
Mitochondria-Endoplasmic Reticulum Crosstalk in Parkinson’s Disease: The Role of Brain Renin Angiotensin System Components


 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Parkinson’s Disease
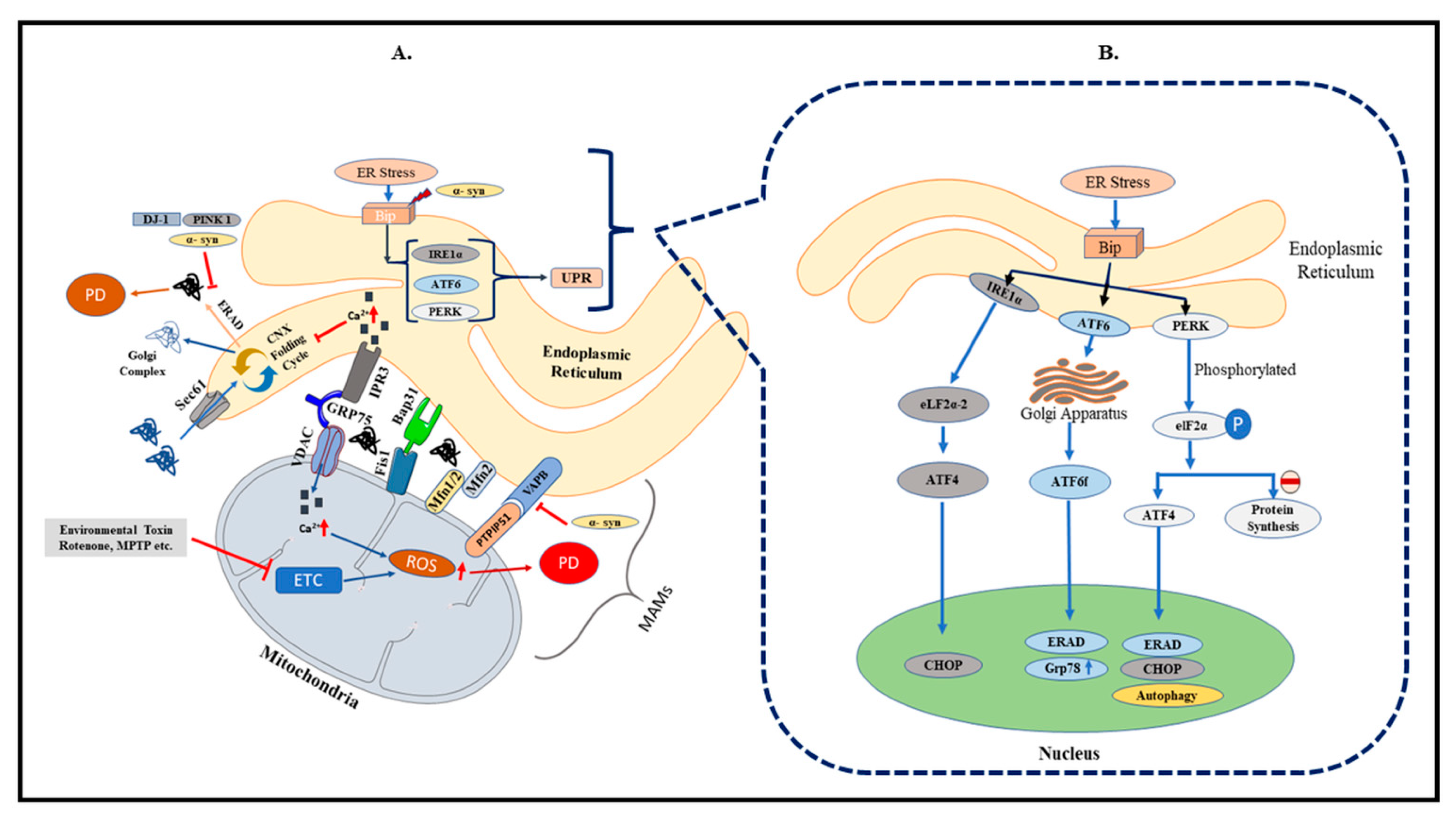
2. Endoplasmic Reticulum and PD
3. Mitochondria and PD
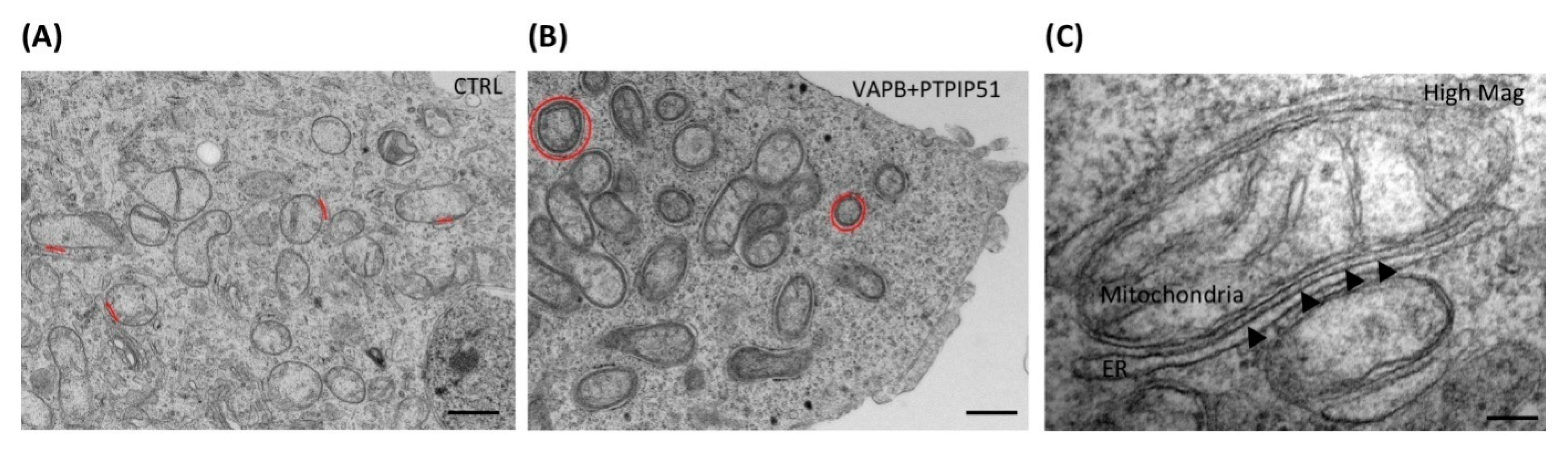
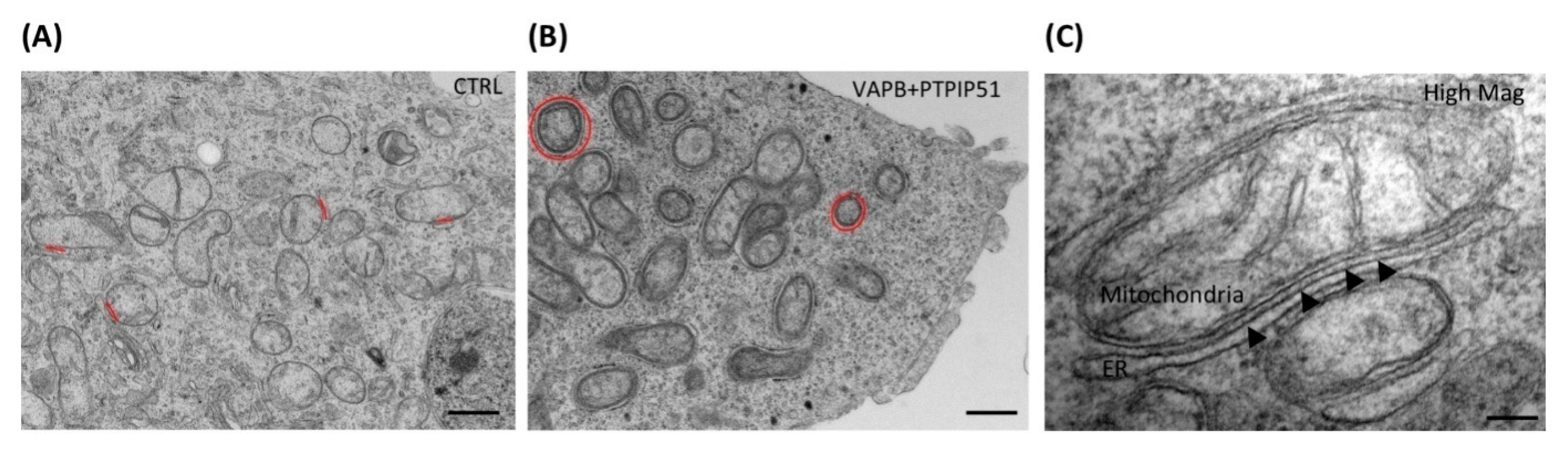
4. MAM in PD
5. PD-Related Gene Mutations and MAM
5.1. α-Syn
5.2. DJ 1
5.3. PINK1 or Parkin
5.4. LRRK2
6. Regulation of Calcium Signaling in MAM
7. Role of MAM in Neuroinflammation
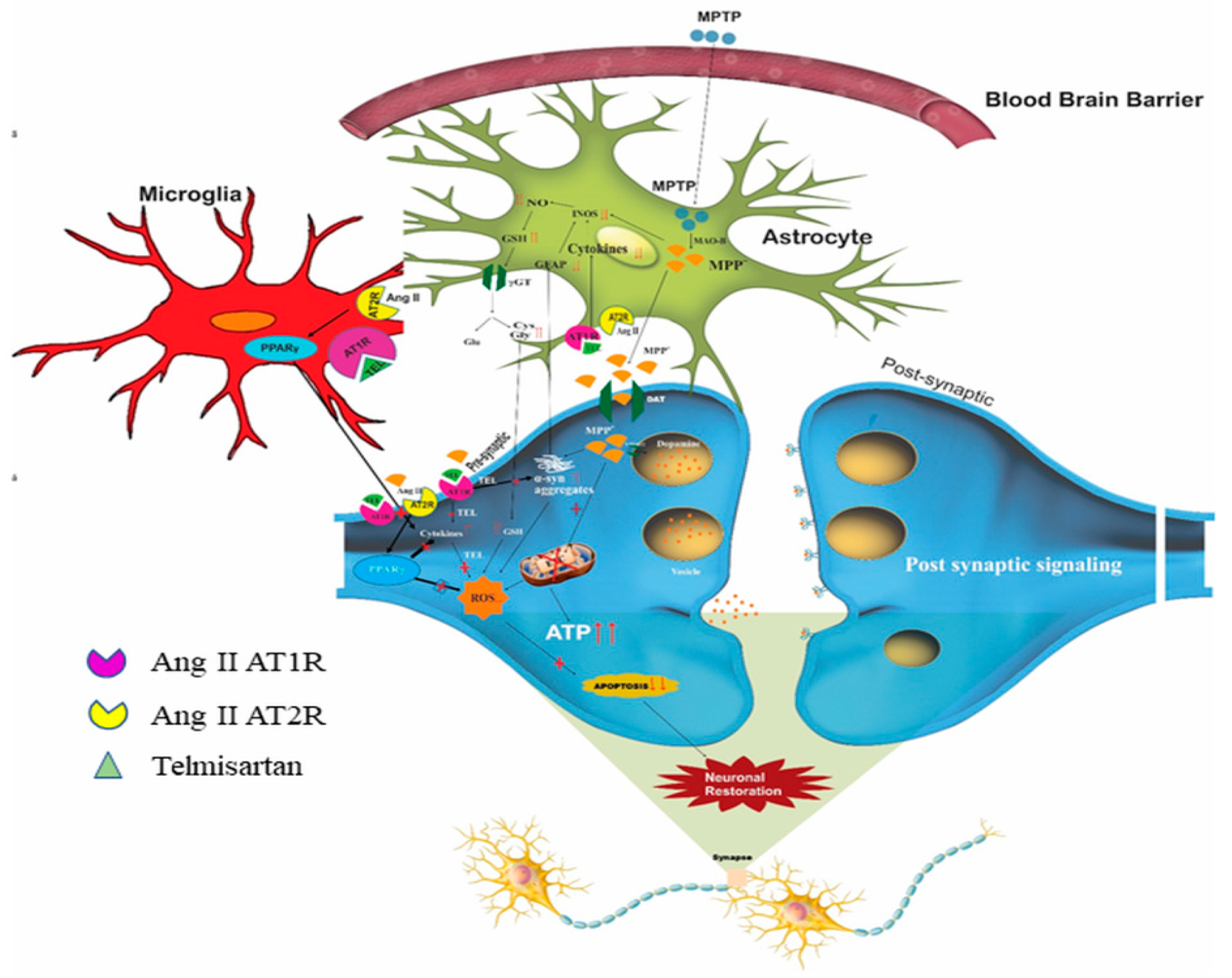
8. Brain RAS
9. Possible Impact of RAS on Mitochondria, ER, and MAM Interaction and Its Link to PD
10. Limitations
11. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| α-Syn | α-Synuclein |
| ACE | Angiotensin Converting Enzyme |
| AD | Alzheimer’s Disease |
| AMPK | 5′ Adenosine Monophosphate-Activated Protein Kinase |
| Ang II | Angiotensin II |
| ASC | Apoptosis Associated Speck-Like Protein Containing CARD |
| ASK1 | Apoptosis Signal-Regulating Kinase 1 |
| AT1/2 R | Angiotensin Receptors |
| ATF6 | Activating Transcription Factor 6 |
| ATP | Adenosine Triphosphate |
| AVV | Adeno-Associated Virus |
| Aβ | Amyloid β |
| Bap 31 | B Cell Receptor Associated Protein 31 |
| BAX | Bcl-2-Associated X Protein |
| BBB | Blood Brain Barrier |
| BDNF | Brain Derived Neurotrophic Factor |
| CHOP | C/EBP Homologous Protein |
| CNX | Calnexin |
| D1/D2 receptors | Dopaminergic |
| DAMPs | Damage Associated Molecular Patterns |
| DDIT3 | DNA Damage-Inducible Transcript 3 |
| Drp1 | Dynamine Related Protein 1 |
| eIF2a | Eukaryotic Translation Initiation Factor 2A |
| ER | Endoplasmic Reticulum |
| ERAD | ER-Associated Degradation |
| Ero1-α | Endoplasmic Reticulum Oxidoreductin 1-A |
| ETC | Electron Transport Chain |
| FADH2 | Flavin Adenine Dinucleotide |
| Fis 1 | Mitochondrial Fission 1 Protein |
| GADD153 | Growth Arrest and DNA Damage 153 |
| GDNF | Glial Cell-Derived Neurotrophic Factor |
| GII | Glucosidase |
| GRP78 | Glucose-Regulated Protein |
| GSH | Glutathione |
| GT | Glucosil Transferase |
| GTP | Guanosine Triphosphate |
| IL1β | Interleukin 1 β |
| IP3R | Inositol Trisphosphate Receptor |
| IRE-1 | Inositol-Requiring Enzyme 1 |
| JNK | c-Jun N-Terminal Kinase |
| LB | Lewy Body |
| L-DOPA | Levodopa |
| LRRK2 | Leucine-rich Repeat Kinase 2 |
| MAM | Mitochondrial Associated Membrane |
| MAPK | Mitogen-Activated Protein Kinase |
| MARCH5 | Membrane-Associated Ring Finger 5 |
| MFN1/2 | Mitofusins |
| MITOL | Mitochondrial Ubiquitin Ligase |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mPTP | Mitochondrial Permeability Transition Pore |
| mtDNA | Mitochondrial DNA |
| MULAN | Mitochondrial Ubiquitin Ligase Activator of NF-kB |
| NADH | Nicotinamide Adenine dinucleotide (NAD) + hydrogen (H) |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-kB | Nuclear Factor Kappa B |
| NLR | Nod Like Receptors |
| NLRP3 | (NOD)-like Receptor Protein 3 |
| NM | Neuromelanin |
| NMDA | N-Methyl-D Aspartate |
| NOX | NADPH Oxidase |
| OMM | Outer Mitochondrial Membrane |
| OPA1 | Protein Optic Atrophy 1 |
| OS | Oxidative Stress |
| PACS-2 | Phosphofurin Acidic Cluster Sorting Protein 2 |
| PAMP | Pathogen Associated Molecular Patters |
| PD | Parkinson’s Disease |
| PDI | Protein Disulfide Isomerise |
| PERK | Protein Kinase R-Like ER Kinase |
| PGC-1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1α |
| PINK 1 | PTEN-Induced Putative Kinase |
| p-Tau | Phosphorylated Tau |
| PTP1B | Protein Tyrosine Phosphatase1b |
| PTPIP51 | Tyrosine Phosphatase- Interacting Protein 51 |
| PUMA | p53 Upregulated Modulator of Apoptosis |
| RAS | Renin–Angiotensin System |
| RLR | RIG- I Receptors |
| ROS | Reactive oxygen Species |
| S1R | Sigma 1 Receptors |
| siRNA | Small interfering RNA |
| SNpc | Substantia Nigra Pars Compacta |
| SNPs | Single Neucleotide Polymorphism |
| SOD | Super Oxide Dismutase |
| TH | Tyosine Hydroxylase |
| TLR | Toll Like Receptors |
| TNF-α | Tumour Necrosis Factor α |
| TXNIP | Thioredoxin Interacting Protein |
| UCHL-1 | Ubiquitin Carboxy-Terminal Hydrolase L1 |
| UPR | Unfolded Protein Response |
| UPS | Ubiquitin Proteasome System |
| VAPB | Vesicle Associated Protein |
