
Probing the Influence of Single-Site Mutations in the Central Cross-β Region of Amyloid β (1–40) Peptides

 , , ,
, , ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Peptides Synthesis
2.2. Sample Preparation for NMR Spectroscopy, X-ray Diffraction Measurements and Transmission Electron Microscopy of Aβ Fibrils
2.3. Sample Preparation for NMR Spectroscopy of Aβ Oligomers
2.4. Fibrillation Kinetics Measurements by ThT-Fluorescence Assay
2.5. Circular Dichroism Spectroscopy
2.6. Transmission Electron Microscopy (TEM)
2.7. X-ray Diffraction Measurements
2.8. Solid-State NMR Spectroscopy
2.9. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid (MTT) Assay
3. Results
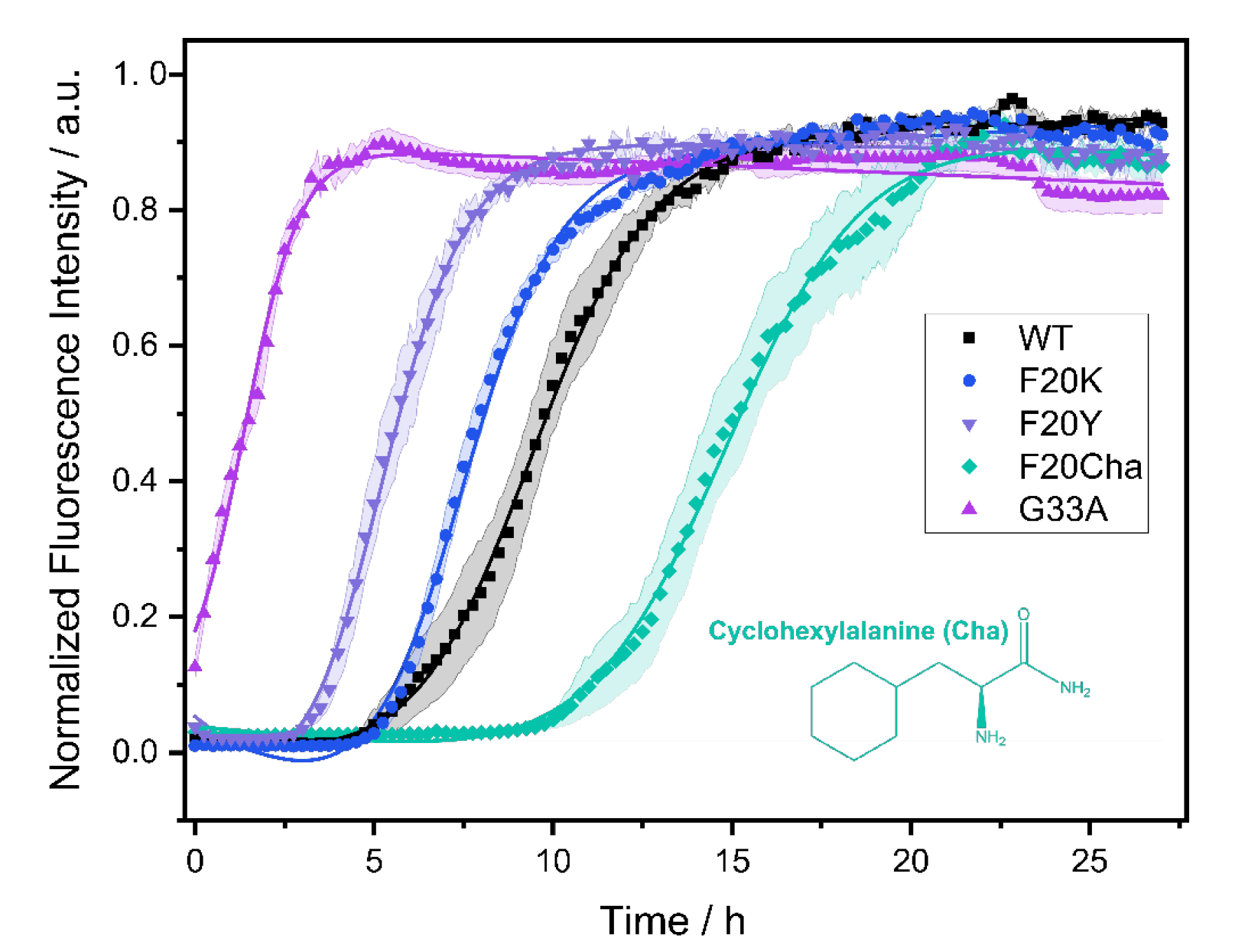
3.1. Fibrillation Kinetics
3.2. Transmission Electron Microscopy
3.3. X-ray Diffraction
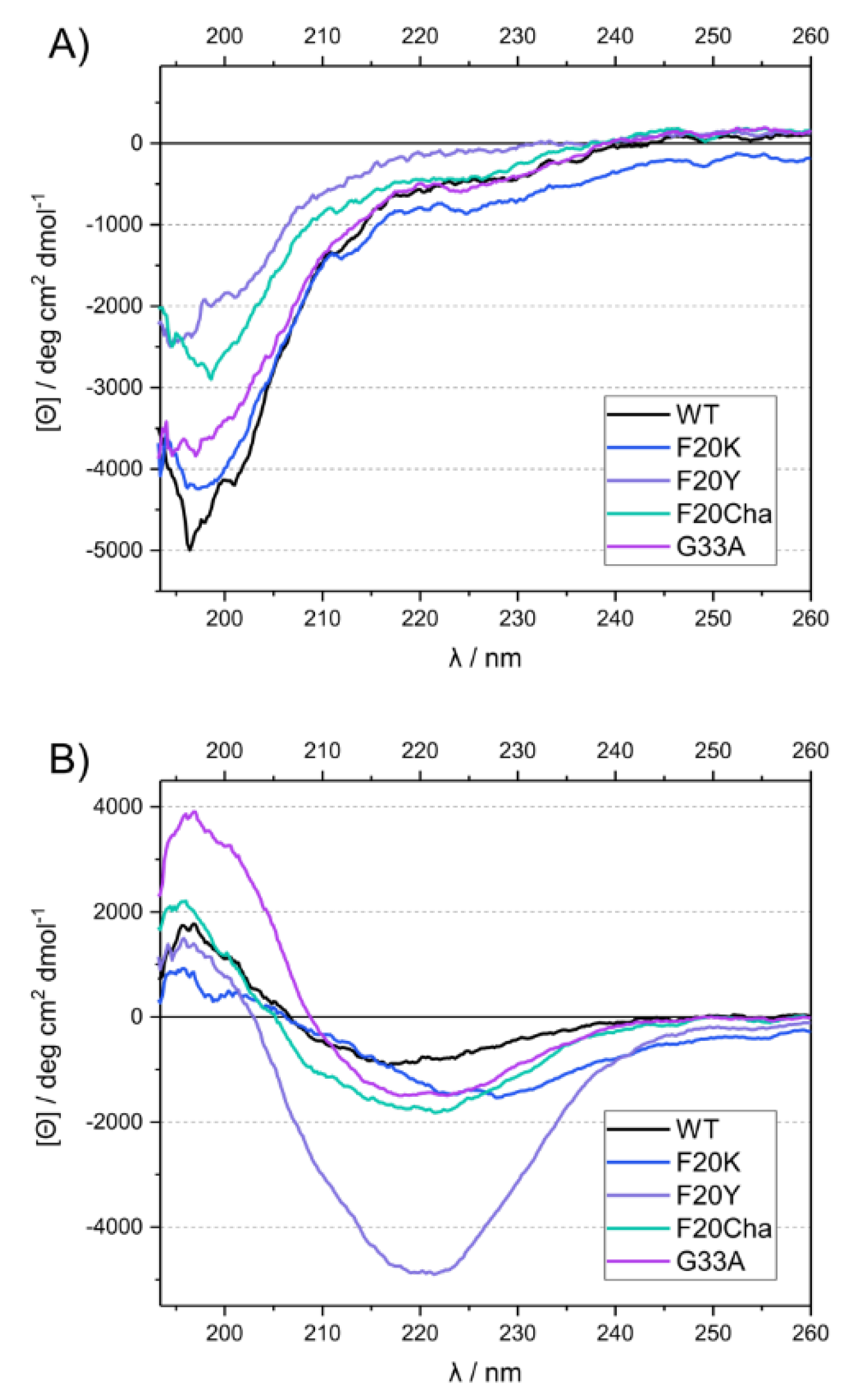
3.4. Circular Dichroism Spectroscopy
3.5. Local Structure of the Aβ40 Fibrils by 13C NMR Chemical Shift Measurements
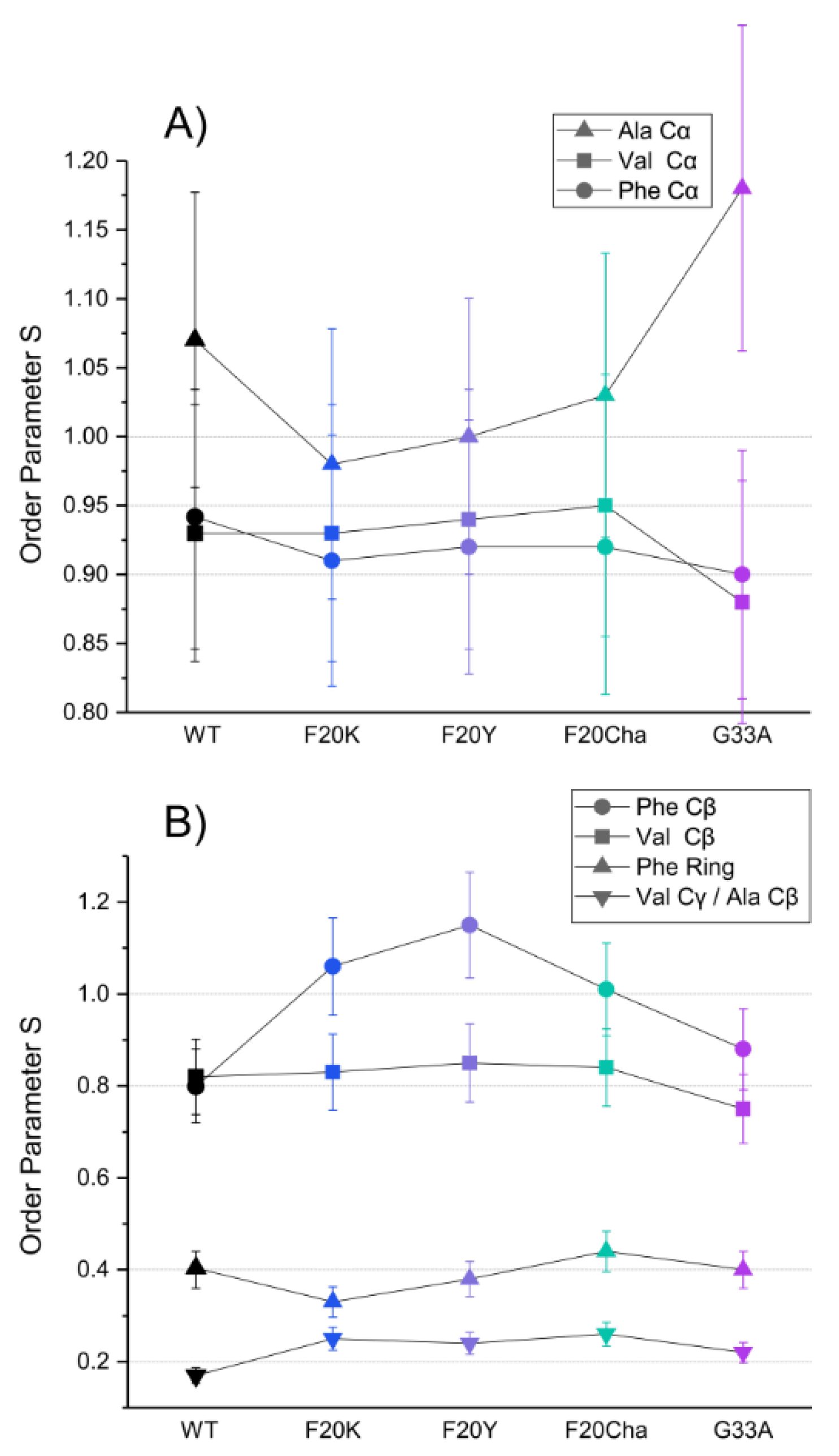
3.6. Investigation of the Molecular Dynamics of the Mutated Aβ40 Fibrils
3.7. Cell Toxicity of the Aβ40 Peptides
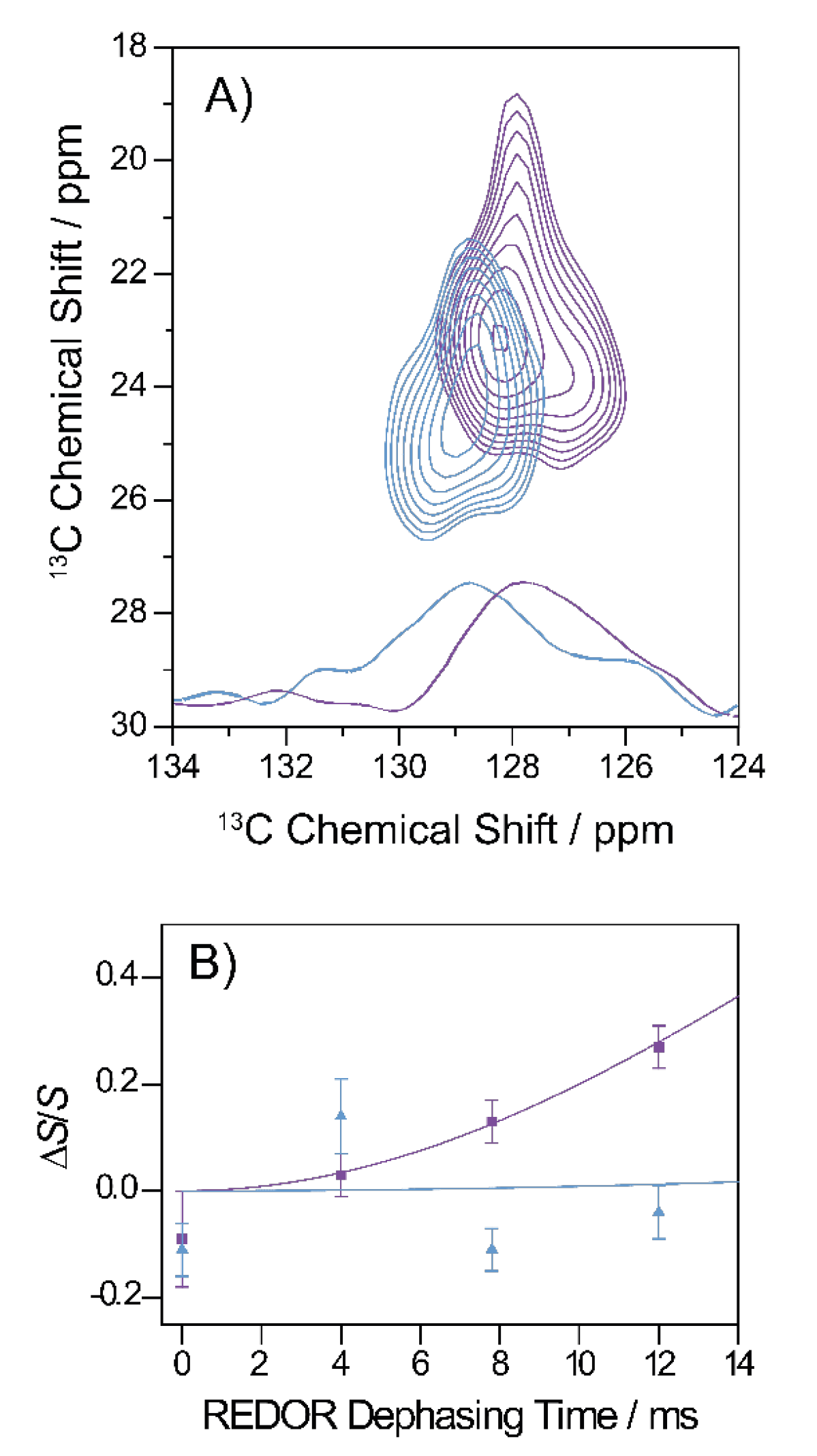
3.8. Specific Structural Investigations on the Gly33Ala Variant
4. Discussion
- (i)
- The equatorial reflection in XRD data that corresponds to the distance between the opposing β-strands suggests a slight increase of the intersheet distance but certainly not to an extent to explain the diameter alteration observed in the TEM images. The fact that Phe20 according to most Aβ40 models [19,44] points to the outside renders a strong influence on this distance unlikely. The moderate increase can be explained by very small packing differences between the opposing β-strands due to alterations in the interaction of the side chains inside the fibril;
- (ii)
