
Heterologous Expression and Assembly of Human TLR Signaling Components in Saccharomyces cerevisiae

 , , and
, , and 
Abstract
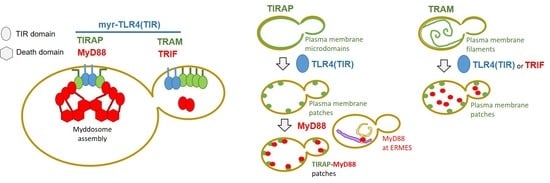
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains, Culture Media, and Growth Conditions
2.2. Plasmid Construction
2.3. Microscopy Techniques, Image Processing, and Statistical Analysis
2.4. Immunodetection by Western Blotting
2.5. Protein Co-Purification Assays
3. Results
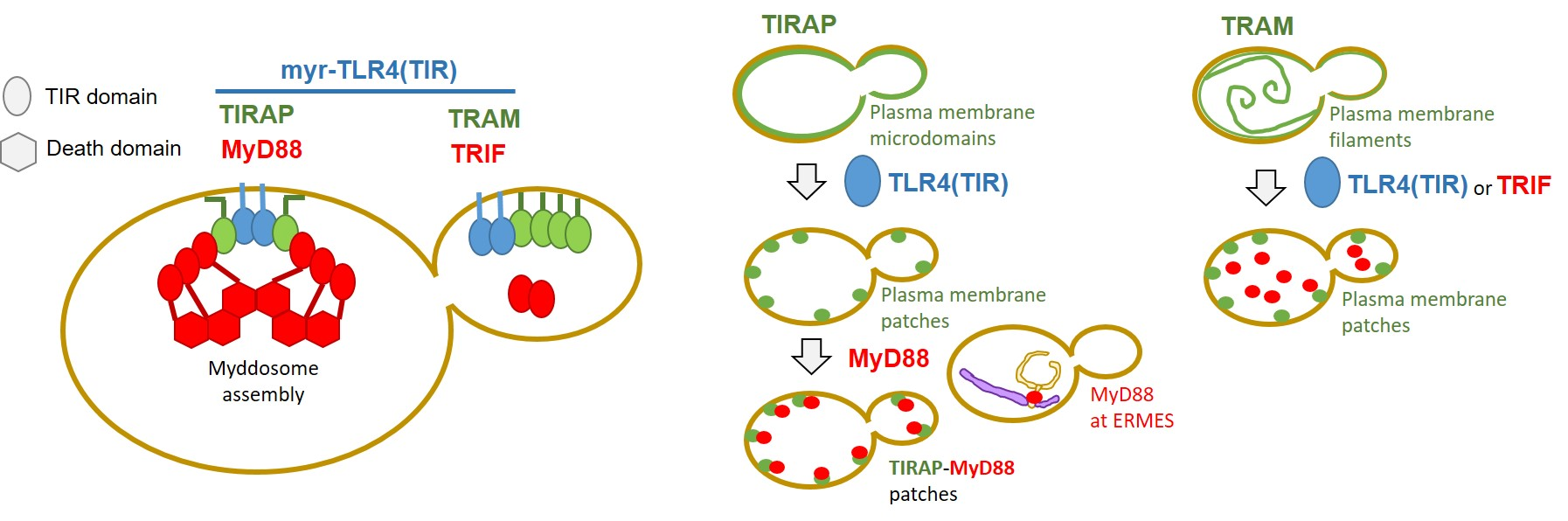
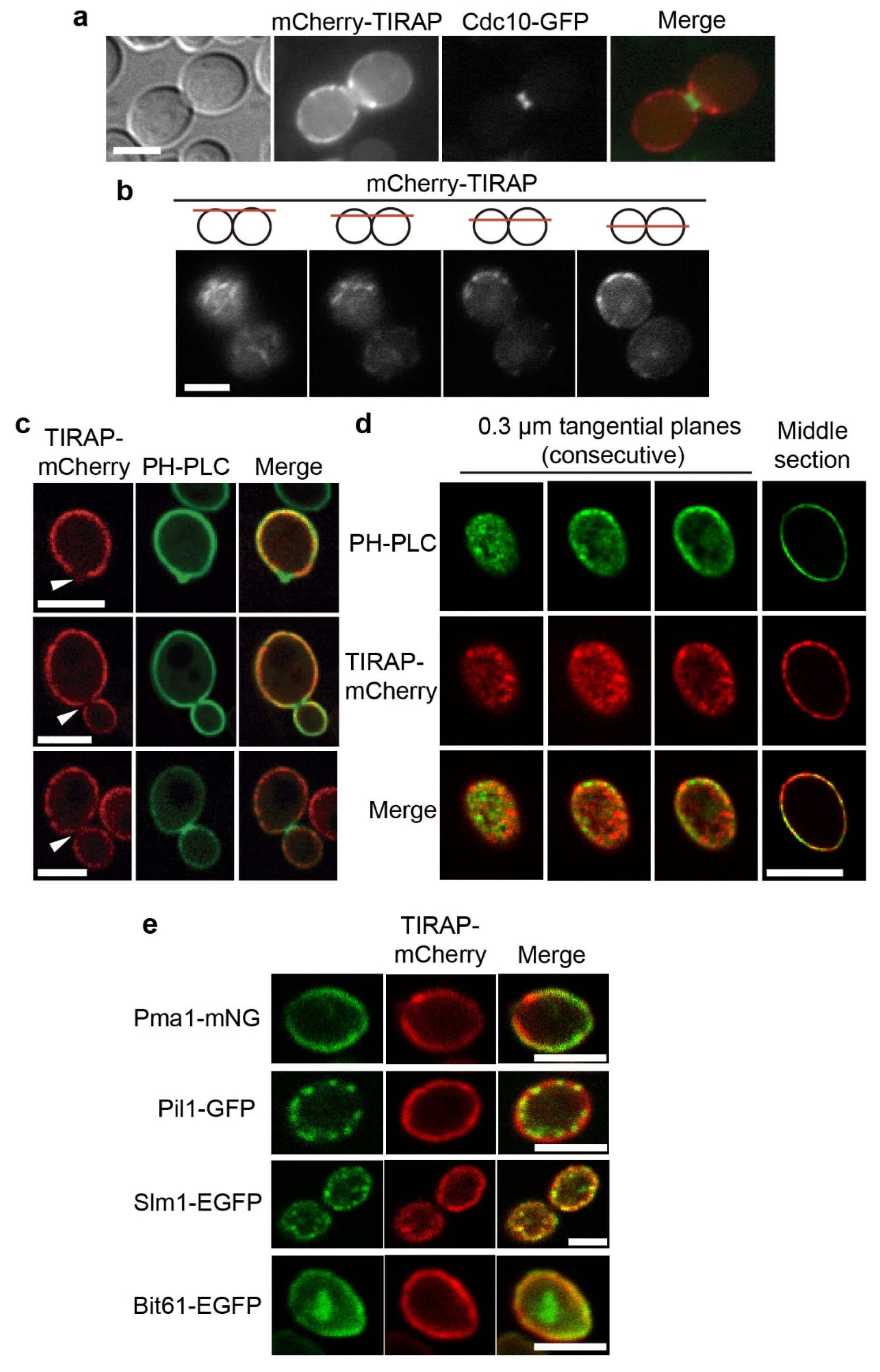
3.1. The TIRAP Adaptor Tightly Localizes to the Yeast PM
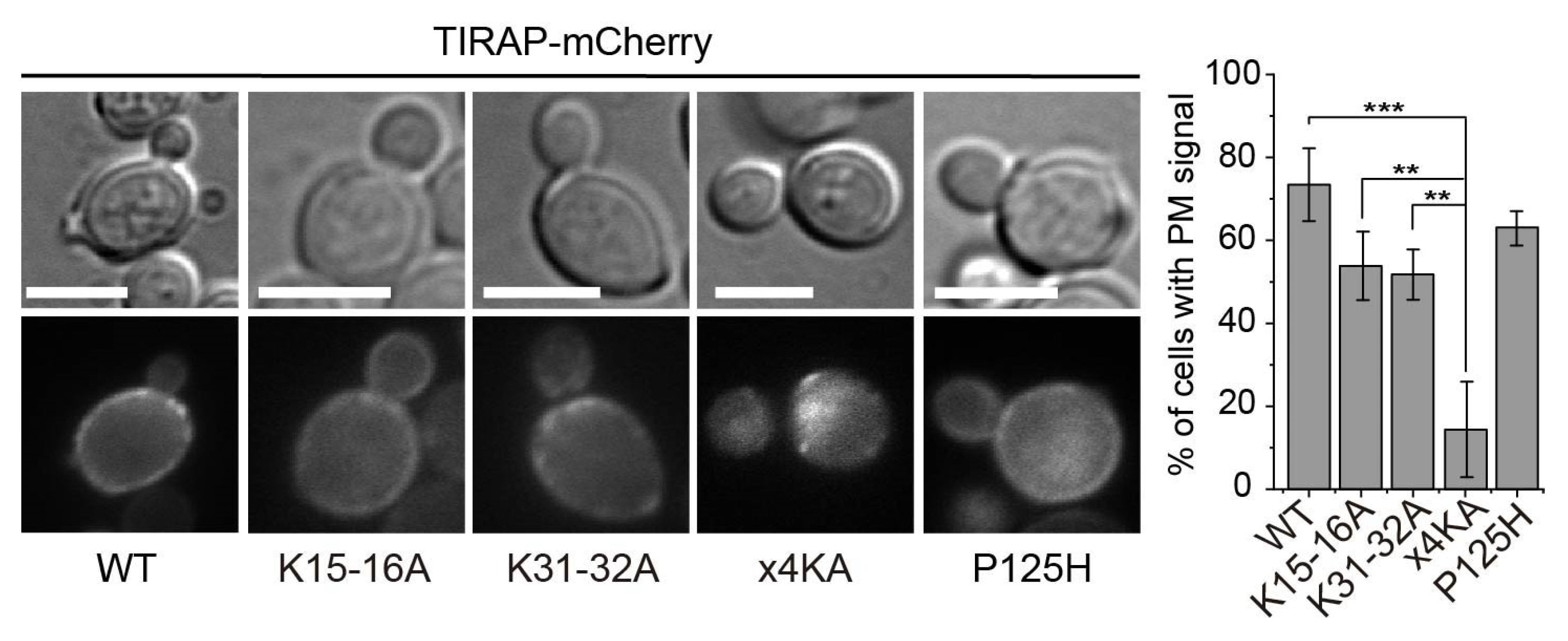
3.2. Lysine Residues in the Polybasic Motif of TIRAP Determine PM Localization in Yeast
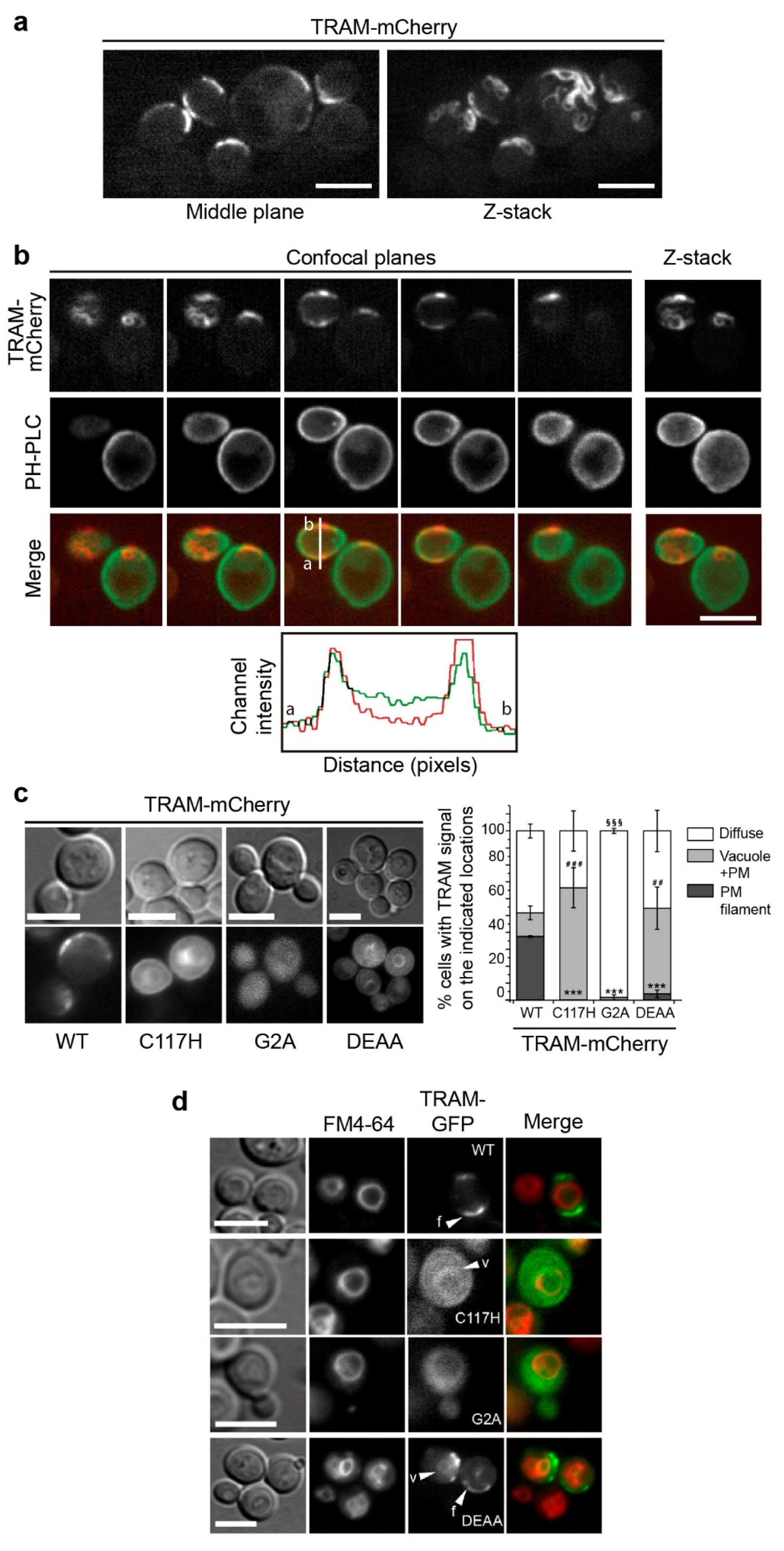
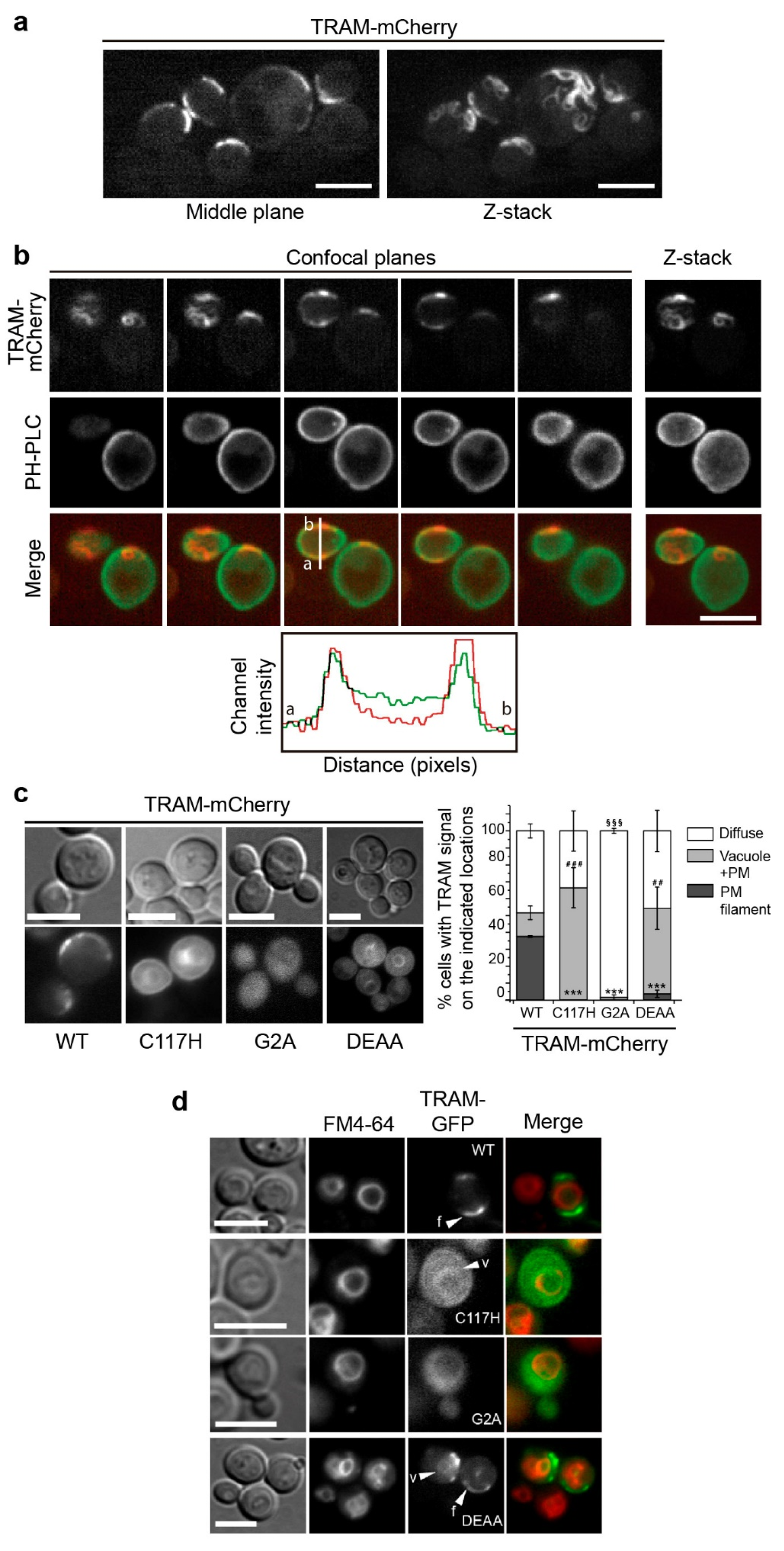
3.3. The TRAM Adaptor Forms Long PM-Tethered Filaments When Expressed in Yeast
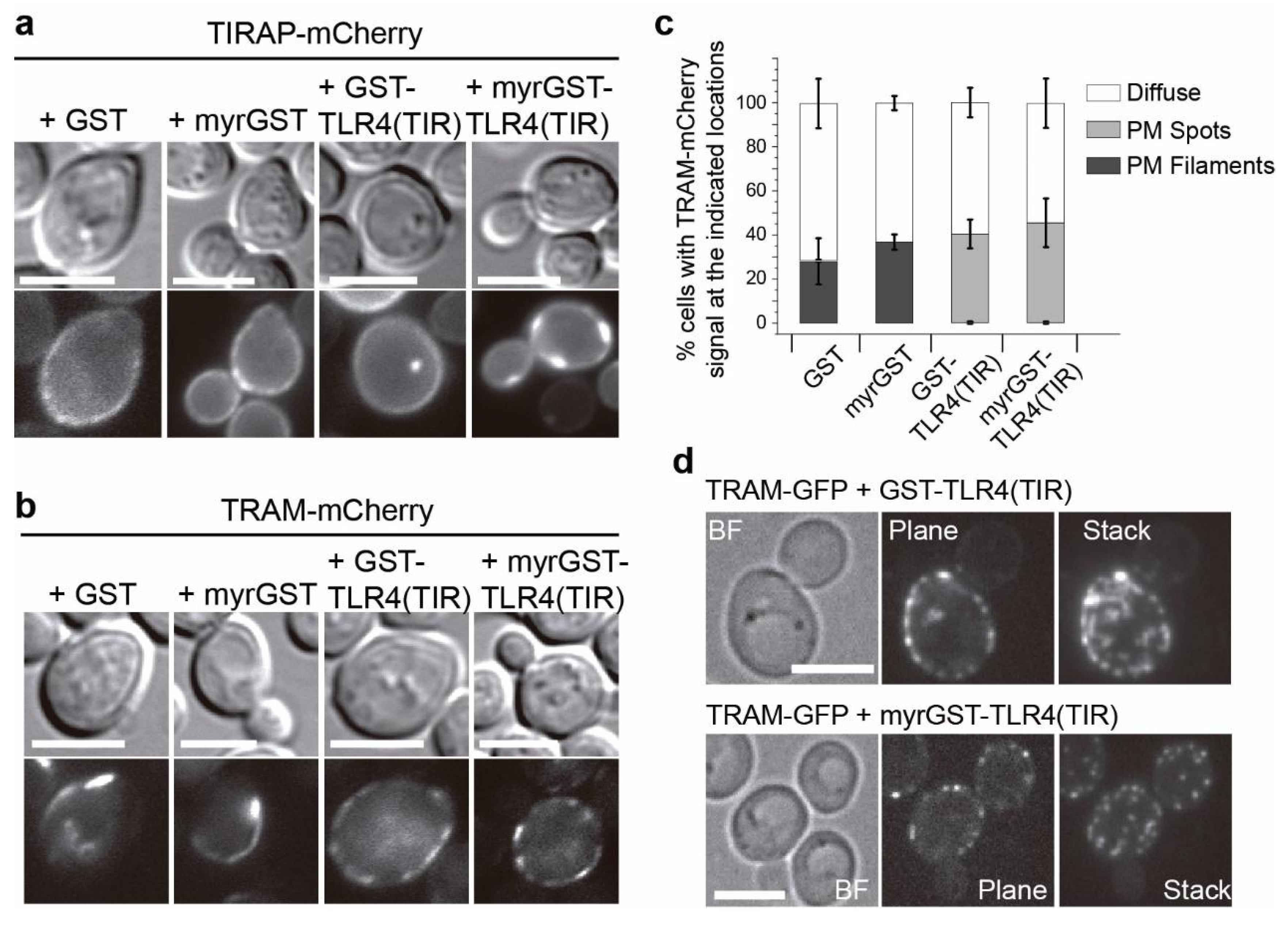
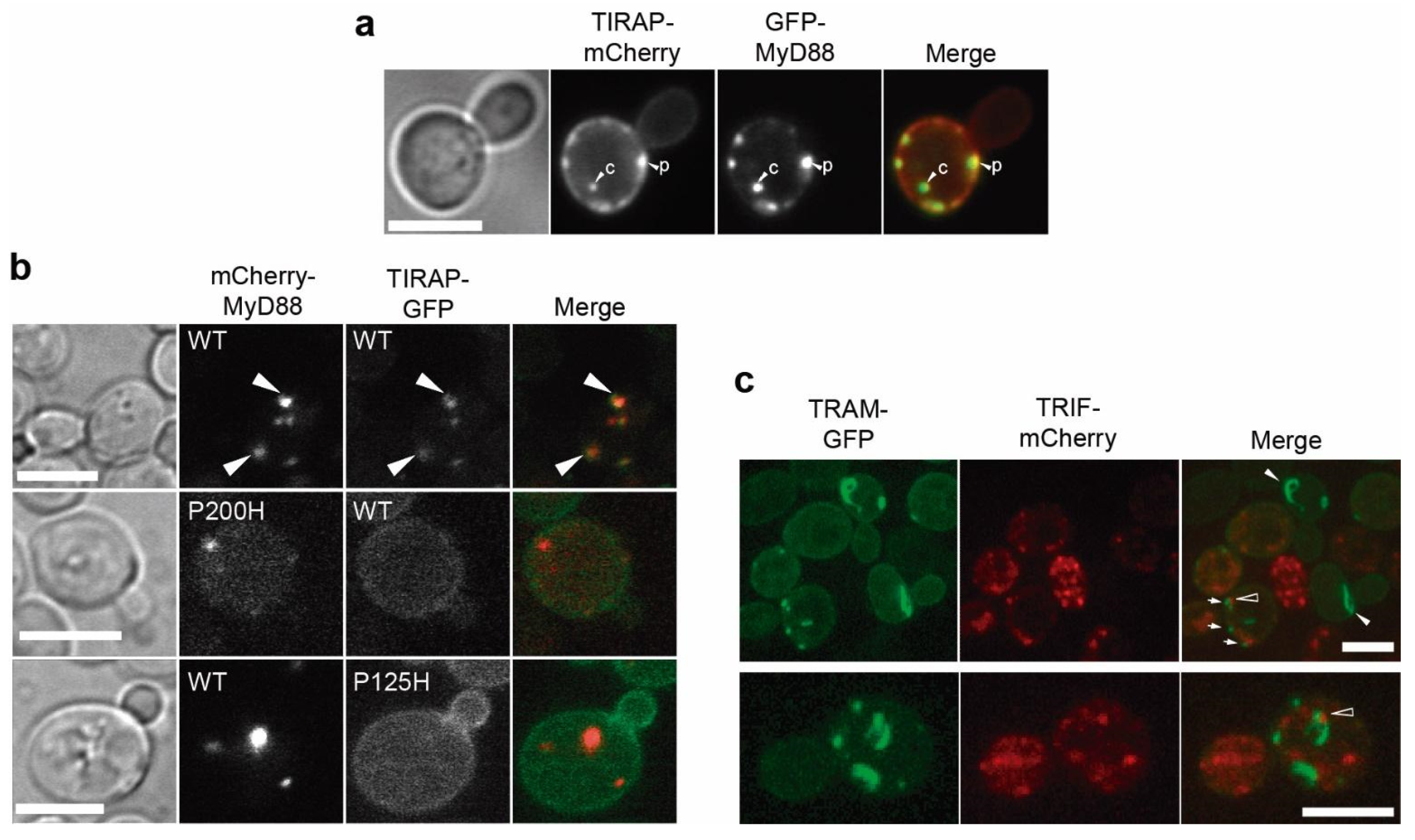
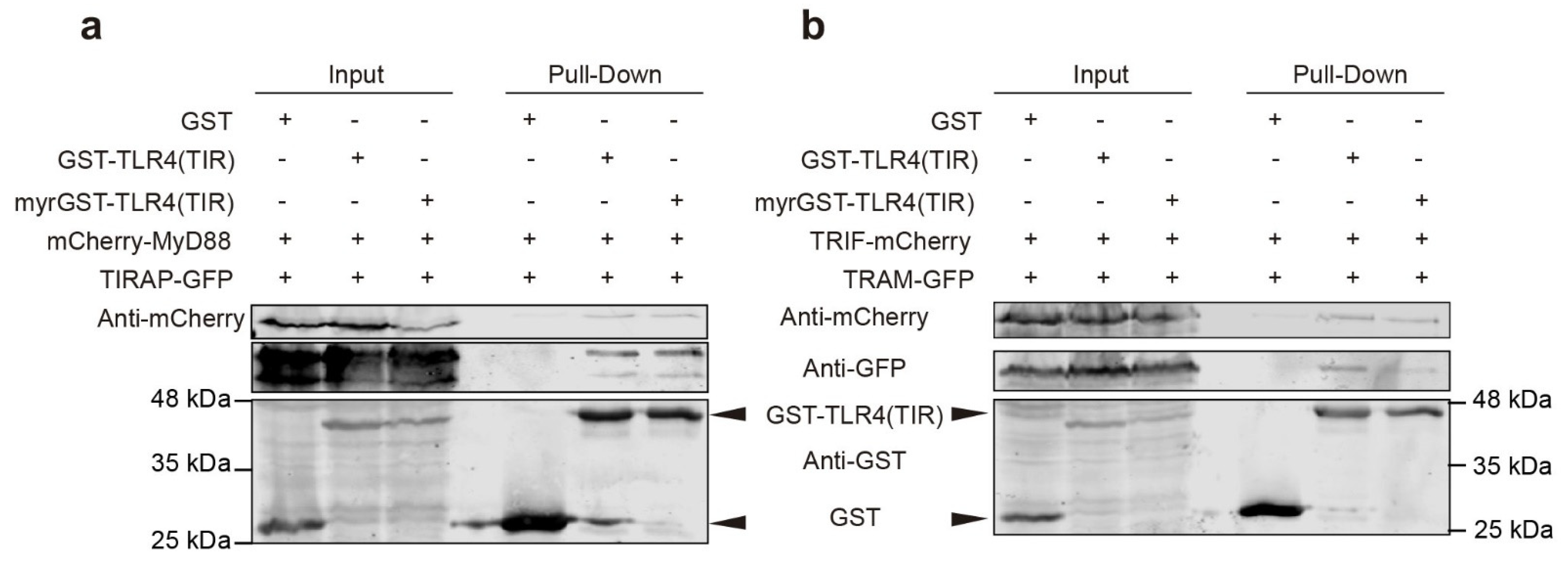
3.4. Co-Expression of TLR4 TIR Domain in Yeast Leads to Nucleation of TIRAP and TRAM In Vivo
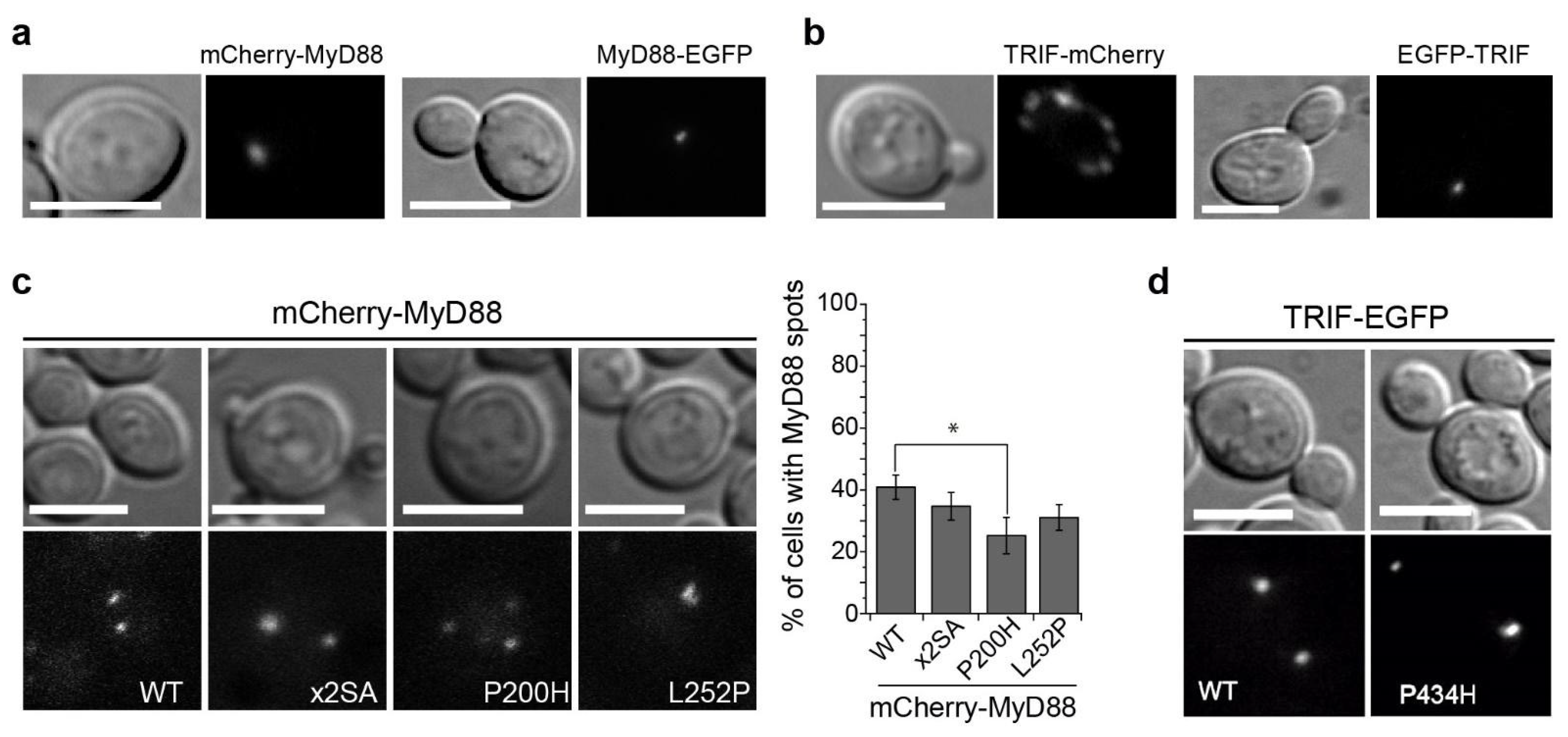
3.5. MyD88 and TRIF Nucleate at Cytoplasmic Spots, a Pattern Unaltered by Key TIR Mutations
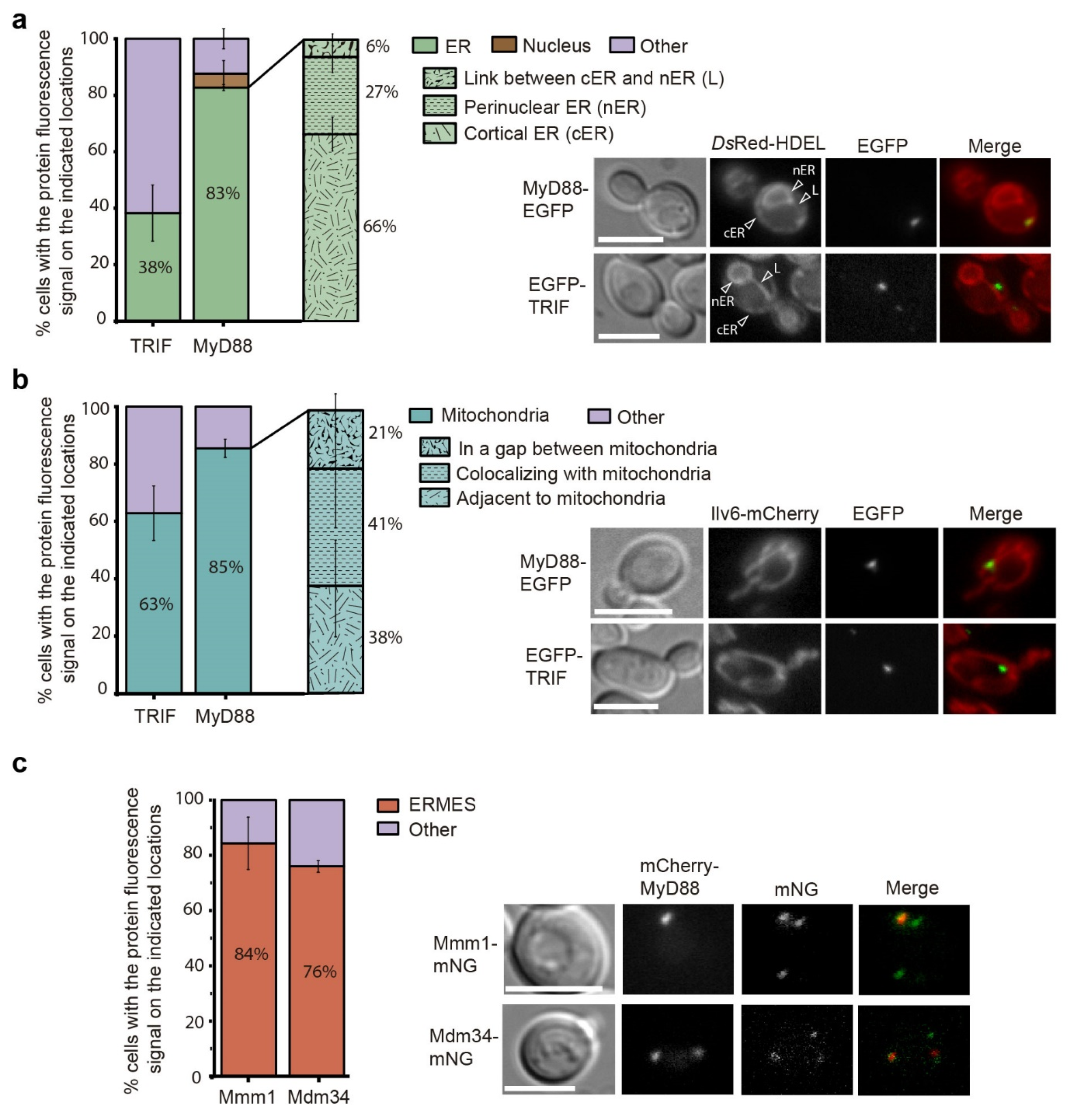
3.6. Human MyD88 Localizes to the Yeast ERMES
3.7. Co-Expression of TIRAP and MyD88 Leads to TIR-Dependent Mutual Recruitment
3.8. TIR-TIR Interactions Involved in SMOC Assembly Can Be Reproduced in Yeast
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kagan, J.C.; Magupalli, V.G.; Wu, H. SMOCs: Supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 2014, 14, 821–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Kagan, J.C. Innate Immune Signaling Organelles Display Natural and Programmable Signaling Flexibility. Cell 2019, 177, 384–398.e311. [Google Scholar] [CrossRef] [Green Version]
