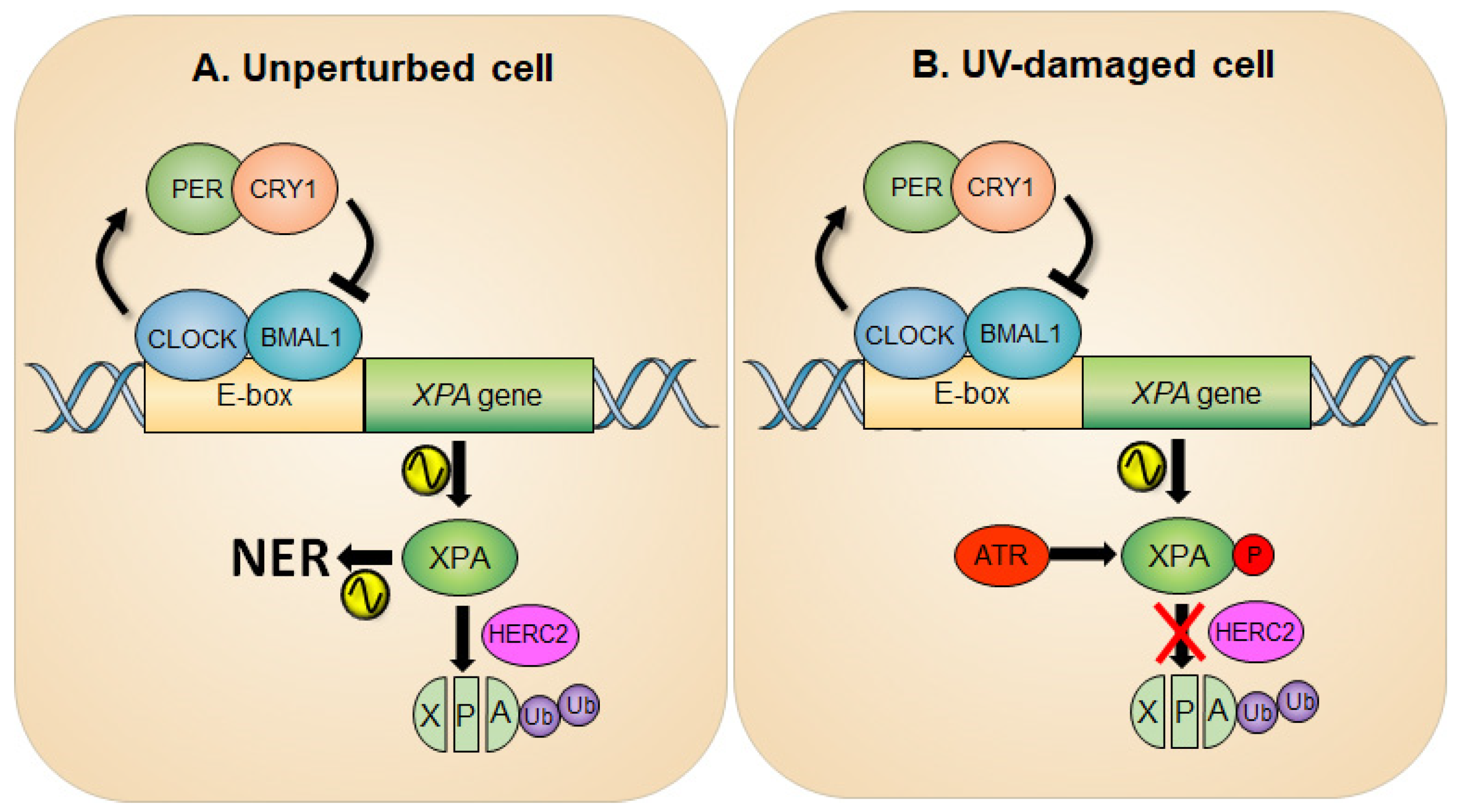
2. Circadian Oscillation of XPA and the NER Activity
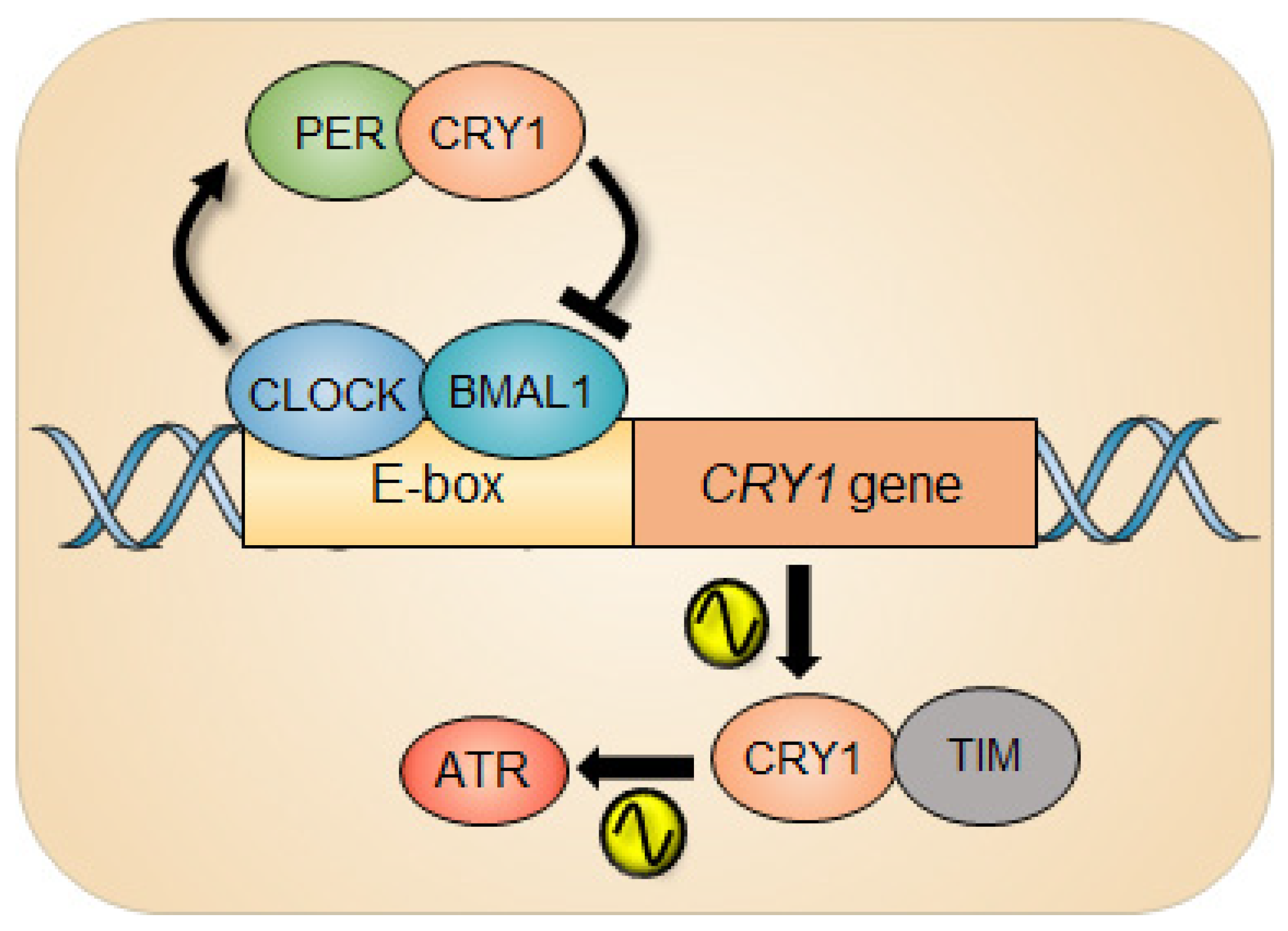
3. Cryptochrome Control of the ATR-Mediated Checkpoint Pathway
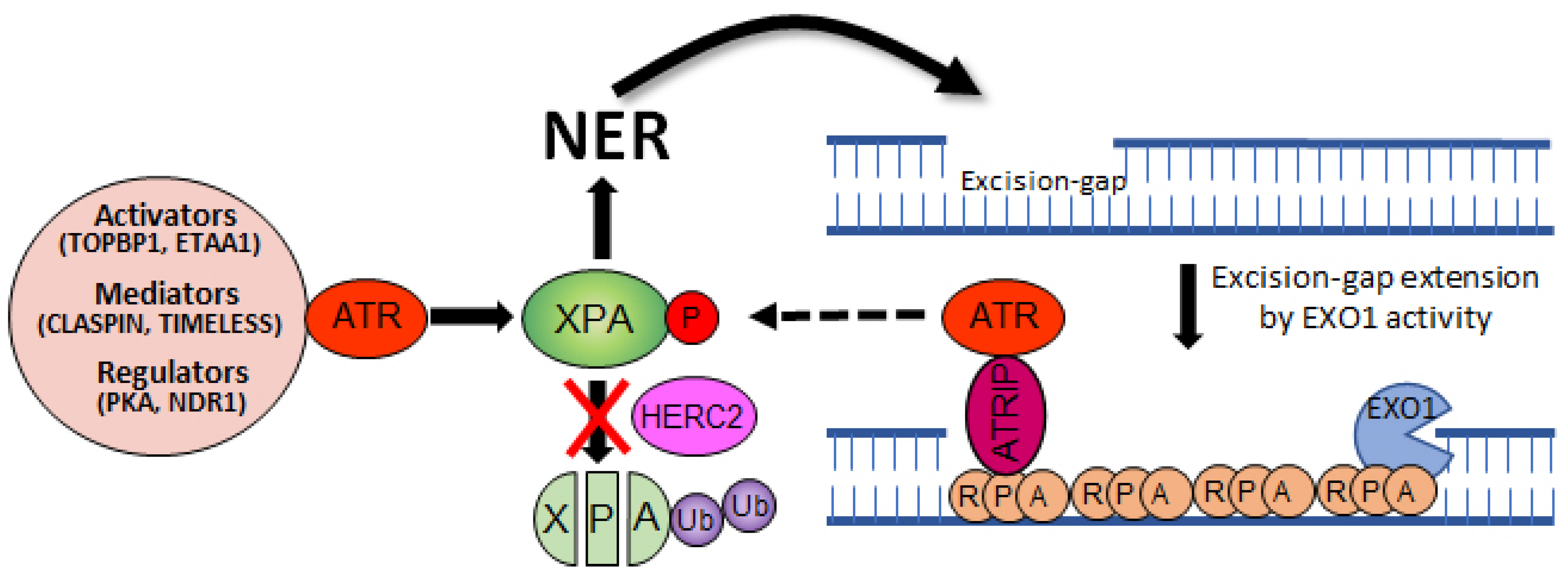
4. Crosstalk between NER and ATR Pathways
5. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hut, R.A.; Beersma, D.G.M. Evolution of time-keeping mechanisms: Early emergence and adaptation to photoperiod. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2141–2154. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J.S. Circadian Integration of Metabolism and Energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Partch, C.L.; Green, C.B.; Takahashi, J.S. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014, 24, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef]
- Sulli, G.; Manoogian, E.N.; Taub, P.R.; Panda, S. Training the Circadian Clock, Clocking the Drugs, and Drugging the Clock to Prevent, Manage, and Treat Chronic Diseases. Trends Pharmacol. Sci. 2018, 39, 812–827. [Google Scholar] [CrossRef]
- Park, J.-M.; Kang, T.-H. Transcriptional and Posttranslational Regulation of Nucleotide Excision Repair: The Guardian of the Genome against Ultraviolet Radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Travers, J.B.; Kemp, M.G. Roles of UVA radiation and DNA damage responses in melanoma pathogenesis. Environ. Mol. Mutagen. 2018, 59, 438–460. [Google Scholar] [CrossRef]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Hoeijmakers, J.H.J.; Vermeulen, W.; Marteijn, J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019, 20, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- de Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Patke, A.; Young, M.W.; Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 2020, 21, 67–84. [Google Scholar] [CrossRef]
- Sarkar, S.; Gaddameedhi, S. Solar ultraviolet-induced DNA damage response: Melanocytes story in transfor-mation to environmental melanomagenesis. Environ. Mol. Mutagen. 2020, 61, 736–751. [Google Scholar] [CrossRef]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768. [Google Scholar] [CrossRef]
- Pulzová, L.B.; Ward, T.A.; Chovanec, M. XPA: DNA Repair Protein of Significant Clinical Importance. Int. J. Mol. Sci. 2020, 21, 2182. [Google Scholar] [CrossRef]