- Current Aβ40 fibril models assume that the fibrils are formed of multiples (usually 2–8) of these opposing β-strands termed protofilaments. The protofilaments line up parallel to each other. The mutations to Phe20 may cause alterations of this agglomeration of the protofilaments. In the literature, different diameters for Aβ40 WT fibrils are reported depending on preparation protocols. These diameters could be associated with assumed numbers of protofilaments [44,45]. Although it is difficult to assess this further with the data provided by our experiments, we assume that the arrangement of the altered side chains in position 20 affects the alignment of the protofilaments.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Cawood, E.E.; Karamanos, T.K.; Wilson, A.J.; Radford, S.E. Visualizing and trapping transient oligomers in amyloid assembly pathways. Biophys. Chem. 2021, 268, 106505. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Modi, P.; Sharma, G.; Deep, S. Kinetics theories to understand the mechanism of aggregation of a protein and to design strategies for its inhibition. Biophys. Chem. 2021, 278, 106665. [Google Scholar] [CrossRef]
- Vignaud, H.; Bobo, C.; Lascu, I.; Sorgjerd, K.M.; Zako, T.; Maeda, M.; Salin, B.; Lecomte, S.; Cullin, C. A structure-toxicity study of Abeta42 reveals a new anti-parallel aggregation pathway. PLoS ONE 2013, 8, e80262. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zlatic, C.O.; Griffin, M.D.; Howlett, G.J.; Todorova, N.; Yarovsky, I.; Gooley, P.R. Hydrogen/Deuterium Exchange and Molecular Dynamics Analysis of Amyloid Fibrils Formed by a D69K Charge-Pair Mutant of Human Apolipoprotein C-II. Biochemistry 2015, 54, 4805–4814. [Google Scholar] [CrossRef]