- Chuenchor, W.; Jin, T.; Ravilious, G.; Xiao, T.S. Structures of pattern recognition receptors reveal molecular mechanisms of autoinhibition, ligand recognition and oligomerization. Curr. Opin. Immunol. 2014, 26, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guven-Maiorov, E.; Keskin, O.; Gursoy, A.; VanWaes, C.; Chen, Z.; Tsai, C.J.; Nussinov, R. The Architecture of the TIR Domain Signalosome in the Toll-like Receptor-4 Signaling Pathway. Sci. Rep. 2015, 5, 13128. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-Mediated Adaptor Recruitment Controls Toll-like Receptor Signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Patra, M.C.; Choi, S. Insight into Phosphatidylinositol-Dependent Membrane Localization of the Innate Immune Adaptor Protein Toll/Interleukin 1 Receptor Domain-Containing Adaptor Protein. Front. Immunol. 2018, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyncke, L.; Bovijn, C.; Pauwels, E.; Van Acker, T.; Ruyssinck, E.; Burg, E.; Tavernier, J.; Peelman, F. Reconstructing the TIR Side of the Myddosome: A Paradigm for TIR-TIR Interactions. Structure 2016, 24, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ve, T.; Vajjhala, P.R.; Hedger, A.; Croll, T.; DiMaio, F.; Horsefield, S.; Yu, X.; Lavrencic, P.; Hassan, Z.; Morgan, G.P.; et al. Structural basis of TIR-domain-assembly formation in MAL- and MyD88-dependent TLR4 signaling. Nat. Struct. Mol. Biol. 2017, 24, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Latty, S.L.; Sakai, J.; Hopkins, L.; Verstak, B.; Paramo, T.; Berglund, N.A.; Cammarota, E.; Cicuta, P.; Gay, N.J.; Bond, P.J.; et al. Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub. Elife 2018, 7, e31377. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ou, J.; Wan, X. LRRC62 attenuates Toll-like receptor signaling by deubiquitinating TAK1 via CYLD. Exp. Cell Res. 2019, 383, 111497. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Doyle, S.L.; McGettrick, A.F.; Hardy, M.; Husebye, H.; Banahan, K.; Gong, M.; Golenbock, D.; Espevik, T.; O’Neill, L.A. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat. Immunol. 2009, 10, 579–586. [Google Scholar] [CrossRef]
