
A Novel One-Pot Enzyme Cascade for the Biosynthesis of Cladribine Triphosphate



Abstract
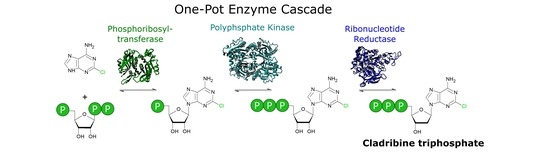
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Enzyme Expression and Purification
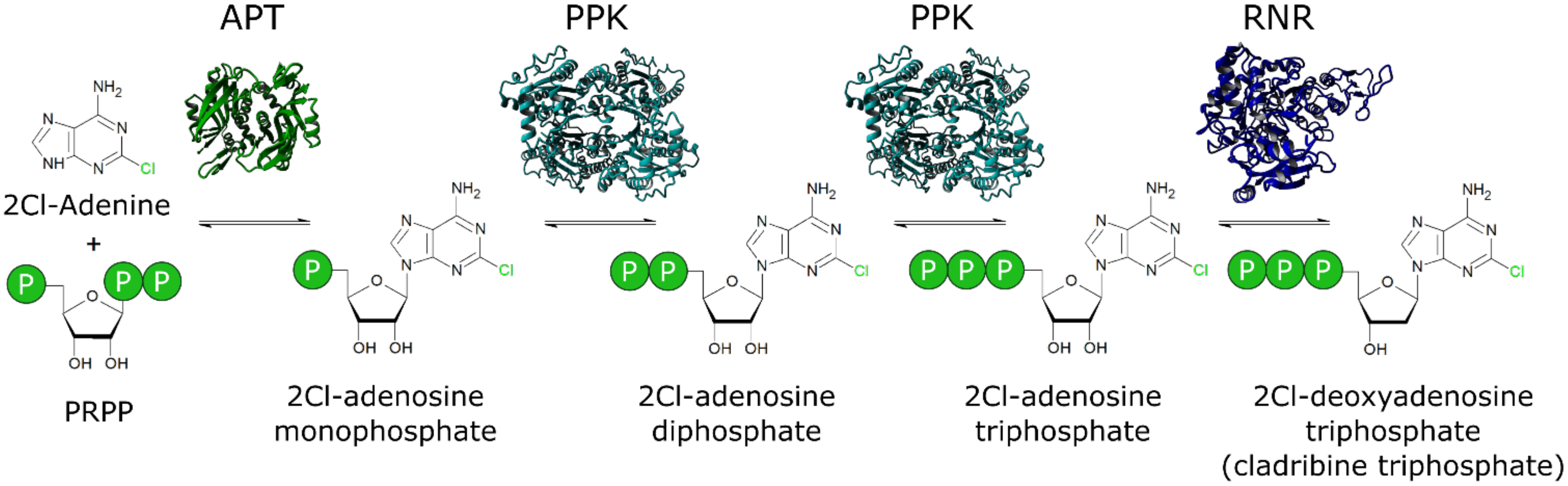
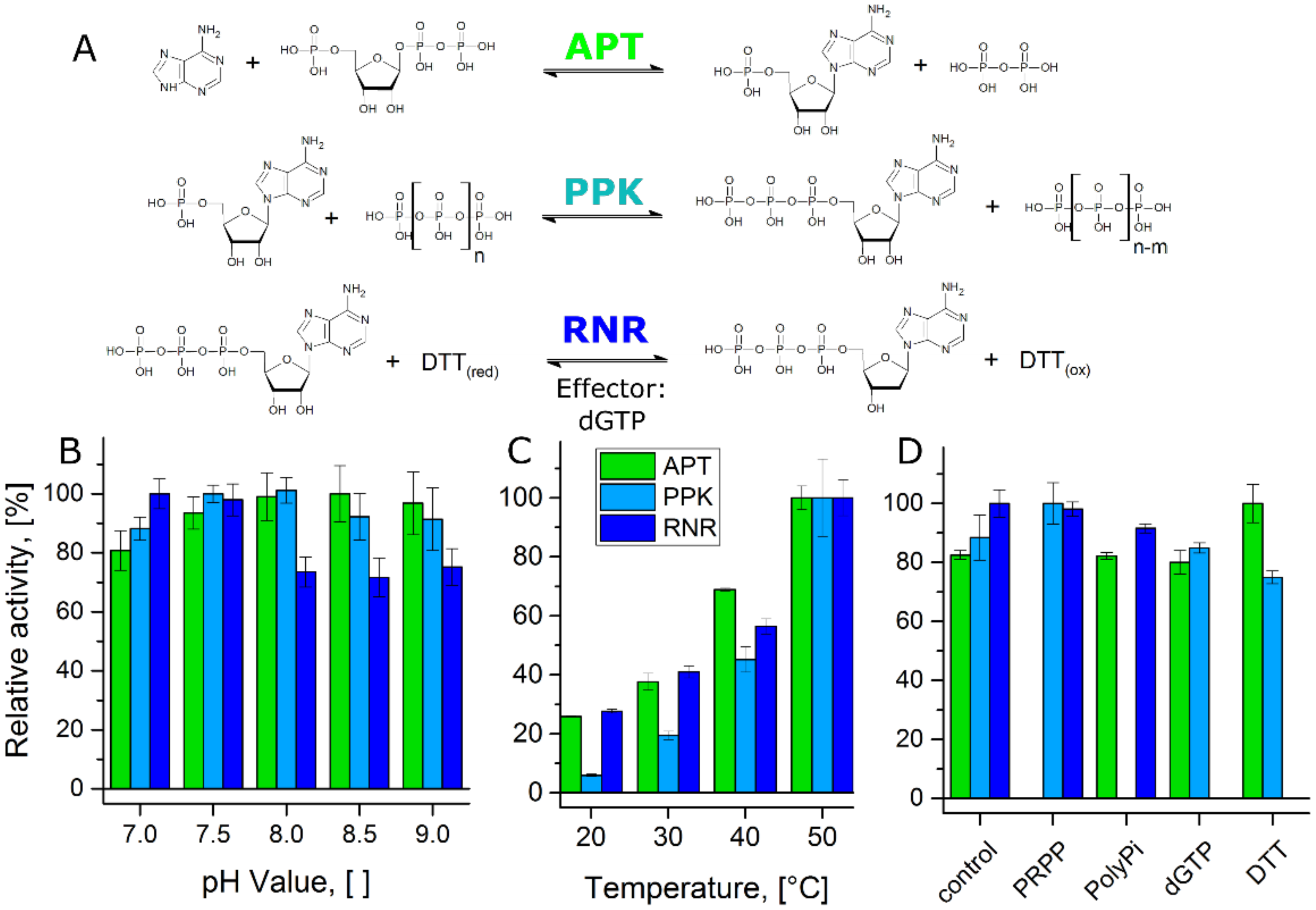
2.3. Enzyme Activity Assays
2.4. Enzyme Cascade Reactions
2.5. HPLC-Analytics
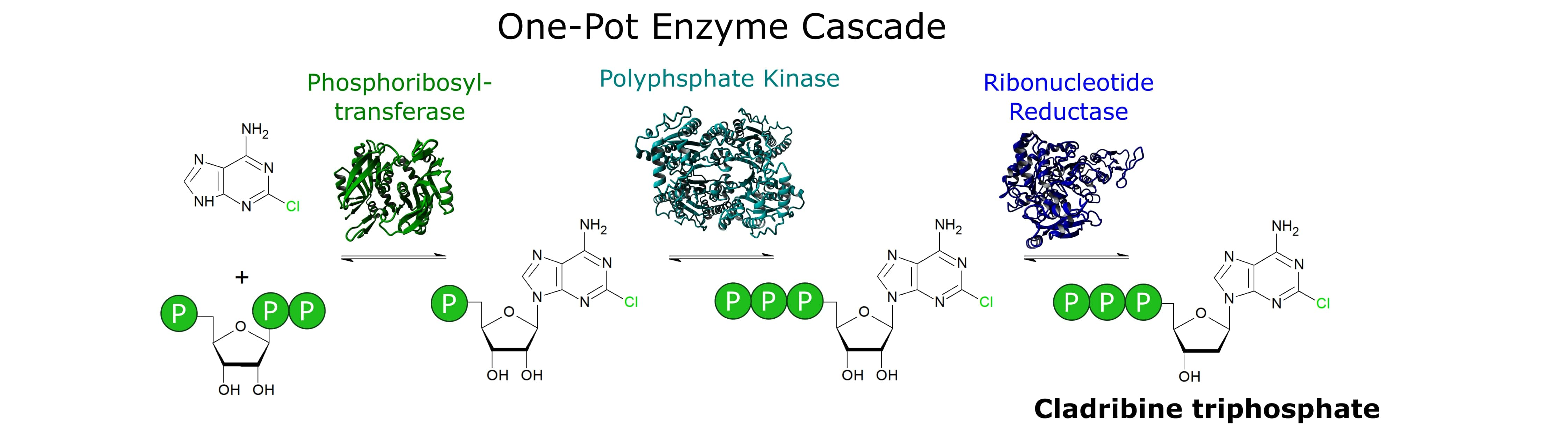