- Mao, Y.; Teoh, C.L.; Yang, S.; Zlatic, C.O.; Rosenes, Z.K.; Gooley, P.R.; Howlett, G.J.; Griffin, M.D. Charge and charge-pair mutations alter the rate of assembly and structural properties of apolipoprotein C-II amyloid fibrils. Biochemistry 2015, 54, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.D.; Shivaprasad, S.; Wetzel, R. Alanine scanning mutagenesis of Abeta(1-40) amyloid fibril stability. J. Mol. Biol. 2006, 357, 1283–1294. [Google Scholar] [CrossRef]
- Wetzel, R.; Shivaprasad, S.; Williams, A.D. Plasticity of amyloid fibrils. Biochemistry 2007, 46, 1–10. [Google Scholar] [CrossRef]
- Adler, J.; Scheidt, H.A.; Kruger, M.; Thomas, L.; Huster, D. Local interactions influence the fibrillation kinetics, structure and dynamics of Abeta(1-40) but leave the general fibril structure unchanged. Phys. Chem. Chem. Phys. 2014, 16, 7461–7471. [Google Scholar] [CrossRef]
- Adler, J.; Baumann, M.; Voigt, B.; Scheidt, H.A.; Bhowmik, D.; Haupl, T.; Abel, B.; Madhu, P.K.; Balbach, J.; Maiti, S.; et al. A Detailed Analysis of the Morphology of Fibrils of Selectively Mutated Amyloid beta (1-40). Chem. Phys. Chem. 2016, 17, 2744–2753. [Google Scholar] [CrossRef]
- Das, A.K.; Rawat, A.; Bhowmik, D.; Pandit, R.; Huster, D.; Maiti, S. An Early Folding Contact between Phe19 and Leu34 is Critical for Amyloid-beta Oligomer Toxicity. ACS Chem. Neurosci. 2015, 6, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Adler, J.; Chandra, B.; Mote, K.R.; Bekcioglu-Neff, G.; Sebastiani, D.; Huster, D. Perturbation of the F19-L34 Contact in Amyloid beta (1-40) Fibrils Induces Only Local Structural Changes but Abolishes Cytotoxicity. J. Phys. Chem. Lett. 2017, 8, 4740–4745. [Google Scholar] [CrossRef]
- Korn, A.; McLennan, S.; Adler, J.; Krueger, M.; Surendran, D.; Maiti, S.; Huster, D. Amyloid beta (1-40) Toxicity Depends on the Molecular Contact between Phenylalanine 19 and Leucine 34. ACS Chem. Neurosci. 2018, 9, 790–799. [Google Scholar] [CrossRef]