| VDAC | Voltage Dependent Anion Channel |
| XBP1 | X-box Binding Protein 1 |
References
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solari, N.; Bonito-Oliva, A.; Fisone, G.; Brambilla, R. Understanding cognitive deficits in Parkinson’s disease: Lessons from preclinical animal models. Learn. Mem. 2013, 20, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruck, A.; Kurki, T.; Kaasinen, V.; Vahlberg, T.; Rinne, J.O. Hippocampal and prefrontal atrophy in patients with early non-demented Parkinson’s disease is related to cognitive impairment. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1467–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, N.; Strafella, A.P. The neurobiology and neural circuitry of cognitive changes in Parkinson’s disease revealed by functional neuroimaging. Mov. Disord. 2012, 27, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huot, P.; Sgambato-Faure, V.; Fox, S.H.; McCreary, A.C. Serotonergic Approaches in Parkinson’s Disease: Translational Perspectives, an Update. ACS Chem. Neurosci. 2017, 8, 973–986. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell. Mol. Life Sci. 2018, 75, 3159–3180. [Google Scholar] [CrossRef] [Green Version]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, 2. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3281589/ (accessed on 15 January 2019). [CrossRef] [Green Version]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Arima, K.; Hirai, S.; Sunohara, N.; Aoto, K.; Izumiyama, Y.; Uéda, K.; Ikeda, K.; Kawai, M. Cellular co-localization of phosphorylated tau- and NACP/α-synuclein-epitopes in Lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res. 1999, 843, 53–61. [Google Scholar] [CrossRef]
- Clark, L.N.; Wang, Y.; Karlins, E.; Saito, L.; Mejia-Santana, H.; Harris, J.; Louis, E.D.; Cote, L.J.; Andrews, H.; Fahn, S.; et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 2006, 67, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-García, J.L.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Valenzuela, R.; Borrajo, A.; Rodríguez-Perez, A.I. Brain renin-angiotensin system and dopaminergic cell vulnerability. Front Neuroanat. 2014, 8, 67. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4086395/ (accessed on 22 October 2020). [CrossRef] [PubMed]
- Garrido-Gil, P.; Rodriguez-Perez, A.I.; Fernandez-Rodriguez, P.; Lanciego, J.L.; Labandeira-Garcia, J.L. Expression of angiotensinogen and receptors for angiotensin and prorenin in the rat and monkey striatal neurons and glial cells. Brain Struct. Funct. 2017, 222, 2559–2571. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Fewell, S.W.; Travers, K.J.; Weissman, J.S.; Brodsky, J.L. The Action of Molecular Chaperones in the Early Secretory Pathway. Annu. Rev. Genet. 2001, 35, 149–191. [Google Scholar] [CrossRef]
- Walter, P.; Blobel, G. Mechanism of protein translocation across the endoplasmic reticulum. Biochem. Soc. Symp. 1982, 47, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Takahashi, R. Expanding Insights on the Involvement of Endoplasmic Reticulum Stress in Parkinson’s Disease. Antioxid. Redox Signal. 2007, 9, 553–561. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Marciniak, S.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Hoozemans, J.; van Haastert, E.; Eikelenboom, P.; de Vos, R.; Rozemuller, J.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Duan, W.; Mattson, M.P. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. Res. 1999, 57, 195–206. [Google Scholar] [CrossRef]
- Baek, J.-H.; Whitfield, D.; Howlett, D.R.; Francis, P.T.; Bereczki, E.; Ballard, C.; Hortobágyi, T.; Attems, J.; Aarsland, D. Unfolded protein response is activated in Lewy body dementias. Neuropathol. Appl. Neurobiol. 2015, 42, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Hashida, K.; Kitao, Y.; Sudo, H.; Awa, Y.; Maeda, S.; Mori, K.; Takahashi, R.; Iinuma, M.; Hori, O. ATF6alpha Promotes Astroglial Activation and Neuronal Survival in a Chronic Mouse Model of Parkinson’s Disease. PLoS ONE 2012, 7, e47950. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3480445/ (accessed on 5 November 2021).