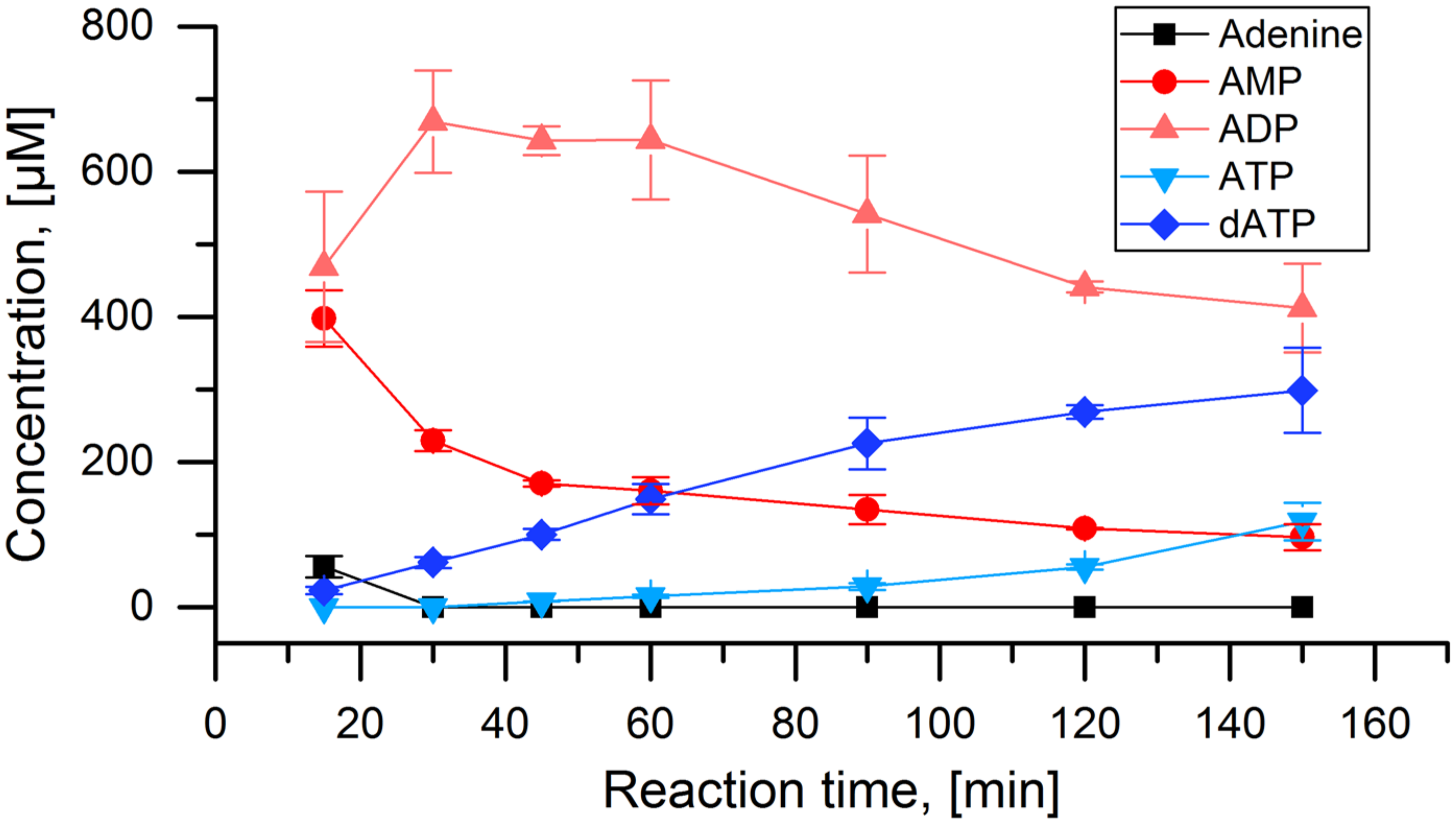
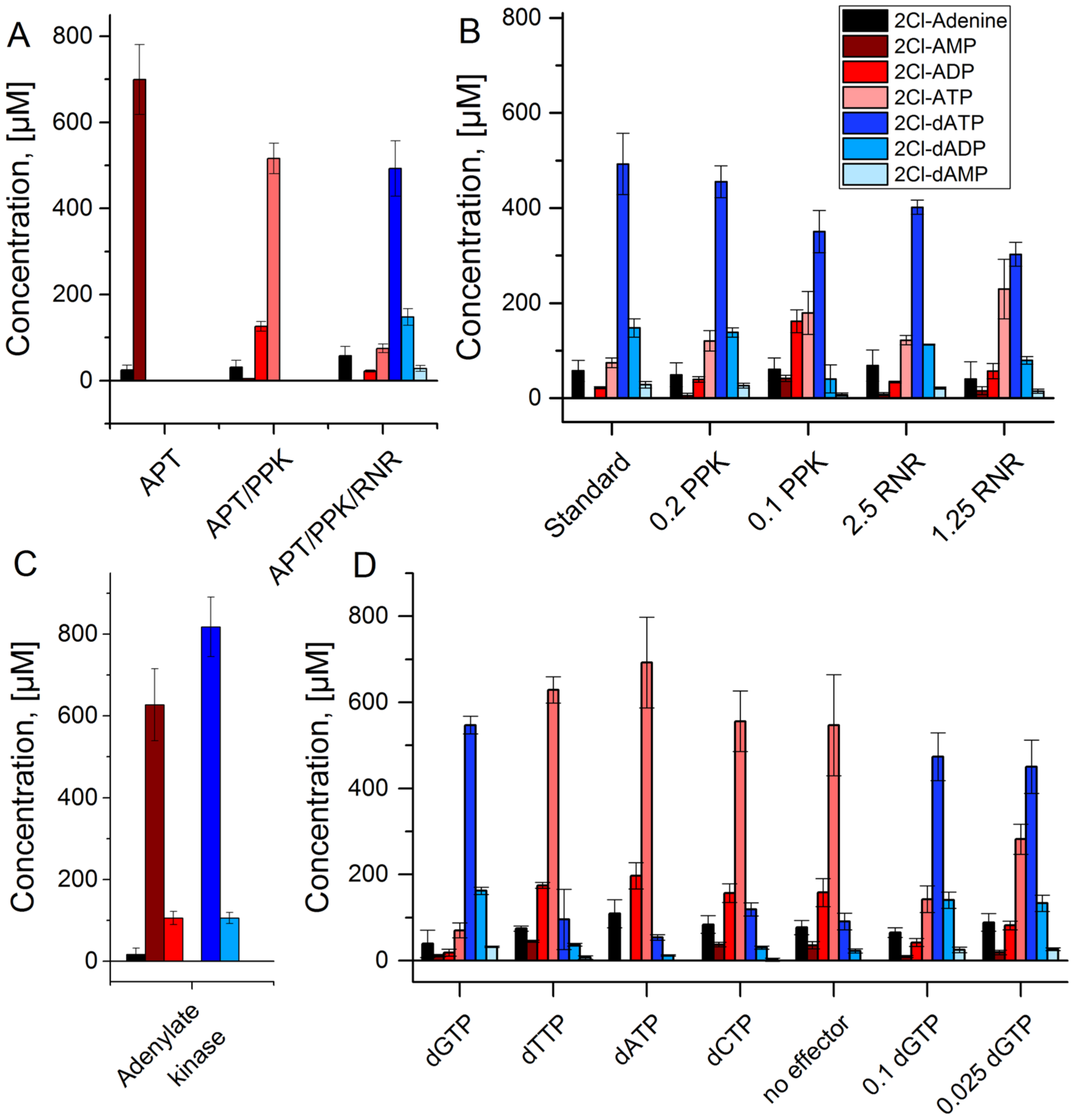
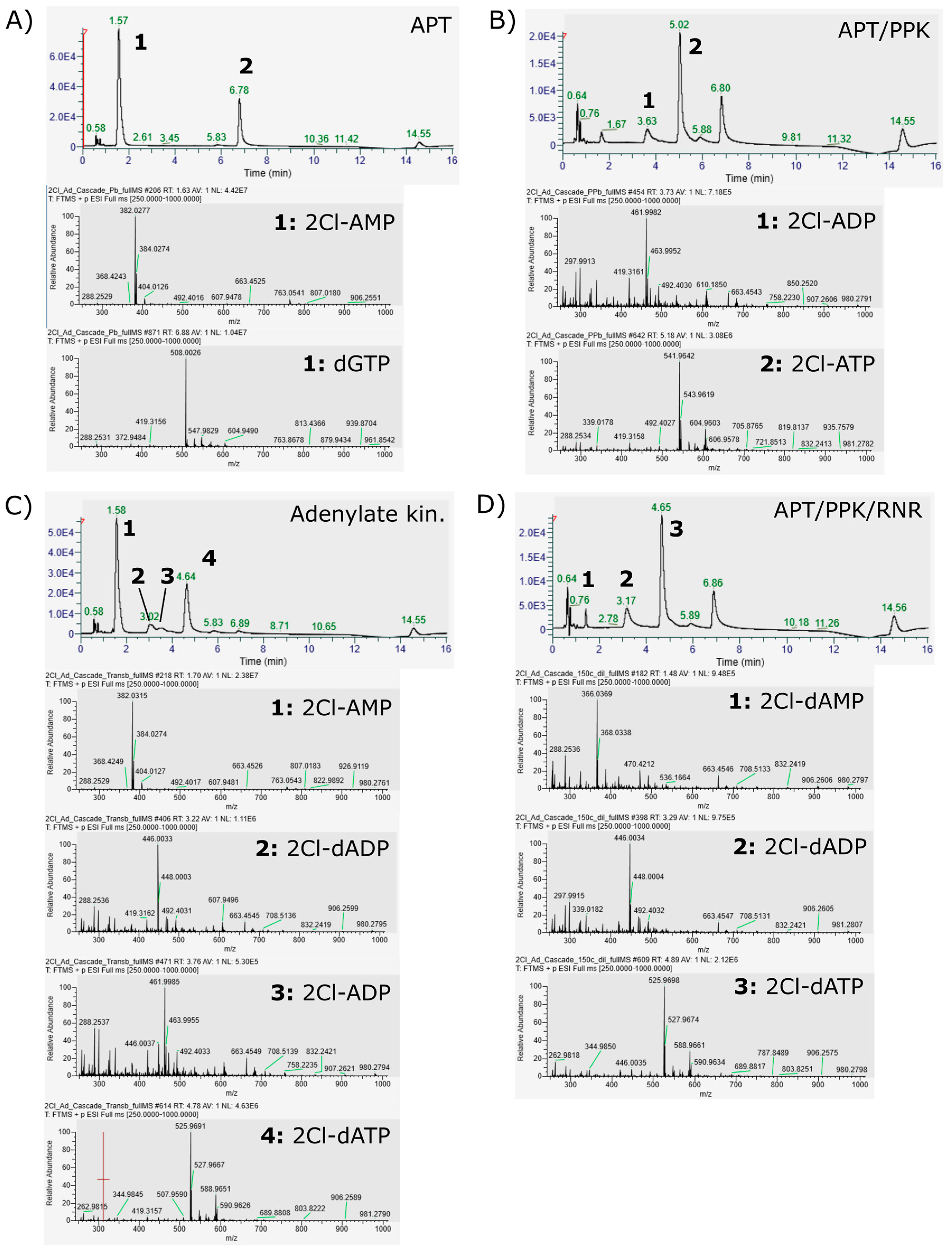
3. Results
4. Discussion
- (1)
- PRPP as substrate is an expensive co-substrate. Although the value of the synthesized cladribine triphosphate may justify that, an in situ generation of PRPP would render the process more feasible. Ribokinases and PRPP synthetases may be used for this end as described in other studies, given that these enzymes are compatible with the current cascade [15,18,20,38].
- (2)
- The adenylate kinase reaction of the PPK that was used in this cascade has been demonstrated for the reaction ATP + AMP → 2 ADP before [28]. This reaction seems to be the main reason for the formation of side product 2Cl-deoxyadenosine diphosphate (Figure 4C and Figure 5). Since it is a competing reaction to the polyphosphate dependent phosphorylation, it may be possible to reduce this side reaction by optimization of PPK and polyphosphate concentrations. Enzyme engineering of the PPK would be a possible way to reduce this side reaction as well. Formation of 2Cl-dADP by RNR catalyzed 2CL-ADP reduction can be excluded as a source for dADP generation, since the applied enzyme is a strict nucleoside triphosphate reducing RNR [32].