- Korn, A.; Surendran, D.; Krueger, M.; Maiti, S.; Huster, D. Ring structure modifications of phenylalanine 19 increase fibrillation kinetics and reduce toxicity of amyloid beta (1–40). Chem. Commun. 2018, 54, 5430–5433. [Google Scholar] [CrossRef] [PubMed]
- Korn, A.; Hofling, C.; Zeitschel, U.; Krueger, M.; Rossner, S.; Huster, D. Incorporation of the Nonproteinogenic Amino Acid beta-Methylamino-alanine Affects Amyloid beta Fibril Properties and Toxicity. ACS Chem. Neurosci. 2020, 11, 1038–1047. [Google Scholar] [CrossRef]
- Vivekanandan, S.; Brender, J.R.; Lee, S.Y.; Ramamoorthy, A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011, 411, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Gonnelli, L.; Luchinat, C.; Mao, J.; Nesi, A. A new structural model of Abeta40 fibrils. J. Am. Chem. Soc. 2011, 133, 16013–16022. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Qahwash, I.; Leapman, R.D.; Meredith, S.C.; Tycko, R. Seeded growth of beta-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. USA 2009, 106, 7443–7448. [Google Scholar] [CrossRef]
- Lu, J.X.; Qiang, W.; Yau, W.M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular Structure of beta-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, U.; Thurber, K.R.; Yau, W.M.; Tycko, R. Molecular structure of a prevalent amyloid-beta fibril polymorph from Alzheimer’s disease brain tissue. Proc. Natl. Acad. Sci. USA 2021, 118, e2023089118. [Google Scholar] [CrossRef]
- Sarkar, B.; Mithu, V.S.; Chandra, B.; Mandal, A.; Chandrakesan, M.; Bhowmik, D.; Madhu, P.K.; Maiti, S. Significant structural differences between transient amyloid-beta oligomers and less-toxic fibrils in regions known to harbor familial Alzheimer’s mutations. Angew. Chem. Int. Ed. 2014, 53, 6888–6892. [Google Scholar] [CrossRef] [PubMed]