- Zanoni, I.; Ostuni, R.; Marek Lorri, R.; Barresi, S.; Barbalat, R.; Barton Gregory, M.; Granucci, F.; Kagan Jonathan, C. CD14 Controls the LPS-Induced Endocytosis of Toll-like Receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marongiu, L.; Gornati, L.; Artuso, I.; Zanoni, I.; Granucci, F. Below the surface: The inner lives of TLR4 and TLR9. J. Leukoc. Biol. 2019, 106, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Rowe, D.C.; McGettrick, A.F.; Latz, E.; Monks, B.G.; Gay, N.J.; Yamamoto, M.; Akira, S.; O’Neill, L.A.; Fitzgerald, K.A.; Golenbock, D.T. The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc. Natl. Acad. Sci. USA 2006, 103, 6299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.O.; Sweet, M.J.; Mansell, A.; Kellie, S.; Kobe, B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J. Leukoc. Biol. 2016, 100, 27–45. [Google Scholar] [CrossRef]
- Matsumoto, M.; Funami, K.; Oshiumi, H.; Seya, T. Toll-IL-1-receptor-containing adaptor molecule-1: A signaling adaptor linking innate immunity to adaptive immunity. Prog. Mol. Biol. Transl. Sci. 2013, 117, 487–510. [Google Scholar]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romerio, A.; Peri, F. Increasing the chemical variety of small-molecule-based TLR4 modulators: An overview. Front. Immunol. 2020, 11, 1210. [Google Scholar] [CrossRef]
- Khurana, V.; Lindquist, S. Modelling neurodegeneration in Saccharomyces cerevisiae: Why cook with baker’s yeast? Nat. Rev. Neurosci. 2010, 11, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Acero, T.; Rodríguez-Escudero, I.; Molina, M.; Cid, V.J. The yeast cell wall integrity pathway signals from recycling endosomes upon elimination of phosphatidylinositol (4,5)-bisphosphate by mammalian phosphatidylinositol 3-kinase. Cell. Signal. 2015, 27, 2272–2284. [Google Scholar] [CrossRef] [PubMed]