- Gorbatyuk, M.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.; Muzyczka, N.; Gorbatyuk, O.S. Glucose Regulated Protein 78 Diminishes α-Synuclein Neurotoxicity in a Rat Model of Parkinson Disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Ammal Kaidery, N.; Thomas, B. Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem. Int. 2018, 117, 91–113. [Google Scholar] [CrossRef]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.S.; Chandel, N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free. Radic. Biol. Med. 2009, 46, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaad, C.A.; Klann, E. Reactive Oxygen Species in the Regulation of Synaptic Plasticity and Memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [Green Version]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennet, J.P., Jr. Parkinson’s Disease Brain Mitochondrial Complex I Has Oxidatively Damaged Subunits and Is Functionally Impaired and Misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Müftüoglu, M.; Elibol, B.; Dalmızrak, Ö.; Ercan, A.; Kulaksız, G.; Ögüs, H.; Dalkara, T.; Ozer, N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord. 2004, 19, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Götz, M.; Dirr, A.; Kupsch, A.; Janetzky, B.; Oertel, W.; Sautter, J.; Schwarz, J.; Reichmann, H.; Riederer, P. Acute MPTP treatment produces no changes in mitochondrial complex activities and indices of oxidative damage in the common marmoset ex vivo one week after exposure to the toxin. Neurochem. Int. 1996, 28, 41–49. [Google Scholar] [CrossRef]
- Lin, M.T.; Cantuti-Castelvetri, I.; Zheng, K.; Ba, K.E.J.; Tan, Y.B.; Arzberger, T.; Lees, A.J.; Betensky, R.A.; Beal, M.F.; Simon, D.K. Somatic mitochondrial DNA mutations in early parkinson and incidental lewy body disease. Ann. Neurol. 2012, 71, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Cali’, T.; Ottolini, D.; Brini, M. Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. BioFactors 2011, 37, 228–240. [Google Scholar] [CrossRef]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in parkin-deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [Green Version]
- Rosen, K.M.; Veereshwarayya, V.; Moussa, C.E.H.; Fu, Q.; Goldberg, M.S.; Schlossmacher, M.G.; Shen, J.; Querfurth, H.W. Parkin protects against mitochondrial toxins and beta-amyloid accumulation in skeletal muscle cells. J. Biol. Chem. 2006, 281, 12809–12816. [Google Scholar] [CrossRef] [Green Version]
- Eschbach, J.; von Einem, B.; Müller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of PGC-1α deregulation and α-synuclein oligomerization. Ann. Neurol. 2015, 77, 15–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at? Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2016, 22, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870. [Google Scholar] [CrossRef] [PubMed]
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 2016, 6, 32656. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Reed, J.R.; Brignac-Huber, L.M.; Backes, W.L. Cytochrome P450 system proteins reside in different regions of the endoplasmic reticulum. Biochem. J. 2014, 464, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Rusiñol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [CrossRef]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Stone, S.J.; Levin, M.C.; Zhou, P.; Han, J.; Walther, T.C.; Farese, R.V. The endoplasmic reticulum enzyme DGAT2 is found in mito-chondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 2009, 284, 5352–5361. [Google Scholar] [CrossRef] [Green Version]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.; Mueller, S.; Miller, T.; Miller, C.C. There’s Something Wrong with my MAM; the ER–Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Min, K.-T. The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endo-plasmic reticulum and mitochondrial Ca2+ channels. J. Cell. Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2010, 30, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arduíno, D.M.; Esteves, A.R.; Cardoso, S.M.; Oliveira, C.R. Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: Relevance to Parkinson’s disease. Neurochem. Int. 2009, 55, 341–348. [Google Scholar] [CrossRef]