- Vassar, P.S.; Culling, C.F. Fluorescent stains, with special reference to amyloid and connective tissues. Arch. Pathol. 1959, 68, 487–498. [Google Scholar] [PubMed]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, T.; Veglia, G. Dual acquisition magic-angle spinning solid-state NMR-spectroscopy: Simultaneous acquisition of multidimensional spectra of biomacromolecules. Angew. Chem. Int. Ed. 2012, 51, 2731–2735. [Google Scholar] [CrossRef]
- Munowitz, M.G.; Griffin, R.G.; Bodenhausen, G.; Huang, T.H. Two-dimensional rotational spin-echo nuclear magnetic resonance in solids: Correlation of chemical shift and dipolar interactions. J. Am. Chem. Soc. 1981, 103, 2529–2533. [Google Scholar] [CrossRef]
- Bielecki, A.; Kolbert, A.C.; Levitt, M.H. Frequency-switched pulse sequences: Homonuclear decoupling and dilute spin NMR in solids. Chem. Phys. Lett. 1989, 155, 341–345. [Google Scholar] [CrossRef]
- Barré, P.; Zschörnig, O.; Arnold, K.; Huster, D. Structural and dynamical changes of the bindin B18 peptide upon binding to lipid membranes. A solid-state NMR study. Biochemistry 2003, 42, 8377–8386. [Google Scholar] [CrossRef]
- Jaroniec, C.P.; Tounge, B.A.; Herzfeld, J.; Griffin, R.G. Frequency selective heteronuclear dipolar recoupling in rotating solids: Accurate (13)C-(15)N distance measurements in uniformly (13)C,(15)N-labeled peptides. J. Am. Chem. Soc. 2001, 123, 3507–3519. [Google Scholar] [CrossRef]
- Fatafta, H.; Khaled, M.; Owen, M.C.; Sayyed-Ahmad, A.; Strodel, B. Amyloid-beta peptide dimers undergo a random coil to beta-sheet transition in the aqueous phase but not at the neuronal membrane. Proc. Natl. Acad. Sci. USA 2021, 118, e2106210118. [Google Scholar] [CrossRef] [PubMed]
- Spera, S.; Bax, A. Empirical correlation between protein backbone conformation and Ca and Cb 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 1991, 113, 5490–5492. [Google Scholar] [CrossRef]