- (3)
- The RNR requires an allosteric effector molecule to reach high activities for the reduction of a specific nucleotide. The effector, being a dNTP itself, is a rather expensive additive to the reaction cascade. Reduction of the concentration is possible for the price of a reduced RNR activity. One possibility to circumvent this issue would be the immobilization of the effector on the enzyme, as shown for NADH on different dehydrogenases [39]. By re-engineering of the effector binding site, the dependence of the enzyme on the allosteric effector might be deleted.
- (1)
- In order to set up the cascade for 2Cl-adenine, it was assembled step by step to establish the required enzyme concentrations. However, each of these reactions could be a valuable biocatalytic process in its own right. Optimizing the reaction shown in Figure 4A could deliver a feasible process for the synthesis of 2Cl-adenosine triphosphate. Using the PPK2-II from Acetinobacter johnsonii, which is slower in the phosphorylation of the nucleoside diphosphate, could be used to synthesize 2Cl-adenosine diphosphate [28,40].
- (2)
- The cascade may not be limited to the synthesis of cladribine triphosphate. Although the APT seems to be quite specific for adenosine, there are different phosphoribosyltransferases that cover different ranges of nucleobases. Examples are uracil phosphoribosyltransferases (UPT) or hypoxanthine(-guanine) phosphoribosyltransferases (H(G)PT) [17,38,41,42]. PPKs and RNRs are capable of the conversion of different natural nucleotides, with both purine and pyrimidine bases [29,32,35]. Therefore, a certain degree of substrate promiscuity towards non-natural nucleotides can be expected from both enzymes.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
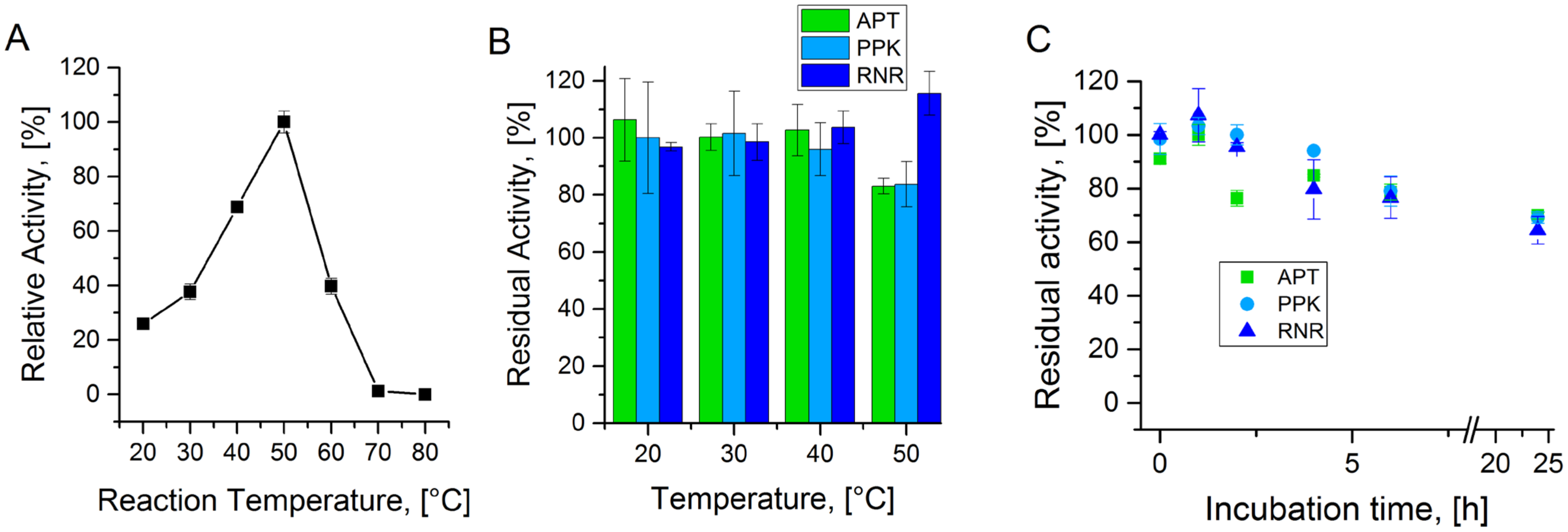
A.1. Temperature Stability of Enzymes from the Cascade

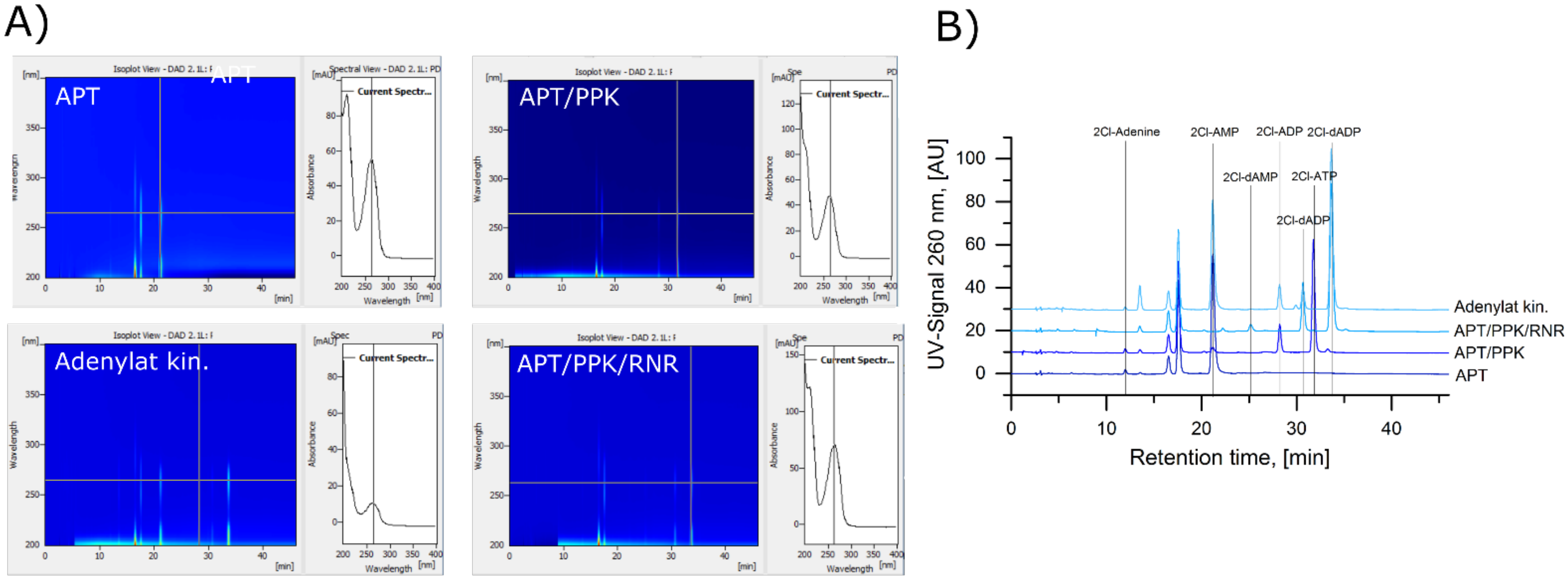
A.2. Identification of 2Cl-Adenosine Reaction Intermediates and Products


A.3. HPLC MS Method
References
- Carson, D.A.; Wasson, D.B.; Beutler, E. Antileukemic and immunosuppressive activity of 2-chloro-2′-deoxyadenosine. Proc. Natl. Acad. Sci. USA 1984, 81, 2232–2236. [Google Scholar] [CrossRef]
- Giovannoni, G.; Comi, G.; Cook, S.; Rammohan, K.; Rieckmann, P.; Sørensen, P.S.; Vermersch, P.; Chang, P.; Hamlett, A.; Musch, B.; et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 416–426. [Google Scholar] [CrossRef]
- Leist, T.P.; Weissert, R. Cladribine: Mode of action and implications for treatment of multiple sclerosis. Clin. Neuropharmacol. 2011, 34, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B. Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leuk. Lymphoma 2011, 52. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; Herrod, S.S.; Schmierer, K. Potential mechanisms of action related to the efficacy and safety of cladribine. Mult. Scler. Relat. Dis. 2019, 30, 176–186. [Google Scholar] [CrossRef]
- Fernandez-Lucas, J. Multienzymatic synthesis of nucleic acid derivatives: A general perspective. Appl. Microbiol. Biotechnol. 2015, 99, 4615–4627. [Google Scholar] [CrossRef]
- Rabuffetti, M.; Bavaro, T.; Semproli, R.; Cattaneo, G.; Massone, M.; Morelli, C.F.; Speranza, G.; Ubiali, D. Synthesis of ribavirin, tecadenoson, and cladribine by enzymatic eransglycosylation. Catalysts 2019, 9, 355. [Google Scholar] [CrossRef]