- Chan, C.; Guzman, J.; Ilijic, E.; Mercer, J.; Rick, C.; Tkach, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochon-dria-associated ER membranes. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tamura, N.; Kono, N.; Shimanaka, Y.; Arai, H.; Yamamoto, H.; Mizushima, N. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 2017, 36, 1719–1735. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+ transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 2013, 1832, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of Novel Proteins Associated with Both α-Synuclein and DJ-1. Mol. Cell. Proteom. 2007, 6, 845–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res. Bull. 2017, 133, 51–59. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol Cell. 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. Available online: https://www.nature.com/articles/nature07534 (accessed on 11 April 2019). [CrossRef]
- Mourier, A.; Motori, E.; Brandt, T.; Lagouge, M.; Atanassov, I.; Galinier, A.; Rappl, G.; Brodesser, S.; Hultenby, K.; Dieterich, C.; et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 2015, 208, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin functionally interacts with PGC-1α to preserve mitochondria and protect dopaminergic neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef] [Green Version]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. A new role for α-synuclein in Parkinson’s disease: Alteration of ER-mitochondrial communication. Mov. Disord. 2015, 30, 1026–1033. [Google Scholar] [CrossRef]
- Li, W.-W.; Yang, R.; Guo, J.-C.; Ren, H.-M.; Zha, X.-L.; Cheng, J.-S.; Cai, D.-F. Localization of α-synuclein to mitochondria within midbrain of mice. NeuroReport 2007, 18, 1543–1546. [Google Scholar] [CrossRef]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid Rafts Mediate the Synaptic Localization of α-Synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [Green Version]
- Mullin, S.; Schapira, A. α-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Suaga, P.; Pedro, J.M.B.-S.; González-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER–mitochondria signaling in Parkinson’s disease. Cell Death Dis. 2018, 9, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Nguyen, J.L.; Hulleman, J.; Li, L.; Rochet, J.-C. Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson’s disease. J. Neurochem. 2008, 105, 2435–2453. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, E.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with au-tosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waak, J.; Weber, S.S.; Görner, K.; Schall, C.; Ichijo, H.; Stehle, T.; Kahle, P.J. Oxidizable Residues Mediating Protein Stability and Cyto-protective Interaction of DJ-1 with Apoptosis Signal-regulating Kinase. J. Biol. Chem. 2009, 284, 14245–14257. [Google Scholar] [CrossRef] [Green Version]
- Blackinton, J.; Lakshminarasimhan, M.; Thomas, K.J.; Ahmad, R.; Greggio, E.; Raza, A.S.; Cookson, M.R.; Wilson, M.A. Formation of a Stabilized Cysteine Sulfinic Acid Is Critical for the Mitochondrial Function of the Parkinsonism Protein DJ-1. J. Biol. Chem. 2009, 284, 6476–6485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Konovalova, E.V.; Lopacheva, O.M.; Grivennikov, I.A.; Lebedeva, O.S.; Dashinimaev, E.B.; Khaspekov, L.G.; Fedotova, E.Y.; Illarioshkin, S. Mutations in the Parkinson’s Disease-Associated PARK2 Gene are Accompanied by Imbalance in Programmed Cell Death Systems. Acta Naturae 2015, 7, 146–149. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelmetti, V.; de Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mito-chondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jy, Y.; Wy, Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4, 2428. [Google Scholar]
- Van Laar, V.S.; Roy, N.; Liu, A.; Rajprohat, S.; Arnold, B.; Dukes, A.A.; Holbein, C.D.; Berman, S.B. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 2015, 74, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef] [Green Version]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Malluci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegen-eration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N.; et al. MITOL Regulates Endoplasmic Reticu-lum-Mitochondria Contacts via Mitofusin2. Mol. Cell. 2013, 51, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Yamano, K.; Kosako, H.; Kimura, Y.; Kimura, M.; Fujiki, Y.; Tanaka, K.; Matsuda, N. Parkin-mediated ubiquitylation redistributes MI-TOL/March5 from mitochondria to peroxisomes. EMBO Rep. 2019, 20, e47728. [Google Scholar] [CrossRef]