- Coronas-Serna, J.M.; Fernández-Acero, T.; Molina, M.; Cid, V.J. A humanized yeast-based toolkit for monitoring phosphatidylinositol 3-kinase activity at both single cell and population levels. Microb. Cell 2018, 5, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.T.; Rhoads, S.N.; Yee, D.S.; Shewmaker, F.P. Yeast Models of Prion-Like Proteins That Cause Amyotrophic Lateral Sclerosis Reveal Pathogenic Mechanisms. Front. Mol. Neurosci. 2018, 11, 453. [Google Scholar] [CrossRef] [Green Version]
- Coronas-Serna, J.M.; Valenti, M.; Del Val, E.; Fernández-Acero, T.; Rodríguez-Escudero, I.; Mingo, J.; Luna, S.; Torices, L.; Pulido, R.; Molina, M.; et al. Modeling human disease in yeast: Recreating the PI3K-PTEN-Akt signaling pathway in Saccharomyces cerevisiae. Int. Microbiol. 2020, 23, 75–87. [Google Scholar] [CrossRef]
- Cervelli, T.; Galli, A. Yeast as a tool to understand the significance of human disease-associated gene variants. Genes 2021, 12, 1303. [Google Scholar] [CrossRef]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Janke, C.; Magiera, M.M.; Rathfelder, N.; Taxis, C.; Reber, S.; Maekawa, H.; Moreno-Borchart, A.; Doenges, G.; Schwob, E.; Schiebel, E.; et al. A versatile toolbox for PCR-based tagging of yeast genes: New fluorescent proteins, more markers and promoter substitution cassettes. Yeast 2004, 21, 947–962. [Google Scholar] [CrossRef]
- Alberti, S.; Gitler, A.D.; Lindquist, S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 2007, 24, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Escudero, I.; Roelants, F.M.; Thorner, J.; Nombela, C.; Molina, M.; Cid, V.J. Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast. Biochem. J. 2005, 390, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Vida, T.A.; Emr, S.D. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 1995, 128, 779–792. [Google Scholar] [CrossRef]
- Valenti, M.; Molina, M.; Cid, V.J. Heterologous Expression and Auto-Activation of Human Pro-Inflammatory Caspase-1 in Saccharomyces cerevisiae and Comparison to Caspase-8. Front. Immunol. 2021, 12, 2798. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Martín, H.; Rodríguez-Pachón, J.M.; Ruiz, C.; Nombela, C.; Molina, M. Regulatory mechanisms for modulation of signaling through the cell integrity Slt2-mediated pathway in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- De Craene, J.-O.; Bertazzi, D.L.; Bär, S.; Friant, S. Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 634. [Google Scholar] [CrossRef]
- Strahl, T.; Thorner, J. Synthesis and function of membrane phosphoinositides in budding yeast, Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1771, 353–404. [Google Scholar] [CrossRef] [Green Version]
- Coronas-Serna, J.M.; Louche, A.; Rodríguez-Escudero, M.; Roussin, M.; Imbert, P.R.C.; Rodríguez-Escudero, I.; Terradot, L.; Molina, M.; Gorvel, J.P.; Cid, V.J.; et al. The TIR-domain containing effectors BtpA and BtpB from Brucella abortus impact NAD metabolism. PLoS Pathog. 2020, 16, e1007979. [Google Scholar] [CrossRef] [Green Version]
- Garrenton, L.S.; Stefan, C.J.; McMurray, M.A.; Emr, S.D.; Thorner, J. Pheromone-induced anisotropy in yeast plasma membrane phosphatidylinositol-4,5-bisphosphate distribution is required for MAPK signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 11805–11810. [Google Scholar] [CrossRef] [Green Version]
- Athanasopoulos, A.; André, B.; Sophianopoulou, V.; Gournas, C. Fungal plasma membrane domains. FEMS Microbiol. Rev. 2019, 43, 642–673. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Piccolis, M.; Chiaruttini, N.; Riezman, I.; Riezman, H.; Roux, A.; Walther, T.C.; Loewith, R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 2012, 14, 542–547. [Google Scholar] [CrossRef]
- Funami, K.; Matsumoto, M.; Oshiumi, H.; Inagaki, F.; Seya, T. Functional interfaces between TICAM-2/TRAM and TICAM-1/TRIF in TLR4 signaling. Biochem. Soc. Trans. 