- Zhou, X.; Szeker, K.; Janocha, B.; Bohme, T.; Albrecht, D.; Mikhailopulo, I.A.; Neubauer, P. Recombinant purine nucleoside phosphorylases from thermophiles: Preparation, properties and activity towards purine and pyrimidine nucleosides. FEBS J. 2013, 280, 1475–1490. [Google Scholar] [CrossRef]
- Fernandez-Lucas, J.; Acebal, C.; Sinisterra, J.V.; Arroyo, M.; de la Mata, I. Lactobacillus reuteri 2′-deoxyribosyltransferase, a novel biocatalyst for tailoring of nucleosides. Appl. Environ. Microbiol. 2010, 76, 1462–1470. [Google Scholar] [CrossRef]
- Laumann, A.S.L.; Britos, C.N.; Cappa, V.A.; Rivero, C.W.; Trelles, J.A. Biotransformation of cladribine by a magnetic immobilizated biocatalyst of lactobacillus animalis. Biotechnol. Lett. 2020, 42, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Pérez, E.; Sánchez-Murcia, P.A.; Jordaan, J.; Blanco, M.D.; Mancheño, J.M.; Gago, F.; Fernández-Lucas, J. Enzymatic synthesis of therapeutic nucleosides using a highly versatile purine nucleoside 2′-deoxyribosyl transferase from trypanosoma brucei. ChemCatChem 2018, 10, 4406–4416. [Google Scholar] [CrossRef]
- Acosta, J.; del Arco, J.; Martinez-Pascual, S.; Clemente-Suárez, V.; Fernández-Lucas, J. One-pot multi-enzymatic production of purine derivatives with application in pharmaceutical and food industry. Catalysts 2018, 8, 9. [Google Scholar] [CrossRef]
- Del Arco, J.; Martinez, M.; Donday, M.; Clemente-Suarez, V.J.; Fernández-Lucas, J. Cloning, expression and biochemical characterization of xanthine and adenine phosphoribosyltransferases from thermus thermophilus hb8. Biocatal. Biotransform. 2017, 36, 216–223. [Google Scholar] [CrossRef]
- Donini, S.; Ferraris, D.M.; Miggiano, R.; Massarotti, A.; Rizzi, M. Structural investigations on orotate phosphoribosyltransferase from mycobacterium tuberculosis, a key enzyme of the de novo pyrimidine biosynthesis. Sci. Rep. 2017, 7, 1180. [Google Scholar] [CrossRef]
- Esipov, R.S.; Abramchik, Y.A.; Fateev, I.V.; Konstantinova, I.D.; Kostromina, M.A.; Muravyova, T.I.; Artemova, K.G.; Miroshnikov, A.I. A cascade of thermophilic enzymes as an approach to the synthesis of modified nucleotides. Acta Nat. 2016, 8, 82–90. [Google Scholar] [CrossRef]
- Fateev, I.V.; Sinitsina, E.V.; Bikanasova, A.U.; Kostromina, M.A.; Tuzova, E.S.; Esipova, L.V.; Muravyova, T.I.; Kayushin, A.L.; Konstantinova, I.D.; Esipov, R.S. Thermophilic phosphoribosyltransferases thermus thermophilus hb27 in nucleotide synthesis. Beilstein J. Org. Chem. 2018, 14, 3098–3105. [Google Scholar] [CrossRef]
- Scism, R.A.; Stec, D.F.; Bachmann, B.O. Synthesis of nucleotide analogues by a promiscuous phosphoribosyltransferase. Org. Lett. 2007, 9, 4179–4182. [Google Scholar] [CrossRef]
- DaCosta, C.P.; Fedor, M.J.; Scott, L.G. 8-azaguanine reporter of purine ionization states in structured rnas. J. Am. Chem. Soc. 2007, 129, 3426–3432. [Google Scholar] [CrossRef] [PubMed]
- Hennig, M.; Scott, L.G.; Sperling, E.; Bermel, W.; Williamson, J.R. Synthesis of 5-fluoropyrimidine nucleotides as sensitive nmr probes of rna structure. J. Am. Chem. Soc. 2007, 129, 14911–14921. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.G.; Geierstanger, B.H.; Williamson, J.R.; Hennig, M. Enzymatic synthesis and 19f nmr studies of 2-fluoroadenine-substituted rna. J. Am. Chem. Soc. 2004, 126, 1177–11777. [Google Scholar] [CrossRef] [PubMed]