- Kang, T.-H.; Reardon, J.T.; Sancar, A. Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic Acids Res. 2010, 39, 3176–3187. [Google Scholar] [CrossRef]
- Kang, T.-H.; Reardon, J.T.; Kemp, M.; Sancar, A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. USA 2009, 106, 2864–2867. [Google Scholar] [CrossRef] [PubMed]
- Hogenesch, J.B. It’s all in a day’s work: Regulation of DNA excision repair by the circadian clock. Proc. Natl. Acad. Sci. USA 2009, 106, 2481–2482. [Google Scholar] [CrossRef] [PubMed]
- Gaddameedhi, S.; Selby, C.P.; Kaufmann, W.K.; Smart, R.C.; Sancar, A. Control of skin cancer by the circadian rhythm. Proc. Natl. Acad. Sci. USA 2011, 108, 18790–18795. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-H.; Lindsey-Boltz, L.A.; Reardon, J.T.; Sancar, A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2010, 107, 4890–4895. [Google Scholar] [CrossRef]
- Altman, B.J.; Hsieh, A.L.; Sengupta, A.; Krishnanaiah, S.Y.; Stine, Z.E.; Walton, Z.E.; Gouw, A.M.; Venkataraman, A.; Li, B.; Goraksha-Hicks, P.; et al. MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab. 2015, 22, 1009–1019. [Google Scholar] [CrossRef]
- Liu, L.; Lee, J.; Zhou, P. Navigating the nucleotide excision repair threshold. J. Cell. Physiol. 2010, 224, 585–589. [Google Scholar] [CrossRef]
- Levi, F.; Schibler, U. Circadian Rhythms: Mechanisms and Therapeutic Implications. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 593–628. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Lee, T.H.; Park, J.-M.; Leem, S.-H.; Kang, T.-H. Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 2014, 33, 19–25. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Danielsen, J.R.; Fugger, K.; Gromova, I.; Nerstedt, A.; Lukas, C.; Bartek, J.; Lukas, J.; Mailand, N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 2009, 12, 80–86. [Google Scholar] [CrossRef]
- Izawa, N.; Wu, W.; Sato, K.; Nishikawa, H.; Kato, A.; Boku, N.; Itoh, F.; Ohta, T. HERC2 Interacts with Claspin and Regulates DNA Origin Firing and Replication Fork Progression. Cancer Res. 2011, 71, 5621–5625. [Google Scholar] [CrossRef]
- Yoo, N.J.; Park, S.W.; Lee, S.H. Frameshift mutations of ubiquitination-related genes HERC2, HERC3, TRIP12, UBE2Q1 and UBE4B in gastric and colorectal carcinomas with microsatellite instability. Pathology 2011, 43, 753–755. [Google Scholar] [CrossRef]