- Bardien, S.; Lesage, S.; Brice, A.; Carr, J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Park. Relat. Disord. 2011, 17, 501–508. [Google Scholar] [CrossRef]
- Krumova, P.; Reyniers, L.; Meyer, M.; Lobbestael, E.; Stauffer, D.; Gerrits, B.; Muller, L.; Hoving, S.; Kaupmann, K.; Voshol, J.; et al. Chemical genetic approach identifies microtubule affinity-regulating kinase 1 as a leucine-rich repeat kinase 2 substrate. FASEB J. 2015, 29, 2980–2992. [Google Scholar] [CrossRef] [Green Version]
- Taymans, J.-M.; Nkiliza, A.; Chartier-Harlin, M.-C. Deregulation of protein translation control, a potential game-changing hy-pothesis for Parkinson’s disease pathogenesis. Trends Mol. Med. 2015, 21, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stafa, K.; Tsika, E.; Moser, R.; Musso, A.; Glauser, L.; Jones, A.; Biskup, S.; Xiong, Y.; Bandopadhyay, R.; Dawson, V.L.; et al. Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 2014, 23, 2055–2077. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.A.; Jansson, J.; Rocha, E.M.; Osborn, T.; Hallett, P.J.; Isacson, O. Fibroblast Biomarkers of Sporadic Parkinson’s Disease and LRRK2 Kinase Inhibition. Mol. Neurobiol. 2016, 53, 5161–5177. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 regulates endoplasmic reticulum–mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 2019, 39, e100875. [Google Scholar]
- Biagioli, M.; Pifferi, S.; Ragghianti, M.; Bucci, S.; Rizzuto, R.; Pinton, P. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 2008, 43, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.-L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [Green Version]
- Lebiedzinska, M.; Szabadkai, G.; Jones, A.W.; Duszynski, J.; Wieckowski, M.R. Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int. J. Biochem. Cell Biol. 2009, 41, 1805–1816. [Google Scholar] [CrossRef]
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucose-regulated protein 75 determines ER–mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017, 3, 17076. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittenhouse, A.R.; Zigmond, R.E. Role of N- and L-type calcium channels in depolarization-induced activation of tyrosine hy-droxylase and release of norepinephrine by sympathetic cell bodies and nerve terminals. J. Neurobiol. 1999, 40, 137–148. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different Species of -Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef]
- Chan, S.L.; Mattson, M.P. Caspase and calpain substrates: Roles in synaptic plasticity and cell death. J. Neurosci. Res. 1999, 58, 167–190. [Google Scholar] [CrossRef]
- Samantaray, S.; Ray, S.K.; Banik, N.L. Calpain as a potential therapeutic target in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2008, 7, 305–312. [Google Scholar] [CrossRef]
- Allen, A.S.; Satten, G.A. A novel haplotype-sharing approach for genome-wide case-control association studies implicates the calpastatin gene in Parkinson’s disease. Genet. Epidemiol. 2009, 33, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.-C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol. Aging 2007, 28, 639–647. [Google Scholar] [CrossRef] [PubMed]
- McGeer, E.G.; McGeer, P.L. The Role of Anti-Inflammatory Agents in Parkinson’s Disease. CNS Drugs 2007, 21, 789–797. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Guthrie, L.N.; Abiraman, K.; Plyler, E.S.; Sprenkle, N.T.; Gibson, S.A.; McFarland, B.C.; Rajbhandari, R.; Rowse, A.L.; Benveniste, E.N.; Meares, G.P. Attenuation of PKR-like ER Kinase (PERK) Signaling Selectively Controls Endoplasmic Reticulum Stress-induced Inflammation Without Compromising Immunological Responses. J. Biol. Chem. 2016, 291, 15830–15840. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Duffy, P.E.; Tennyson, V.M. Phase and Electron Microscopic Observations of Lewy Bodies and Melanin Granules in the Substantia Nigra and Locus Caeruleus in Parkinson’s Disease. J. Neuropathol. Exp. Neurol. 1965, 24, 398–414. [Google Scholar] [CrossRef]