- Fehlau, M.; Kaspar, F.; Hellendahl, K.F.; Schollmeyer, J.; Neubauer, P.; Wagner, A. Modular enzymatic cascade synthesis of nucleotides using a (d)atp regeneration system. Front. Bioeng. Biotechnol. 2020, 8, 854. [Google Scholar] [CrossRef]
- Andexer, J.N.; Richter, M. Emerging enzymes for atp regeneration in biocatalytic processes. ChemBioChem 2015, 16, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Mordhorst, S.; Andexer, J.N. Round, round we go—Strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333. [Google Scholar] [CrossRef]
- Mordhorst, S.; Siegrist, J.; Muller, M.; Richter, M.; Andexer, J.N. Catalytic alkylation using a cyclic s-adenosylmethionine regeneration system. Angew. Chem. Int. Ed. Engl. 2017, 56, 4037–4041. [Google Scholar] [CrossRef]
- Murata, K.; Uchida, T.; Kato, J.; Chibata, I. Polyphosphate kinase: Distribution, some properties and its application as an atp regeneration system. Agr. Biol. Chem. 1988, 52, 1471–1477. [Google Scholar]
- Resnick, S.M.; Zehnder, A.J. In vitro atp regeneration from polyphosphate and amp by polyphosphate:Amp phosphotransferase and adenylate kinase from acinetobacter johnsonii 210a. Appl. Environ. Microbiol. 2000, 66, 2045–2051. [Google Scholar] [CrossRef]
- Shimane, M.; Sugai, Y.; Kainuma, R.; Natsume, M.; Kawaide, H. Mevalonate-dependent enzymatic synthesis of amorphadiene driven by an atp-regeneration system using polyphosphate kinase. Biosci. Biotechnol. Biochem. 2012, 76, 1558–1560. [Google Scholar] [CrossRef][Green Version]
- Mordhorst, S.; Singh, J.; Mohr, M.K.F.; Hinkelmann, R.; Keppler, M.; Jessen, H.J.; Andexer, J.N. Several polyphosphate kinase 2 enzymes catalyse the production of adenosine 5’-polyphosphates. ChemBioChem A Eur. J. Chem. Biol. 2019, 20, 1019–1022. [Google Scholar] [CrossRef]
- Motomura, K.; Hirota, R.; Okada, M.; Ikeda, T.; Ishida, T.; Kuroda, A. A new subfamily of polyphosphate kinase 2 (class iii ppk2) catalyzes both nucleoside monophosphate phosphorylation and nucleoside diphosphate phosphorylation. Appl. Environ. Microbiol. 2014, 80, 2602–2608. [Google Scholar] [CrossRef] [PubMed]
- Parnell, A.E.; Mordhorst, S.; Kemper, F.; Giurrandino, M.; Prince, J.P.; Schwarzer, N.J.; Hofer, A.; Wohlwend, D.; Jessen, H.J.; Gerhardt, S.; et al. Substrate recognition and mechanism revealed by ligand-bound polyphosphate kinase 2 structures. Proc. Natl. Acad. Sci. USA 2017, 115, 3350–3355. [Google Scholar] [CrossRef] [PubMed]
- Lundin, D.; Berggren, G.; Logan, D.T.; Sjoberg, B.M. The origin and evolution of ribonucleotide reduction. Life 2015, 5, 604–636. [Google Scholar] [CrossRef]
- Loderer, C.; Holmfeldt, K.; Lundin, D. Non-host class ii ribonucleotide reductase in thermus viruses: Sequence adaptation and host interaction. PeerJ 2019, 7, e6700. [Google Scholar] [CrossRef]
