
Cohesin Mutations Induce Chromatin Conformation Perturbation of the H19/IGF2 Imprinted Region and Gene Expression Dysregulation in Cornelia de Lange Syndrome Cell Lines

 , , , , , , , ,
, , , , , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Lymphoblastoid Cell Lines
2.2. Chromatin Conformation Capture Assay (3C)
2.3. DNA Methylation Analysis
2.4. nCounter Analysis
2.5. Statistical Analysis
3. Results
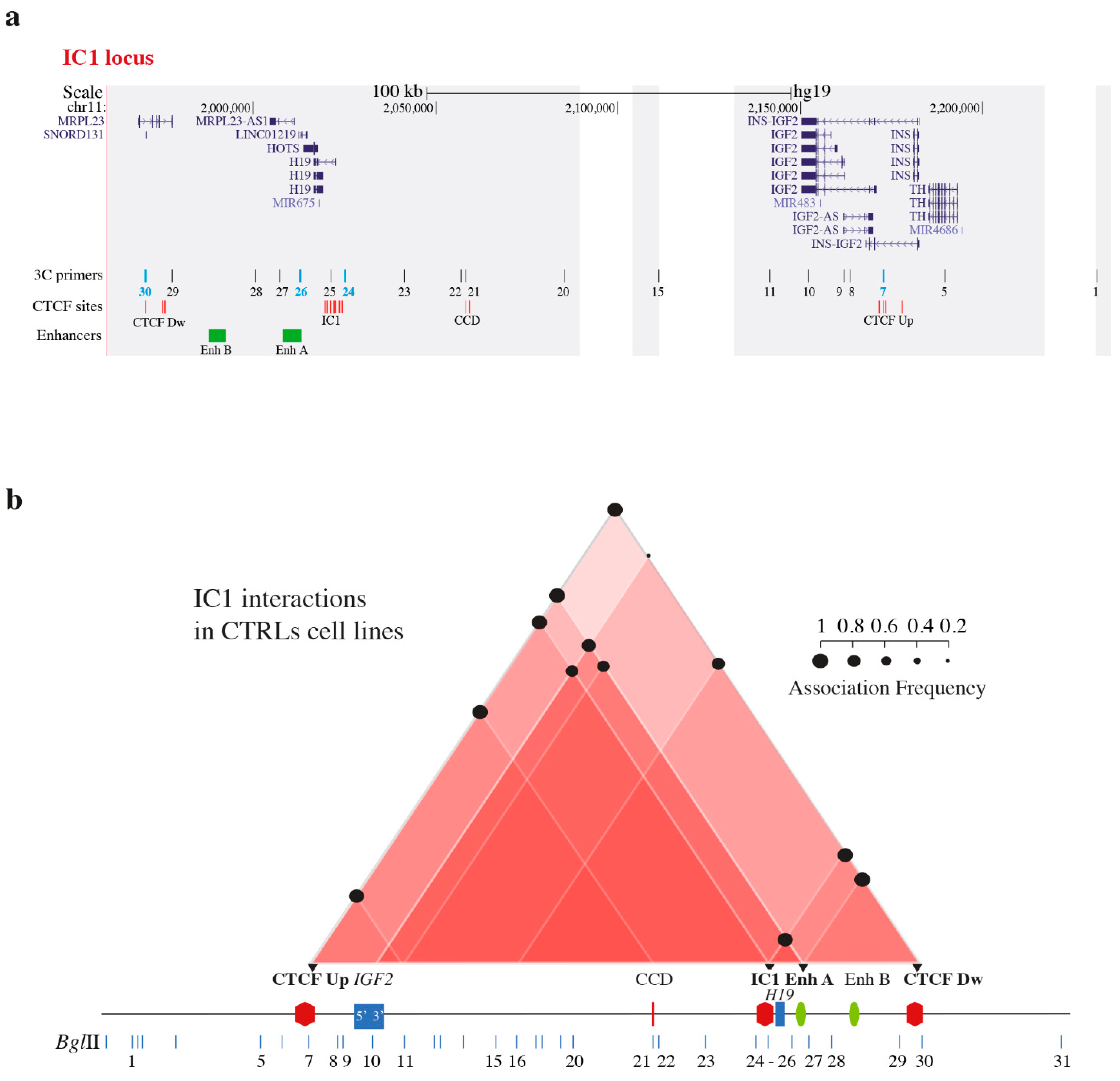
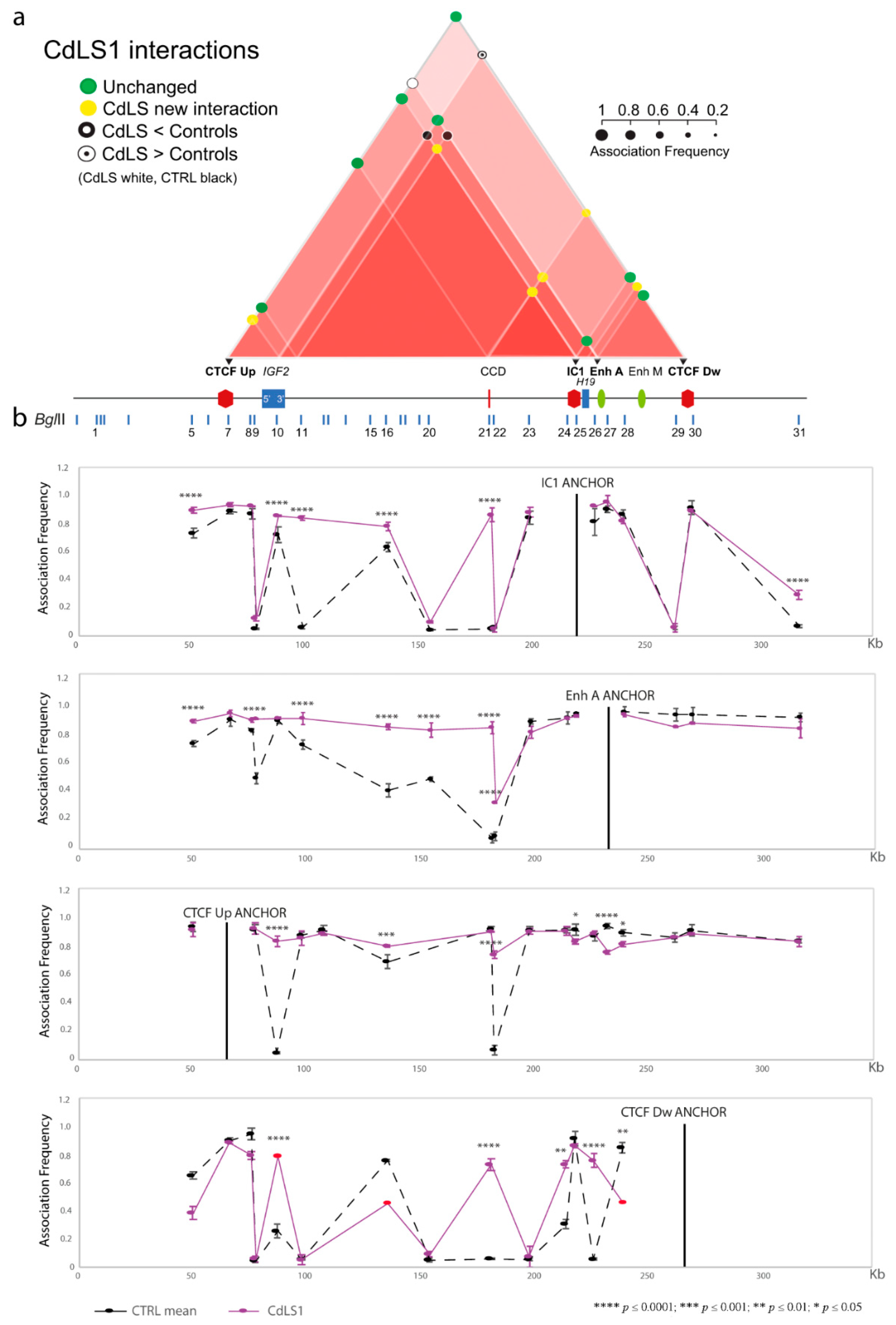
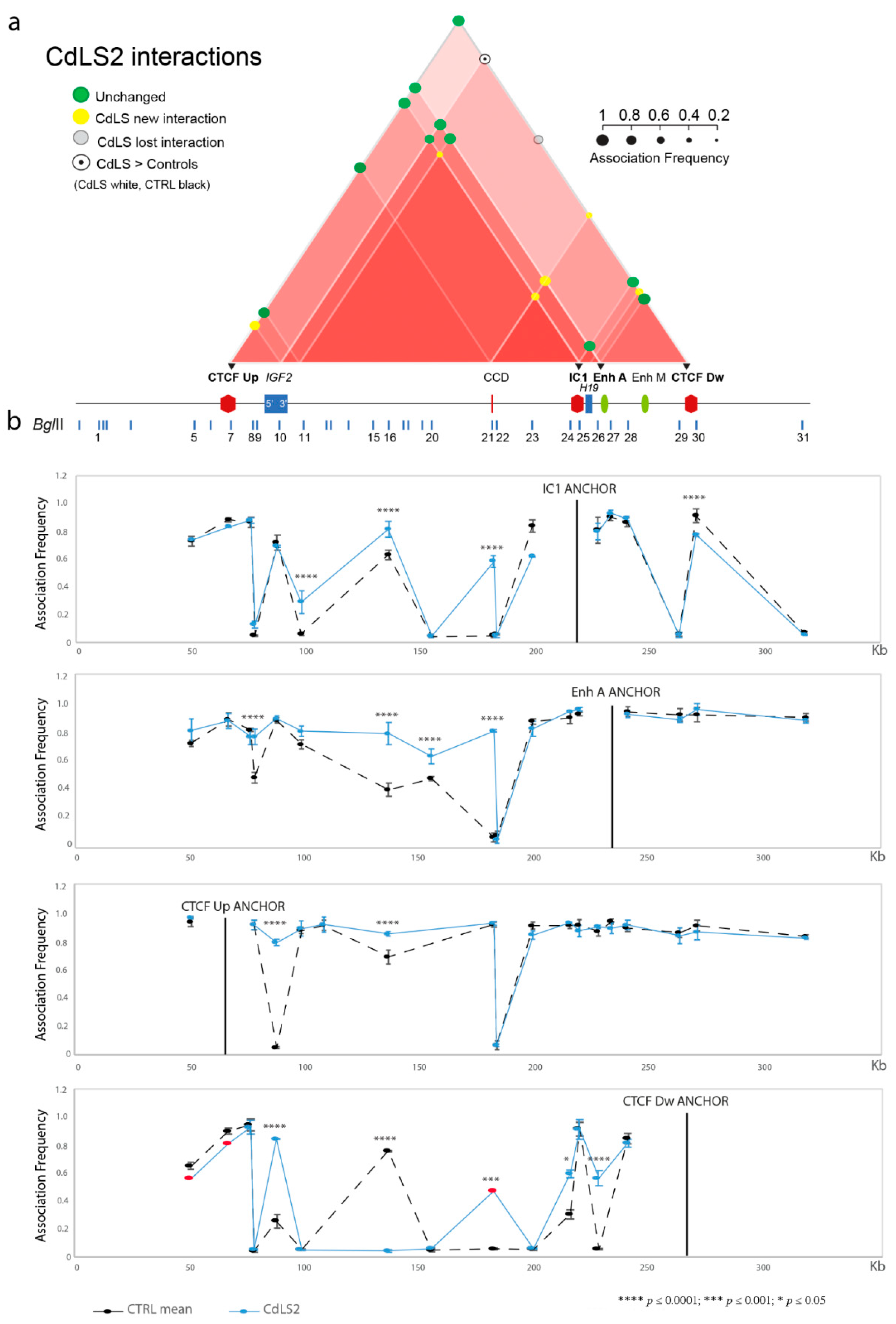