- Wilms, H.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Zecca, L.; Lucius, R. Activation of microglia by human neuromelanin is NF-κB-dependent and involves p38 mitogen-activated protein kinase: Implications for Parkinson’s disease. FASEB J. 2003, 17, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yan, Z.-F.; Gao, J.-H.; Sun, L.; Huang, X.-Y.; Liu, Z.; Yu, S.-Y.; Cao, C.-J.; Zuo, L.-J.; Chen, Z.-J.; et al. Role and Mechanism of Microglial Activation in Iron-Induced Selective and Progressive Dopaminergic Neurodegeneration. Mol. Neurobiol. 2014, 49, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groß, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome Activators Induce Interleukin-1α Secretion via Distinct Pathways with Differential Requirement for the Protease Function of Caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef] [Green Version]
- Hui-Ming, G.; Feng, Z.; Hui, Z.; Wayneho, K.; Wilson, B.; Hong, J.S. Neuroinflammation and α-Synuclein Dysfunction Potentiate Each Other, Driving Chronic Progression of Neurodegeneration in a Mouse Model of Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 807–814. [Google Scholar]
- Sarkar, C.; Basu, B.; Chakroborty, D.; Dasgupta, P.S.; Basu, S. The immunoregulatory role of dopamine: An update. Brain, Behav. Immun. 2010, 24, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
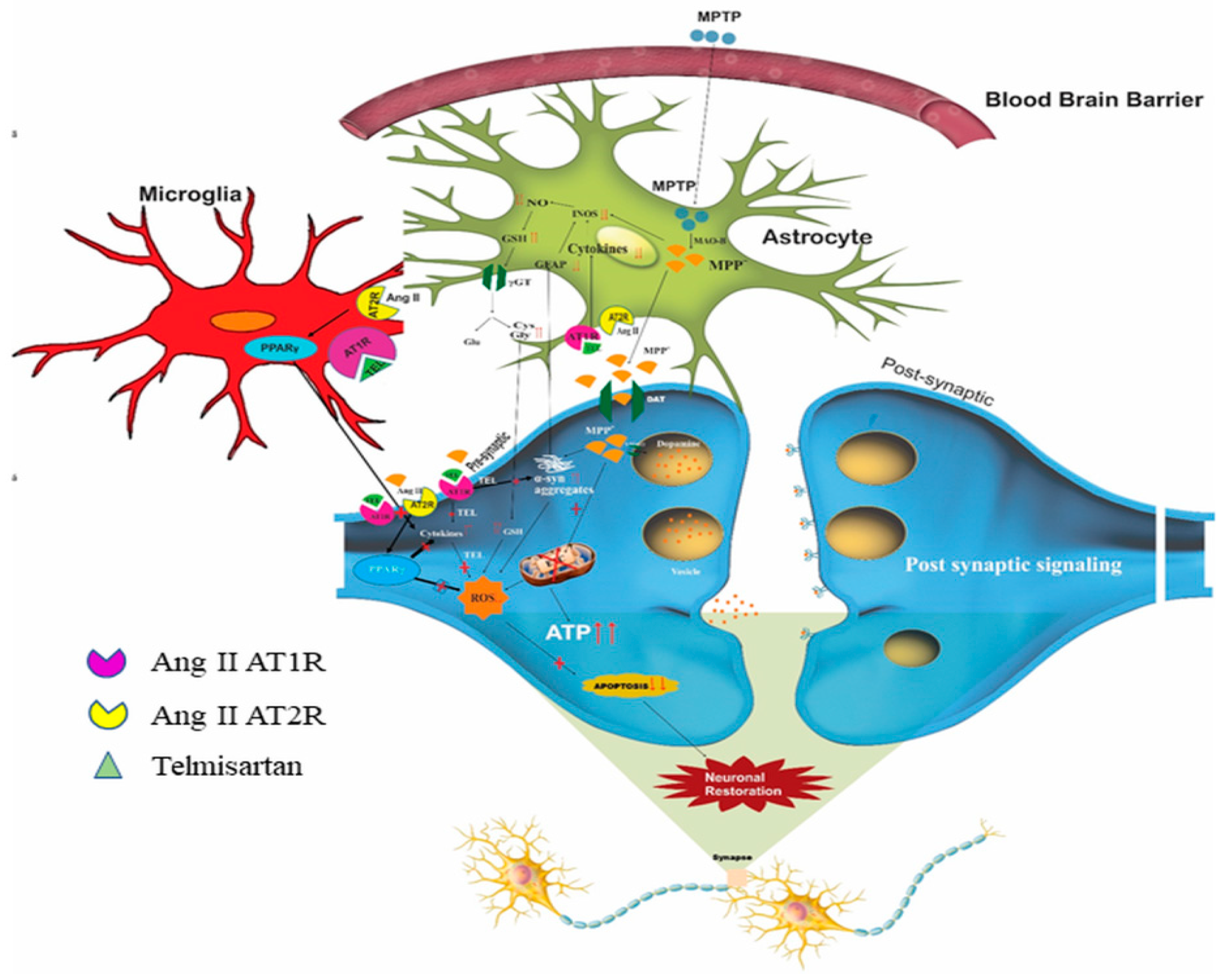
- Joglar, B.; Rodriguez-Pallares, J.; Rodriguez-Perez, A.I.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: Relevance to progression of the disease. J. Neurochem. 2009, 109, 656–669. [Google Scholar] [CrossRef]
- Sathiya, S.; Ranju, V.; Kalaivani, P.; Priya, R.J.; Sumathy, H.; Sunil, A.G.; Babu, S.C. Telmisartan attenuates MPTP induced dopaminergic degeneration and motor dysfunction through regulation of α-synuclein and neurotrophic factors (BDNF and GDNF) ex-pression in C57BL/6J mice. Neuropharmacology 2013, 73, 98–110. [Google Scholar] [CrossRef]
- Lenkei, Z.; Palkovits, M.; Corvol, P.; Cortèsa, C. Expression of Angiotensin Type-1 (AT1) and Type-2 (AT2) Receptor mRNAs in the Adult Rat Brain: A Functional Neuroanatomical Review. Front. Neuroendocr. 1997, 18, 383–439. [Google Scholar] [CrossRef]
- Allen, A.M.; Zhuo, J.; Mendelsohn, F.A. Localization and function of angiotensin AT1 receptors. Am. J. Hypertens. 2000, 13, S31–S38. [Google Scholar] [CrossRef] [Green Version]
- Grammatopoulos, T.N.; Jones, S.M.; Ahmadi, F.A.; Hoover, B.R.; Snell, L.D.; Skoch, J.; Jhaveri, V.V.; Poczobutt, A.M.; Weyhenmeyer, J.A.; Zawada, W.M. Angiotensin type 1 receptor antagonist losartan, reduces MPTP-induced degeneration of dopaminergic neurons in substantia nigra. Mol. Neurodegener. 2007, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, S.; Mani, S.; Rajamani, B.; Manivasagam, T.; Thenmozhi, A.J.; Bhat, A.; Ray, B.; Essa, M.M.; Guillemin, G.J.; Chidambaram, S.B. Telmisartan Ameliorates Astroglial and Dopa-minergic Functions in a Mouse Model of Chronic Parkinsonism. Neurotox. Res. 2018, 34, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Garcia, J.L.; Rodríguez-Perez, A.I.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Lanciego, J.L.; Guerra, M.J. Brain Ren-in-Angiotensin System and Microglial Polarization: Implications for Aging and Neurodegeneration. Front Aging Neurosci. 2017, 9, 129. Available online: https://www.frontiersin.org/articles/10.3389/fnagi.2017.00129/full (accessed on 5 March 2019). [CrossRef] [Green Version]
- Rogerson, F.M.; Schlawe, I.; Paxinos, G.; Chai, S.Y.; McKinley, M.J.; Mendelsohn, F.A. Localization of angiotensin converting enzyme by in vitro autoradiography in the rabbit brain. J. Chem. Neuroanat. 1995, 8, 227–243. [Google Scholar] [CrossRef]
- Li, W.; Peng, H.; Cao, T.; Sato, R.; McDaniels, S.J.; Kobori, H.; Navar, G.; Feng, Y. Brain-Targeted (Pro)renin Receptor Knockdown Attenuates Angiotensin II–Dependent Hypertension. Hypertension 2012, 59, 1188–1194. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. The brain renin–angiotensin system: A diversity of functions and implications for CNS diseases. Pflügers Arch. Eur. J. Physiol. 2013, 465, 133–151. [Google Scholar] [CrossRef]
- Reardon, K.A.; Mendelsohn, F.A.O.; Chai, S.Y.; Horne, M.K. The angiotensin converting enzyme (ACE) inhibitor, perindopril, modifies the clinical features of Parkinson’s disease. Aust. N. Zealand J. Med. 2000, 30, 48–53. [Google Scholar] [CrossRef]