- Ibarrola-Villava, M.; Fernández, L.P.; Pita, G.; Bravo, J.; Floristan, U.; Sendagorta, E.; Feito, M.; Avilés, J.A.; Martin-Gonzalez, M.; Lázaro, P.; et al. Genetic analysis of three important genes in pigmentation and melanoma susceptibility: CDKN2A, MC1R and HERC2/OCA2. Exp. Dermatol. 2010, 19, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Allain, D.; Bernhardt, M.; Gillespie, J.; Peters, S.; Iwenofu, O.; Nelson, H.; Arron, S.; Toland, A. Variants at theOCA2/HERC2locus affect time to first cutaneous squamous cell carcinoma in solid organ transplant recipients collected using two different study designs. Br. J. Dermatol. 2017, 177, 1066–1073. [Google Scholar] [CrossRef]
- Wu, W.; Sato, K.; Koike, A.; Nishikawa, H.; Koizumi, H.; Venkitaraman, A.R.; Ohta, T. HERC2 Is an E3 Ligase That Targets BRCA1 for Degradation. Cancer Res. 2010, 70, 6384–6392. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhao, H.; Liao, J.; Xu, X. HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability. Nucleic Acids Res. 2014, 42, 13074–13081. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Deng, M.; Li, Y.; Yin, P.; Gao, B.; Fang, Y.; Wu, P.; Liu, T.; Lou, Z. HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Res. 2014, 42, 13110–13121. [Google Scholar] [CrossRef]
- Shell, S.M.; Li, Z.; Shkriabai, N.; Kvaratskhelia, M.; Brosey, C.; Serrano, M.A.; Chazin, W.J.; Musich, P.R.; Zou, Y. Checkpoint Kinase ATR Promotes Nucleotide Excision Repair of UV-induced DNA Damage via Physical Interaction with Xeroderma Pigmentosum Group, A. J. Biol. Chem. 2009, 284, 24213–24222. [Google Scholar]
- Wu, X.; Shell, S.M.; Yang, Z.; Zou, Y. Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia tel-angiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res. 2006, 66, 2997–3005. [Google Scholar] [CrossRef] [PubMed]
- Burrows, A.E.; Elledge, S.J. How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes Dev. 2008, 22, 1416–1421. [Google Scholar] [CrossRef]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef]
- Praetorius-Ibba, M.; Wang, Q.-E.; Wani, G.; El-Mahdy, M.A.; Zhu, Q.; Qin, S.; Wani, A.A. Role of Claspin in regulation of nucleotide excision repair factor DDB2. DNA Repair 2007, 6, 578–587. [Google Scholar] [CrossRef]