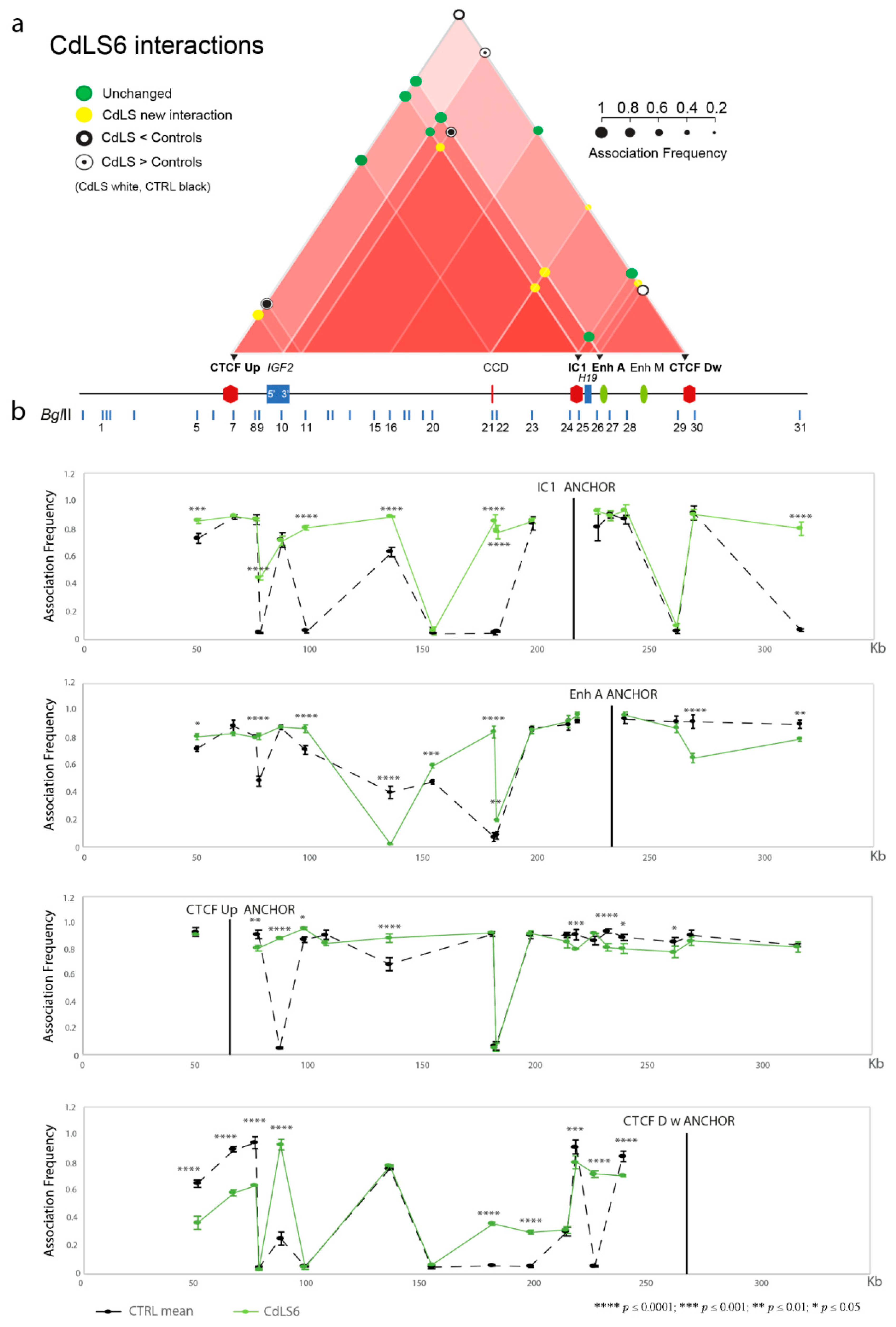
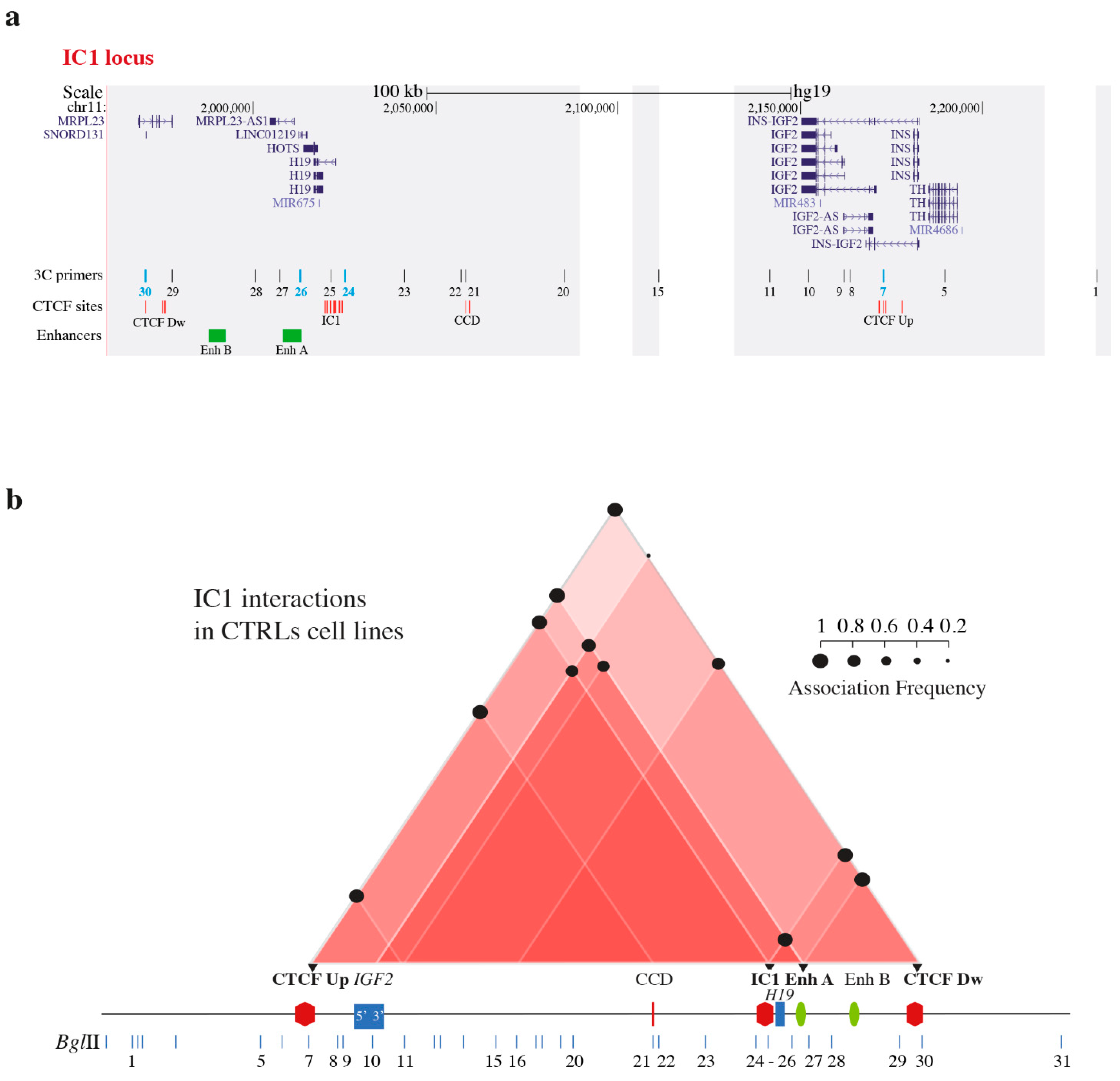
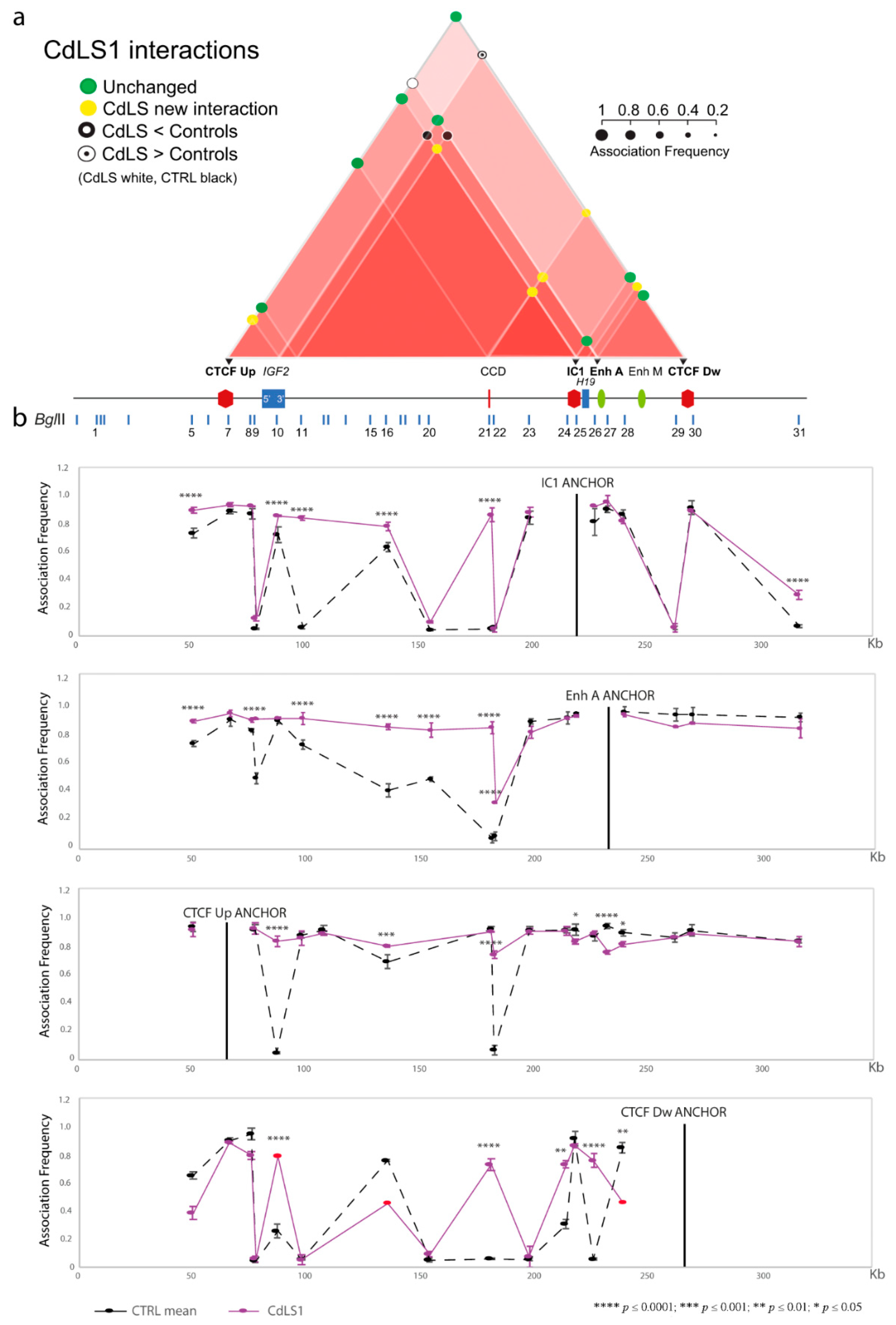