- Farrer, L.A.; Sherbatich, T.; Keryanov, S.A.; Korovaitseva, G.I.; Rogaeva, E.A.; Petruk, S. Association between angioten-sin-converting enzyme and Alzheimer disease. Arch Neurol. 2000, 57, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Gil, P.; Valenzuela, R.; Villar-Cheda, B.; Lanciego, J.L.; Labandeira-Garcia, J.L. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: An intracellular renin–angiotensin system in the nigra. Anat. Embryol. 2012, 218, 373–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadir, P.M. The Frail Renin-Angiotensin System. Clin. Geriatr. Med. 2011, 27, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Cowan, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mech-anisms and Relevance for Cardiovascular Disease. Antioxid. Redox. Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Costa-Besada, A.M.; Iglesias-Gonzalez, J.; Perez-Costas, E.; Villar-Cheda, B.; Garrido-Gil, P.; Melendez-Ferro, M.; Soto-Otero, R.; Lanciego, J.L.; Henrion, D.; et al. Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 2016, 7, e2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar-Cheda, B.; Costa-Besada, M.A.; Valenzuela, R.; Perez-Costas, E.; Melendez-Ferro, M.; Labandeira-Garcia, J.L. The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis. 2017, 8, e3044. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.; Barnes, N.M. Alterations in angiotensin AT1 and AT2 receptor subtype levels in brain regions from patients with neu-rodegenerative disorders. Eur. J. Pharmacol. 1996, 297, 299–306. [Google Scholar] [CrossRef]
- Zawada, W.M.; Mrak, R.E.; Biedermann, J.; Palmer, Q.D.; Gentleman, S.M.; Aboud, O.; Griffin, W.S.T. Loss of angiotensin II receptor expression in dopamine neurons in Parkinson’s disease correlates with pathological progression and is accompanied by increases in Nox4- and 8-OH guanosine-related nucleic acid oxidation and caspase-3 activation. Acta Neuropathol. Commun. 2015, 3, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, A.; Stegbauer, J.; Linker, R.A. Macrophages in neuroinflammation: Role of the renin-angiotensin-system. Pflügers Arch. Eur. J. Physiol. 2017, 469, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Parekh, R.U.; Robidoux, J.; Sriramula, S. Kinin B1 Receptor Blockade Prevents Angiotensin II-induced Neuroinflammation and Oxidative Stress in Primary Hypothalamic Neurons. Cell. Mol. Neurobiol. 2020, 40, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Coleman, C.G.; Chan, J.; Faraco, G.; Marques-Lopes, J.; Milner, T.A.; Guruju, M.R.; Anrather, J.; Davisson, R.L.; Iadecola, C.; et al. Angiotensin II slow-pressor hypertension enhances NMDA currents and NOX2-dependent superoxide production in hypothalamic paraventricular neurons. Am. J. Physiol. Integr. Comp. Physiol. 2013, 304, R1096–R1106. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Real, A.; Rey, P.; Soto-Otero, R.; Méndez-Álvarez, E.; Labandeira-Garcia, J. Angiotensin-converting enzyme inhibition reduces oxidative stress and protects dopaminergic neurons in a 6-hydroxydopamine rat model of Parkinsonism. J. Neurosci. Res. 2005, 81, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Sonsalla, P.K.; Coleman, C.; Wong, L.-Y.; Harris, S.L.; Richardson, J.R.; Gadad, B.S.; Li, W.; German, D.C. The angiotensin converting enzyme inhibitor captopril protects nigrostriatal dopamine neurons in animal models of parkinsonism. Exp. Neurol. 2013, 250, 376–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Pallares, J.; Rey, P.; Parga, J.; Muñoz, A.; Guerra, M.; Labandeira-Garcia, J. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol. Dis. 2008, 31, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, A.I.; Dominguez-Meijide, A.; Lanciego, J.L.; Guerra, M.J.; Labandeira-Garcia, J.L. Dopaminergic degeneration is enhanced by chronic brain hypoperfusion and inhibited by angiotensin receptor blockage. AGE 2013, 35, 1675–1690. [Google Scholar] [CrossRef] [Green Version]