- Luca, S.; Filippov, D.V.; van Boom, J.H.; Oschkinat, H.; de Groot, H.J.; Baldus, M. Secondary chemical shifts in immobilized peptides and proteins: A qualitative basis for structure refinement under magic angle spinning. J. Biomol. NMR 2001, 20, 325–331. [Google Scholar] [CrossRef]
- Hong, M.; Yao, X.; Jakes, K.S.; Huster, D. Investigation of molecular motions by magic-angle cross-polarization NMR spectroscopy. J. Phys. Chem. B 2002, 106, 7355–7364. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Morgado, I.; Rothemund, S.; Huster, D. Dynamics of amyloid beta fibrils revealed by solid-state NMR. J. Biol. Chem. 2012, 287, 2017–2021. [Google Scholar] [CrossRef]
- Reddy, G.; Straub, J.E.; Thirumalai, D. Influence of preformed Asp23-Lys28 salt bridge on the conformational fluctuations of monomers and dimers of Abeta peptides with implications for rates of fibril formation. J. Phys. Chem. B 2009, 113, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Ghosh, A.; Kotler, S.A.; Krishnamoorthy, J.; Bera, S.; Morris, V.; Sil, T.B.; Garai, K.; Reif, B.; Bhunia, A.; et al. Probing transient non-native states in amyloid beta fiber elongation by NMR. Chem. Commun. 2019, 55, 4483–4486. [Google Scholar] [CrossRef]
- Tycko, R. Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 2014, 23, 1528–1539. [Google Scholar] [CrossRef]
- Tycko, R. Molecular structure of amyloid fibrils: Insights from solid-state NMR. Q. Rev. Biophys. 2006, 39, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Park, S.H.; Opella, S.J. Membrane protein structure from rotational diffusion. Biochim. Biophys. Acta 2015, 1848, 229–245. [Google Scholar] [CrossRef]
- Fatafta, H.; Batuhan, K.; Bundschuh, B.F.; Loschwitz, J.; Strodel, B. Disorder-to-order transition of the amyloid-β peptide upon lipid binding. Biophys. Chem. 2021, 280, 106700. [Google Scholar] [CrossRef]