- Kang, T.-H.; Leem, S.-H. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 2014, 42, 4427–4434. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Horrell, E.M.W.; Christian, P.A.; Vanover, J.C.; Boulanger, M.C.; Zou, Y.; D’Orazio, J.A. PKA-Mediated Phosphorylation of ATR Promotes Recruitment of XPA to UV-Induced DNA Damage. Mol. Cell 2014, 54, 999–1011. [Google Scholar] [CrossRef]
- Park, J.-M.; Choi, J.Y.; Yi, J.M.; Chung, J.W.; Leem, S.-H.; Koh, S.S.; Kang, T.-H. NDR1 modulates the UV-induced DNA-damage checkpoint and nucleotide excision repair. Biochem. Biophys. Res. Commun. 2015, 461, 543–548. [Google Scholar] [CrossRef]
- Lee, T.H.; Choi, J.Y.; Park, J.-M.; Kang, T.-H. Posttranscriptional control of the replication stress response via TTP-mediated Claspin mRNA stabilization. Oncogene 2020, 39, 3245–3257. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Luo, J. SIRT1 Regulates UV-Induced DNA Repair through Deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef]
- Choi, J.Y.; Park, J.-M.; Yi, J.M.; Leem, S.-H.; Kang, T.-H. Enhanced nucleotide excision repair capacity in lung cancer cells by preconditioning with DNA-damaging agents. Oncotarget 2015, 6, 22575–22586. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian Control of the NAD+ Salvage Pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.-K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian Clock Feedback Cycle through NAMPT-Mediated NAD+ Biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar]
- Jung, Y.; Lippard, S.J. Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-H.; Sancar, A. Circadian regulation of DNA excision repair: Implications for chrono-chemotherapy. Cell Cycle 2009, 8, 1665–1667. [Google Scholar]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Lindsey-Boltz, L.A. Bringing It All Together: Coupling Excision Repair to the DNA Damage Checkpoint. Photochem. Photobiol. 2016, 93, 238–244. [Google Scholar] [CrossRef]
- Lee, T.-H.; Kang, T.-H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 302, 255–259. [Google Scholar]
- Ünsal-Kaçmaz, K.; Mullen, T.E.; Kaufmann, W.K.; Sancar, A. Coupling of Human Circadian and Cell Cycles by the Timeless Protein. Mol. Cell. Biol. 2005, 25, 3109–3116. [Google Scholar]