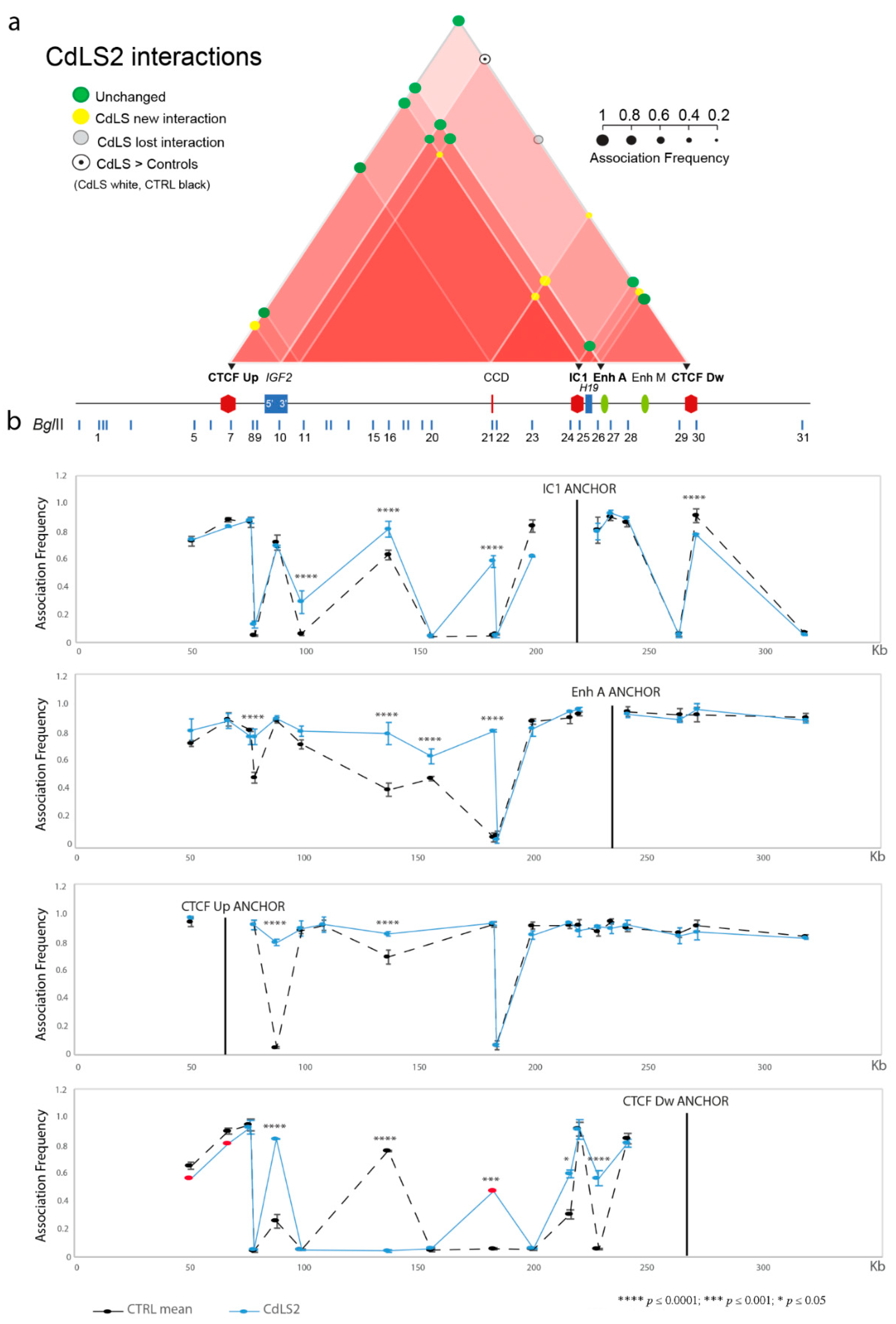
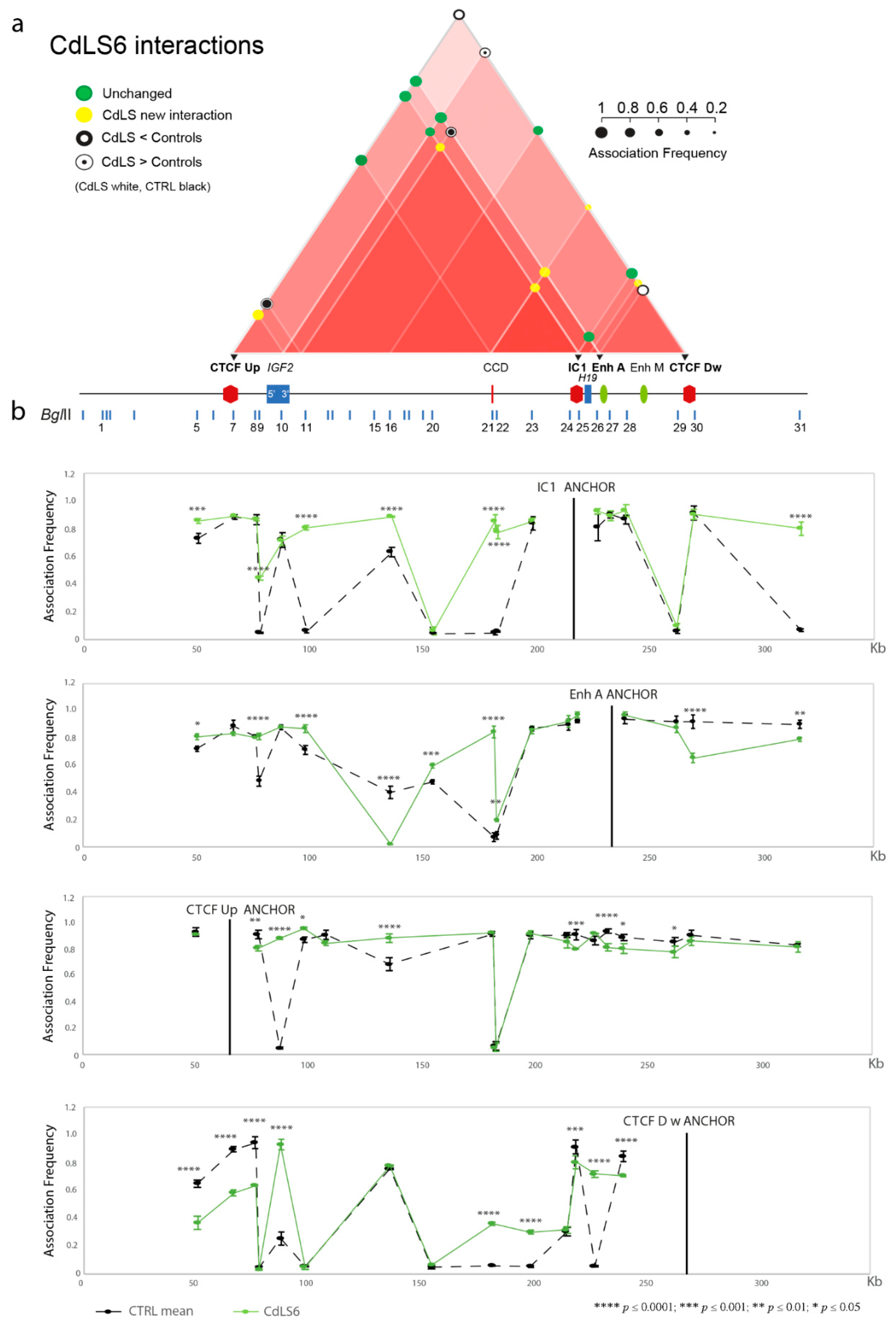
3.1. Chromatin Interactions at the IGF2/H19 Domain in Cells from CdLS Patients