2017, 45, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Liu, C.; Wang, L.; Gunawardena, H.P.; Yu, Y.; Du, R.; Taxman, D.J.; Dai, P.; Yan, Z.; Yu, J.; et al. Protein phosphatase 2A catalytic subunit α plays a MyD88-dependent, central role in the gene-specific regulation of endotoxin tolerance. Cell Rep. 2013, 3, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avbelj, M.; Wolz, O.O.; Fekonja, O.; Benčina, M.; Repič, M.; Mavri, J.; Krüger, J.; Schärfe, C.; Delmiro Garcia, M.; Panter, G.; et al. Activation of lymphoma-associated MyD88 mutations via allostery-induced TIR-domain oligomerization. Blood 2014, 124, 3896–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Zhan, C.; Qi, R.; Wei, G.; Guven-Maiorov, E.; Nussinov, R.; Ma, B. Conformational dynamics of cancer-associated MyD88-TIR domain mutant L252P (L265P) allosterically tilts the landscape toward homo-dimerization. Protein Eng. Des. Sel. 2016, 29, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Domingues, L.; Ismail, A.; Charro, N.; Rodríguez-Escudero, I.; Holden, D.W.; Molina, M.; Cid, V.J.; Mota, L.J. The Salmonella effector SteA binds phosphatidylinositol 4-phosphate for subcellular targeting within host cells. Cell. Microbiol. 2016, 18, 949–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragaz, C.; Pietsch, H.; Urwyler, S.; Tiaden, A.; Weber, S.S.; Hilbi, H. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell. Microbiol. 2008, 10, 2416–2433. [Google Scholar] [CrossRef]
- Weber, S.S.; Ragaz, C.; Reus, K.; Nyfeler, Y.; Hilbi, H. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2006, 2, e46. [Google Scholar] [CrossRef]
- Kornmann, B. The endoplasmic reticulum-mitochondria encounter structure: Coordinating lipid metabolism across membranes. Biol. Chem. 2020, 401, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Malcova, I.; Senohrabkova, L.; Novakova, L.; Hasek, J. eIF3a destabilization and TDP-43 alter dynamics of heat-induced stress granules. Int. J. Mol. Sci. 2021, 22, 5164. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; Yim, A.K.Y.; DiAntonio, A.; Milbrandt, J. TIR Domain Proteins Are an Ancient Family of NAD(+)-Consuming Enzymes. Curr. Biol. 2018, 28, 421–430.e424. [Google Scholar] [CrossRef] [Green Version]