- Choi, J.Y.; Joh, H.M.; Park, J.-M.; Kim, M.J.; Chung, T.H.; Kang, T.-H. Non-thermal plasma-induced apoptosis is modulated by ATR- and PARP1-mediated DNA damage responses and circadian clock. Oncotarget 2016, 7, 32980–32989. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; McNair, C.M.; McCann, J.J.; Alshalalfa, M.; Shostak, A.; Severson, T.M.; Zhu, Y.; Bergman, A.; Gordon, N.; Mandigo, A.C.; et al. The circadian cryptochrome, CRY1, is a pro-tumorigenic factor that rhythmically modulates DNA repair. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, R.V.; Antoch, M.P. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 2007, 17, 311–317. [Google Scholar] [CrossRef]
- Antoch, M.P.; Kondratov, R.V.; Takahashi, J.S. Circadian clock genes as modulators of sensitivity to genotoxic stress. Cell Cycle 2005, 4, 901–907. [Google Scholar] [CrossRef]
- Wood, P.A.; Yang, X.; Taber, A.; Oh, E.-Y.; Ansell, C.; Ayers, S.E.; Al-Assaad, Z.; Carnevale, K.; Berger, F.G.; Peña, M.M.O.; et al. Period 2 Mutation Accelerates ApcMin/+ Tumorigenesis. Mol. Cancer Res. 2008, 6, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Komatsu, N.; Kawamata, N.; Miller, C.W.; Desmond, J.; Virk, R.K.; Marchevsky, A.; McKenna, R.; Taguchi, H.; Koeffler, H.P. Epigenetic silencing of the candidate tumor suppressor gene Per1 in non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 1399–1404. [Google Scholar] [CrossRef]
- Hoffman, A.E.; Yi, C.-H.; Zheng, T.; Stevens, R.G.; Leaderer, D.; Zhang, Y.; Holford, T.R.; Hansen, J.; Paulson, J.; Zhu, Y. CLOCK in Breast Tumorigenesis: Genetic, Epigenetic, and Transcriptional Profiling Analyses. Cancer Res. 2010, 70, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C.C. The circadian gene period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef]
- Ye, R.; Selby, C.P.; Chiou, Y.-Y.; Ozkan-Dagliyan, I.; Gaddameedhi, S.; Sancar, A. Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 2014, 28, 1989–1998. [Google Scholar] [CrossRef]
- Putker, M.; Wong, D.C.S.; Seinkmane, E.; Rzechorzek, N.M.; Zeng, A.; Hoyle, N.P.; Chesham, J.E.; Edwards, M.D.; Feeney, K.A.; Fischer, R.; et al. CRYPTOCHROMES confer robustness, not rhythmicity, to circadian timekeeping. EMBO J. 2021, 40, e106745. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, G.T.; Muijtjens, M.; Kobayashi, K.; Takano, R.; Kanno, S.-I.; Takao, M.; Wit, J.D.; Verkerk, A.; Eker, A.P.M.; Leenen, D.; et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature 1999, 398, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Nardo, T.; Giannattasio, M.; Minuzzo, M.; Stefanini, M.; Plevani, P.; Falconi, M.M. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc. Natl. Acad. Sci. USA 2006, 103, 17325–17330. [Google Scholar] [CrossRef]