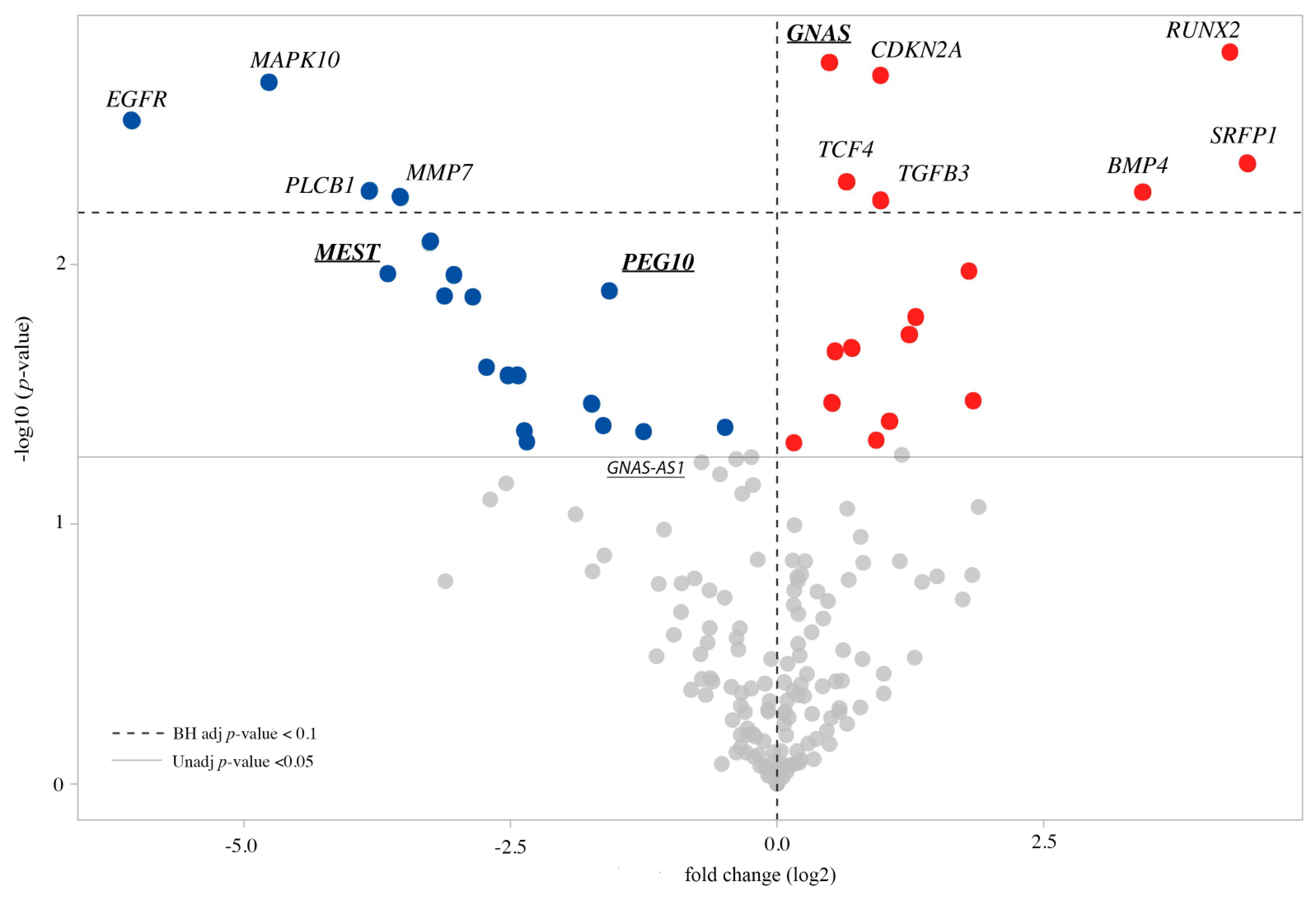
3.2. Expression Profile of the CdLS Cell Lines
3.3. Imprinted and Imprinted-Related Genes Panel