- Bertin, A.; McMurray, M.A.; Thai, L.; Garcia, G., 3rd; Votin, V.; Grob, P.; Allyn, T.; Thorner, J.; Nogales, E. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J. Mol. Biol. 2010, 404, 711–731. [Google Scholar] [CrossRef] [Green Version]
- Enokizono, Y.; Kumeta, H.; Funami, K.; Horiuchi, M.; Sarmiento, J.; Yamashita, K.; Standley, D.M.; Matsumoto, M.; Seya, T.; Inagaki, F. Structures and interface mapping of the TIR domain-containing adaptor molecules involved in interferon signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 19908–19913. [Google Scholar] [CrossRef] [Green Version]
- Mahita, J.; Sowdhamini, R. Integrative modelling of TIR domain-containing adaptor molecule inducing interferon-β (TRIF) provides insights into its autoinhibited state. Biol. Direct 2017, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Funami, K.; Sasai, M.; Ohba, Y.; Oshiumi, H.; Seya, T.; Matsumoto, M. Spatiotemporal mobilization of Toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J. Immunol. 2007, 179, 6867–6872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidugu, V.K.; Pidugu, H.B.; Wu, M.-M.; Liu, C.-J.; Lee, T.-C. Emerging Functions of Human IFIT Proteins in Cancer. Front. Mol. Biosci. 2019, 6, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiya, T.; Kajita, E.; Horinouchi, T.; Nishimoto, A.; Miwa, S. Distinct roles of TIR and non-TIR regions in the subcellular localization and signaling properties of MyD88. FEBS Lett. 2007, 581, 3223–3229. [Google Scholar] [CrossRef] [Green Version]
- Shirane, M.; Wada, M.; Morita, K.; Hayashi, N.; Kunimatsu, R.; Matsumoto, Y.; Matsuzaki, F.; Nakatsumi, H.; Ohta, K.; Tamura, Y.; et al. Protrudin and PDZD8 contribute to neuronal integrity by promoting lipid extraction required for endosome maturation. Nat. Commun. 2020, 11, 4576. [Google Scholar] [CrossRef]
- Avbelj, M.; Panter, G.; Jerala, R. The role of N-terminal segment and membrane association in MyD88-mediated signaling. Biochem. Biophys. Res. Commun. 2018, 495, 878–883. [Google Scholar] [CrossRef]