- Hanasoge, S.; Ljungman, M. H2AX phosphorylation after UV irradiation is triggered by DNA repair interme-diates and is mediated by the ATR kinase. Carcinogenesis 2007, 28, 2298–2304. [Google Scholar] [CrossRef]
- Kemp, M.G. Damage removal and gap filling in nucleotide excision repair. Enzymes 2019, 45, 59–97. [Google Scholar]
- Sertic, S.; Pizzi, S.; Cloney, R.; Lehmann, A.R.; Marini, F.; Plevani, P.; Muzi-Falconi, M. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proc. Natl. Acad. Sci. USA 2011, 108, 13647–13652. [Google Scholar] [CrossRef] [PubMed]
- García-Borrón, J.C.; Sánchez-Laorden, B.L.; Jiménez-Cervantes, C. Melanocortin-1 receptor structure and functional regulation. Pigment. Cell Res. 2005, 18, 393–410. [Google Scholar] [CrossRef]
- Ray, A.; Milum, K.; Battu, A.; Wani, G.; Wani, A.A. NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair 2013, 12, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-A.; Slattery, S.D.; Moon, S.-H.; Darlington, Y.F.; Lu, X.; Donehower, L.A. The oncogenic phosphatase WIP1 negatively regulates nucleotide excision repair. DNA Repair 2010, 9, 813–823. [Google Scholar] [CrossRef]
- Belanger, F.; Rajotte, V.; Drobetsky, E.A. A Majority of Human Melanoma Cell Lines Exhibits an S Phase-Specific Defect in Excision of UV-Induced DNA Photoproducts. PLoS ONE 2014, 9, e85294. [Google Scholar] [CrossRef]
- Ballesta, A.; Innominato, P.F.; Dallmann, R.; Rand, D.A.; Lévi, F.A. Systems Chronotherapeutics. Pharmacol. Rev. 2017, 69, 161–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yoo, S.-H.; Takahashi, J.S. Development and Therapeutic Potential of Small-Molecule Modulators of Circadian Systems. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 231–252. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, T.-H. Circadian Rhythm of NER and ATR Pathways. Biomolecules 2021, 11, 715. https://doi.org/10.3390/biom11050715
Kang T-H. Circadian Rhythm of NER and ATR Pathways. Biomolecules. 2021; 11(5):715. https://doi.org/10.3390/biom11050715
Chicago/Turabian StyleKang, Tae-Hong. 2021. "Circadian Rhythm of NER and ATR Pathways" Biomolecules 11, no. 5: 715. https://doi.org/10.3390/biom11050715
APA StyleKang, T.-H. (2021). Circadian Rhythm of NER and ATR Pathways. Biomolecules, 11(5), 715. https://doi.org/10.3390/biom11050715