- Loderer, C.; Jonna, V.R.C.; Cona, M.; Rozman Grinberg, I.; Sahlin, M.; Hofer, A.; Lundin, D.; Sjöberg, B.M. A unique cysteine-rich zn-finger domain present in a majority of class ii ribonucleotide reductases mediates catalytic turnover. J. Biol. Chem. 2017. [Google Scholar] [CrossRef]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Larsson, K.M.; Jordan, A.; Eliasson, R.; Reichard, P.; Logan, D.T.; Nordlund, P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat. Struct. Mol. Biol. 2004, 11, 1142–1149. [Google Scholar] [CrossRef]
- Hochstadt, J. Adenine phosphoribosyltransferase from escherichia coli. Methods Enzymol. 1978, 51, 558–567. [Google Scholar]
- Alfonzo, J.D.; Sahota, A.; Taylor, M.W. Purification and characterization of adenine phosphoribosyltransferase from saccharomyces cerevisiae. Biochim. Biophys. Acta. 1997, 1341, 173–182. [Google Scholar] [CrossRef]
- Scism, R.A.; Bachmann, B.O. Five-component cascade synthesis of nucleotide analogues in an engineered self-immobilized enzyme aggregate. ChemBioChem A Eur. J. Chem. Biol. 2010, 11, 67–70. [Google Scholar] [CrossRef]
- Schäfer, H.G.; Jacobi, T.; Eichhorn, E.; Woenckhaus, C. Covalent fixation of nad+ to dehydrogenases and properties of the modified enzymes. Biol. Chem. Hoppe Seyler 1986, 367, 969–980. [Google Scholar] [CrossRef]
- Itoh, H.; Shiba, T. Polyphosphate synthetic activity of polyphosphate:Amp phosphotransferase in acinetobacter johnsonii 210a. J. Bacteriol. 2004, 186, 5178–5181. [Google Scholar] [CrossRef]
- Acosta, J.; Del Arco, J.; Del Pozo, M.L.; Herrera-Tapias, B.; Clemente-Suárez, V.J.; Berenguer, J.; Hidalgo, A.; Fernández-Lucas, J. Hypoxanthine-guanine phosphoribosyltransferase/adenylate kinase from zobellia galactanivorans: A bifunctional catalyst for the synthesis of nucleoside-5′-mono-, di- and triphosphates. Front. Bioeng. Biotechnol. 2020, 8, 677. [Google Scholar] [CrossRef]
- Villela, A.D.; Ducati, R.G.; Rosado, L.A.; Bloch, C.; Prates, M.V.; Gonçalves, D.C.; Ramos, C.H.I.; Basso, L.A.; Santos, D.S. Biochemical characterization of uracil phosphoribosyltransferase from mycobacterium tuberculosis. PLoS ONE 2013, 8, e56445. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Name | Source | Uniprot ID | Reference |
|---|---|---|---|---|
| APT | EcAPT | Escherichia coli | P69503 | [36] |
| PPK | MrPPK | Meiothermus ruber | M9XB82 | [28,29,30] |
| RNR | TVNrdJm | Thermus virus TV74-23 | A7XXH5 | [32] |
| Enzyme | Substrate | KM, [µmol L−1] | kcat, [s−1] |
|---|---|---|---|
| APT | adenine | 11.8 ± 5.2 | 119 ± 14 |
| PPK | AMP | 41 ± 6.3 | 17 ± 1.3 |
| PPK | ADP | 144 ± 14 | 0.43 ± 0.02 |
| RNR | ATP | 199 ± 13 | 0.56 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frisch, J.; Maršić, T.; Loderer, C. A Novel One-Pot Enzyme Cascade for the Biosynthesis of Cladribine Triphosphate. Biomolecules 2021, 11, 346. https://doi.org/10.3390/biom11030346
Frisch J, Maršić T, Loderer C. A Novel One-Pot Enzyme Cascade for the Biosynthesis of Cladribine Triphosphate. Biomolecules. 2021; 11(3):346. https://doi.org/10.3390/biom11030346
Chicago/Turabian StyleFrisch, Julia, Tin Maršić, and Christoph Loderer. 2021. "A Novel One-Pot Enzyme Cascade for the Biosynthesis of Cladribine Triphosphate" Biomolecules 11, no. 3: 346. https://doi.org/10.3390/biom11030346
APA StyleFrisch, J., Maršić, T., & Loderer, C. (2021). A Novel One-Pot Enzyme Cascade for the Biosynthesis of Cladribine Triphosphate. Biomolecules, 11(3), 346. https://doi.org/10.3390/biom11030346