- Connerth, M.; Tatsuta, T.; Haag, M.; Klecker, T.; Westermann, B.; Langer, T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 2012, 338, 815–818. [Google Scholar] [CrossRef]
- Lu, J.; Chan, C.; Yu, L.; Fan, J.; Sun, F.; Zhai, Y. Molecular mechanism of mitochondrial phosphatidate transfer by Ups1. Commun. Biol. 2020, 3, 468. [Google Scholar] [CrossRef] [PubMed]
- Clabbers, M.T.B.; Holmes, S.; Muusse, T.W.; Vajjhala, P.R.; Thygesen, S.J.; Malde, A.K.; Hunter, D.J.B.; Croll, T.I.; Flueckiger, L.; Nanson, J.D.; et al. MyD88 TIR domain higher-order assembly interactions revealed by microcrystal electron diffraction and serial femtosecond crystallography. Nat. Commun. 2021, 12, 2578. [Google Scholar] [CrossRef]
- Moncrieffe, M.C.; Bollschweiler, D.; Li, B.; Penczek, P.A.; Hopkins, L.; Bryant, C.E.; Klenerman, D.; Gay, N.J. MyD88 Death-Domain Oligomerization Determines Myddosome Architecture: Implications for Toll-like Receptor Signaling. Structure 2020, 28, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coronas-Serna, J.M.; del Val, E.; Kagan, J.C.; Molina, M.; Cid, V.J. Heterologous Expression and Assembly of Human TLR Signaling Components in Saccharomyces cerevisiae. Biomolecules 2021, 11, 1737. https://doi.org/10.3390/biom11111737
Coronas-Serna JM, del Val E, Kagan JC, Molina M, Cid VJ. Heterologous Expression and Assembly of Human TLR Signaling Components in Saccharomyces cerevisiae. Biomolecules. 2021; 11(11):1737. https://doi.org/10.3390/biom11111737
Chicago/Turabian StyleCoronas-Serna, Julia María, Elba del Val, Jonathan C. Kagan, María Molina, and Víctor J. Cid. 2021. "Heterologous Expression and Assembly of Human TLR Signaling Components in Saccharomyces cerevisiae" Biomolecules 11, no. 11: 1737. https://doi.org/10.3390/biom11111737
APA StyleCoronas-Serna, J. M., del Val, E., Kagan, J. C., Molina, M., & Cid, V. J. (2021). Heterologous Expression and Assembly of Human TLR Signaling Components in Saccharomyces cerevisiae. Biomolecules, 11(11), 1737. https://doi.org/10.3390/biom11111737