- Gilmanshin, R.; Dyer, R.B.; Callender, R.H. Structural heterogeneity of the various forms of apomyoglobin: Implications for protein folding. Protein Sci. 1997, 6, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Kuwajima, K. Rapid formation of a molten globule intermediate in refolding of alpha-lactalbumin. Fold. Des. 1996, 1, 275–287. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef]
- Barnes, C.A.; Robertson, A.J.; Louis, J.M.; Anfinrud, P.; Bax, A. Observation of beta-Amyloid Peptide Oligomerization by Pressure-Jump NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141, 13762–13766. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Morgado, I.; Rothemund, S.; Huster, D.; Fandrich, M. Solid-State NMR Spectroscopic Investigation of Abeta Protofibrils: Implication of a beta-Sheet Remodeling upon Maturation into Terminal Amyloid Fibrils. Angew. Chem. Int. Ed. 2011, 50, 2837–2840. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sahoo, B.R.; Ozawa, D.; Kinoshita, M.; Kang, J.; Lim, M.H.; Okumura, M.; Huh, Y.H.; Moon, E.; Jang, J.H.; et al. Diverse Structural Conversion and Aggregation Pathways of Alzheimer’s Amyloid-beta (1-40). ACS Nano 2019, 13, 8766–8783. [Google Scholar] [CrossRef]
- Chandra, B.; Bhowmik, D.; Maity, B.K.; Mote, K.R.; Dhara, D.; Venkatramani, R.; Maiti, S.; Madhu, P.K. Major reaction coordinates linking transient amyloid-beta oligomers to fibrils measured at atomic level. Biophys. J. 2017, 113, 805–816. [Google Scholar] [CrossRef]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussiere, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-beta. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef]
- Chandra, B.; Haider, S.; Adler, J.; Korn, A.; Huster, D.; Maiti, S. Emerging structural details of transient amyloid-beta oligomers suggest designs for effective small molecule modulators. Chem, Phys. Lett. 2017, 675, 51–55. [Google Scholar] [CrossRef]