- Grammatopoulos, T.N.; Outeiro, T.F.; Hyman, B.T.; Standaert, D.G. Angiotensin II protects against α-synuclein toxicity and reduces protein aggregation in vitro. Biochem. Biophys. Res. Commun. 2007, 363, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Gil, P.; Joglar, B.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of PPAR-γ in the neuroprotective and anti-inflammatory effects of angiotensin type 1 receptor inhibition: Effects of the receptor antagonist telmisartan and receptor deletion in a mouse MPTP model of Parkinson’s disease. J. Neuroinflammation 2012, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Tong, Q.; Wu, L.; Jiang, T.; Ou, Z.; Zhang, Y.; Zhu, D. Inhibition of endoplasmic reticulum stress-activated IRE1α-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of Parkinson’s disease. Eur. J. Pharmacol. 2016, 776, 106–115. [Google Scholar] [CrossRef]
- Ou, Z.; Jiang, T.; Gao, Q.; Tian, Y.-Y.; Zhou, J.-S.; Wu, L.; Shi, J.-Q.; Zhang, Y.-D. Mitochondrial-dependent mechanisms are involved in angiotensin II-induced apoptosis in dopaminergic neurons. J. Renin-Angiotensin-Aldosterone Syst. 2016, 17, 1470320316672349. [Google Scholar] [CrossRef] [Green Version]
- Costa-Besada, M.A.; Valenzuela, R.; Garrido-Gil, P.; Villar-Cheda, B.; Parga, J.A.; Lanciego, J.L.; Labandeira-Garcia, J.L. Paracrine and Intracrine Angi-otensin 1-7/Mas Receptor Axis in the Substantia Nigra of Rodents, Monkeys, and Humans. Mol. Neurobiol. 2018, 55, 5847–5867. [Google Scholar] [CrossRef]
- Rey, P.; Lopez-Real, A.; Sanchez-Iglesias, S.; Muñoz, A.; Soto-Otero, R.; Labandeira-Garcia, J.L. Angiotensin type-1-receptor antag-onists reduce 6-hydroxydopamine toxicity for dopaminergic neurons. Neurobiol. Aging 2007, 28, 555–567. [Google Scholar] [CrossRef]
- Wu, L.; Tian, Y.-Y.; Shi, J.-P.; Xie, W.; Shi, J.-Q.; Lu, J.; Zhang, Y.-D. Inhibition of endoplasmic reticulum stress is involved in the neuroprotective effects of candesartan cilexitil in the rotenone rat model of Parkinson’s disease. Neurosci. Lett. 2013, 548, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal Apoptosis Induced by Endoplasmic Reticulum Stress Is Regulated by ATF4-CHOP-Mediated Induction of the Bcl-2 Homology 3-Only Member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef]
- Lu, J.; Wu, L.; Jiang, T.; Wang, Y.; Zhao, H.; Gao, Q.; Pan, Y.; Tian, Y.; Zhang, Y. Angiotensin AT2 receptor stimulation inhibits activation of NADPH oxidase and ameliorates oxidative stress in rotenone model of Parkinson’s disease in CATH.a cells. Neurotoxicol. Teratol. 2015, 47, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Qu, Y.; Sun, L.; Jia, L.; Bu, J.; Ye, M.; Chen, Z.; Geng, Y.; Zhou, S.; Fang, B. Fuyuan Xingnao Decoction Promotes Angiogenesis Through the Rab1/AT1R Pathway in Diabetes Mellitus Complicated with Cerebral Infarction. Front. Pharmacol. 2021, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Astin, R.; Bentham, R.; Djafarzadeh, S.; Horscroft, J.A.; Kuc, R.E.; Leung, P.S.; Skipworth, J.R.A.; Vicencio, J.M.; Davenport, A.P.; Murray, A.J.; et al. No evidence for a local renin-angiotensin system in liver mitochondria. Sci. Rep. 2013, 3, 2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunanda, T.; Ray, B.; Mahalakshmi, A.M.; Bhat, A.; Rashan, L.; Rungratanawanich, W.; Song, B.-J.; Essa, M.M.; Sakharkar, M.K.; Chidambaram, S.B. Mitochondria-Endoplasmic Reticulum Crosstalk in Parkinson’s Disease: The Role of Brain Renin Angiotensin System Components. Biomolecules 2021, 11, 1669. https://doi.org/10.3390/biom11111669
Sunanda T, Ray B, Mahalakshmi AM, Bhat A, Rashan L, Rungratanawanich W, Song B-J, Essa MM, Sakharkar MK, Chidambaram SB. Mitochondria-Endoplasmic Reticulum Crosstalk in Parkinson’s Disease: The Role of Brain Renin Angiotensin System Components. Biomolecules. 2021; 11(11):1669. https://doi.org/10.3390/biom11111669
Chicago/Turabian StyleSunanda, Tuladhar, Bipul Ray, Arehally M. Mahalakshmi, Abid Bhat, Luay Rashan, Wiramon Rungratanawanich, Byoung-Joon Song, Musthafa Mohamed Essa, Meena Kishore Sakharkar, and Saravana Babu Chidambaram. 2021. "Mitochondria-Endoplasmic Reticulum Crosstalk in Parkinson’s Disease: The Role of Brain Renin Angiotensin System Components" Biomolecules 11, no. 11: 1669. https://doi.org/10.3390/biom11111669
APA StyleSunanda, T., Ray, B., Mahalakshmi, A. M., Bhat, A., Rashan, L., Rungratanawanich, W., Song, B.-J., Essa, M. M., Sakharkar, M. K., & Chidambaram, S. B. (2021). Mitochondria-Endoplasmic Reticulum Crosstalk in Parkinson’s Disease: The Role of Brain Renin Angiotensin System Components. Biomolecules, 11(11), 1669. https://doi.org/10.3390/biom11111669