- Han, J.; Du, Z.; Lim, M.H. Mechanistic Insight into the design of chemical tools to control multiple pathogenic features in Alzheimer’s disease. Acc. Chem. Res. 2021, 54, 3930–3940. [Google Scholar] [CrossRef]
- Gazit, E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Nag, S.; Sarkar, B.; Bandyopadhyay, A.; Sahoo, B.; Sreenivasan, V.K.; Kombrabail, M.; Muralidharan, C.; Maiti, S. Nature of the amyloid-beta monomer and the monomer-oligomer equilibrium. J. Biol. Chem. 2011, 286, 13827–13833. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritzsch, J.; Korn, A.; Surendran, D.; Krueger, M.; Scheidt, H.A.; Mote, K.R.; Madhu, P.K.; Maiti, S.; Huster, D. Probing the Influence of Single-Site Mutations in the Central Cross-β Region of Amyloid β (1–40) Peptides. Biomolecules 2021, 11, 1848. https://doi.org/10.3390/biom11121848
Fritzsch J, Korn A, Surendran D, Krueger M, Scheidt HA, Mote KR, Madhu PK, Maiti S, Huster D. Probing the Influence of Single-Site Mutations in the Central Cross-β Region of Amyloid β (1–40) Peptides. Biomolecules. 2021; 11(12):1848. https://doi.org/10.3390/biom11121848
Chicago/Turabian StyleFritzsch, Jacob, Alexander Korn, Dayana Surendran, Martin Krueger, Holger A. Scheidt, Kaustubh R. Mote, Perunthiruthy K. Madhu, Sudipta Maiti, and Daniel Huster. 2021. "Probing the Influence of Single-Site Mutations in the Central Cross-β Region of Amyloid β (1–40) Peptides" Biomolecules 11, no. 12: 1848. https://doi.org/10.3390/biom11121848
APA StyleFritzsch, J., Korn, A., Surendran, D., Krueger, M., Scheidt, H. A., Mote, K. R., Madhu, P. K., Maiti, S., & Huster, D. (2021). Probing the Influence of Single-Site Mutations in the Central Cross-β Region of Amyloid β (1–40) Peptides. Biomolecules, 11(12), 1848. https://doi.org/10.3390/biom11121848