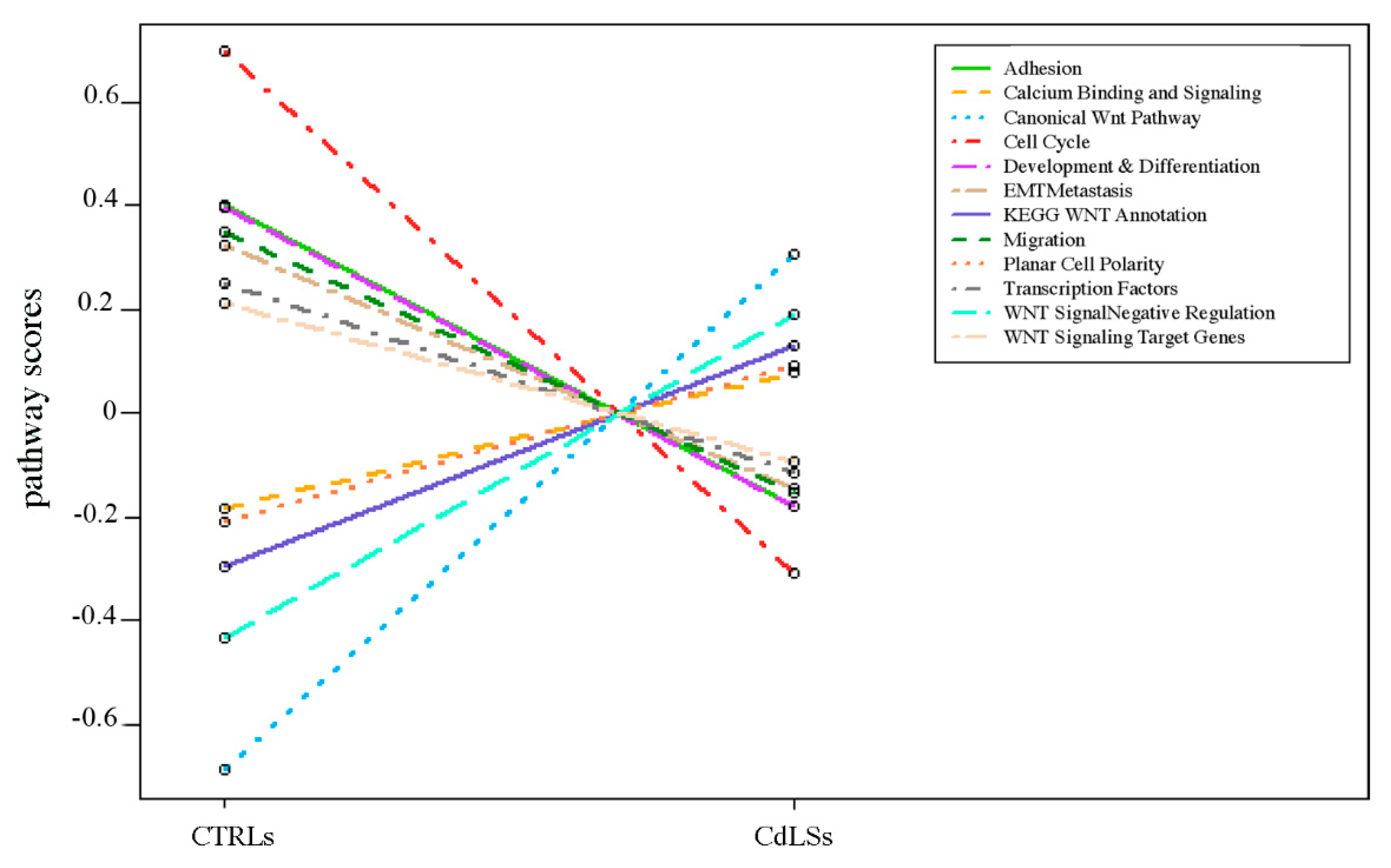
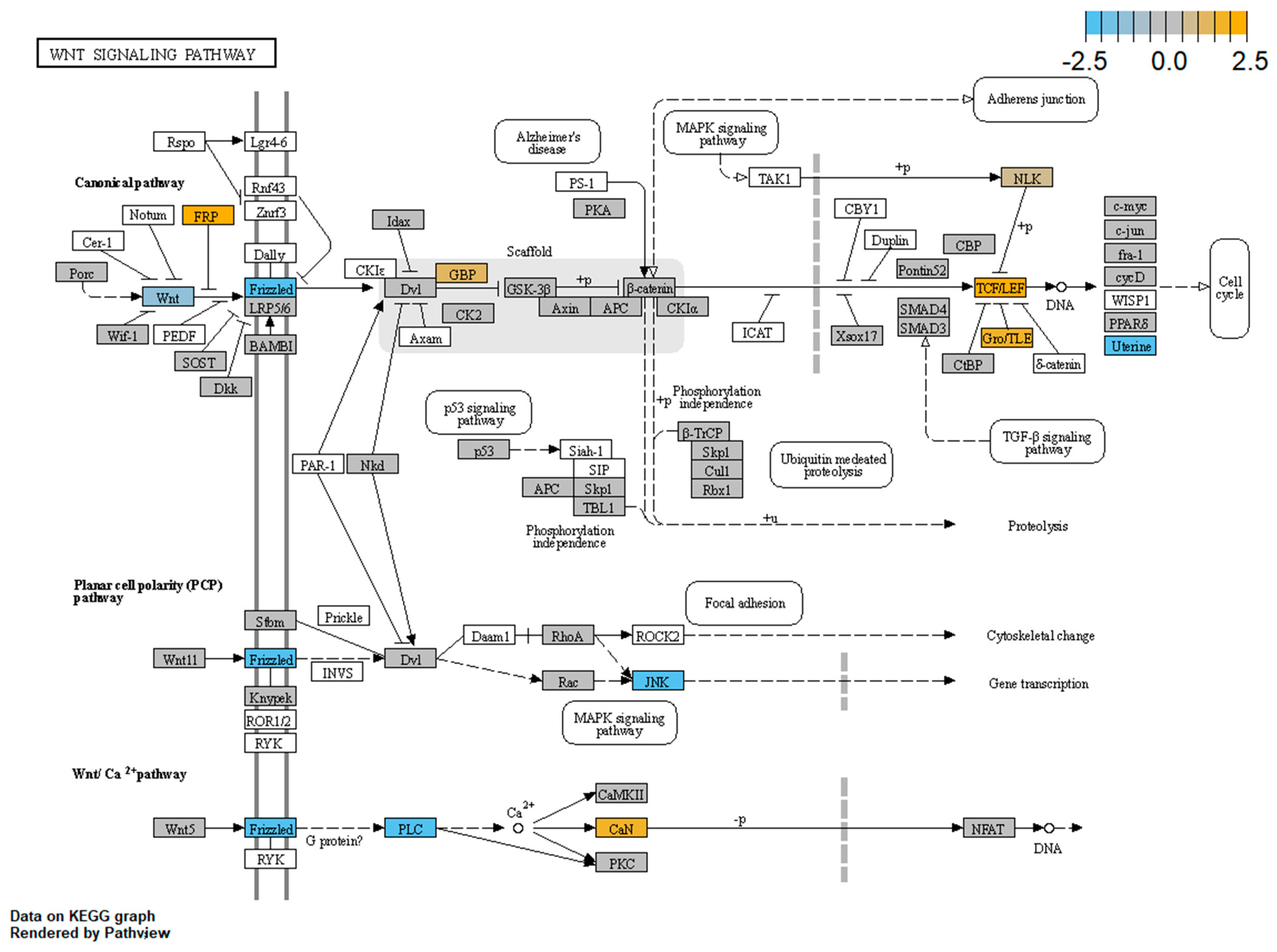
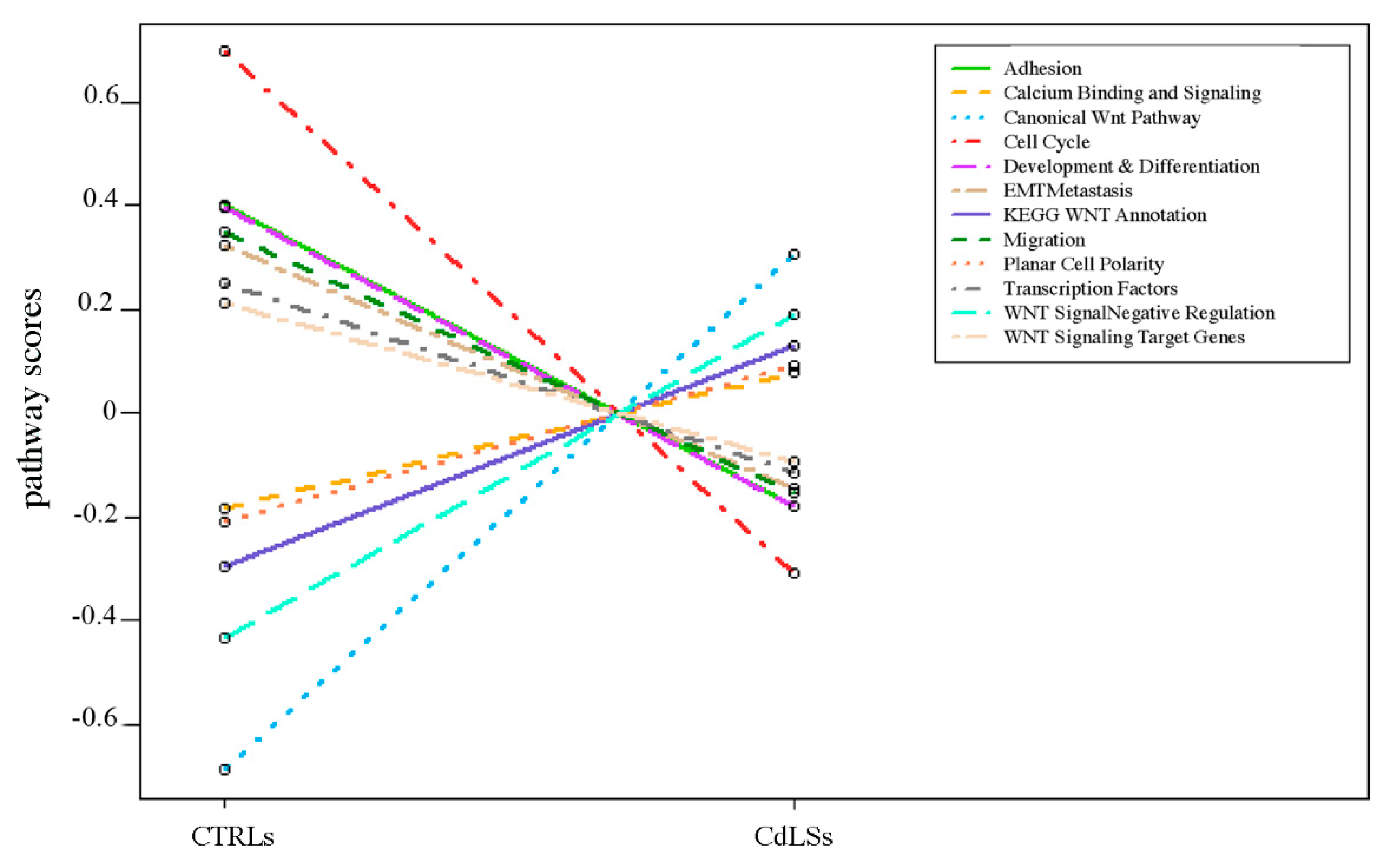
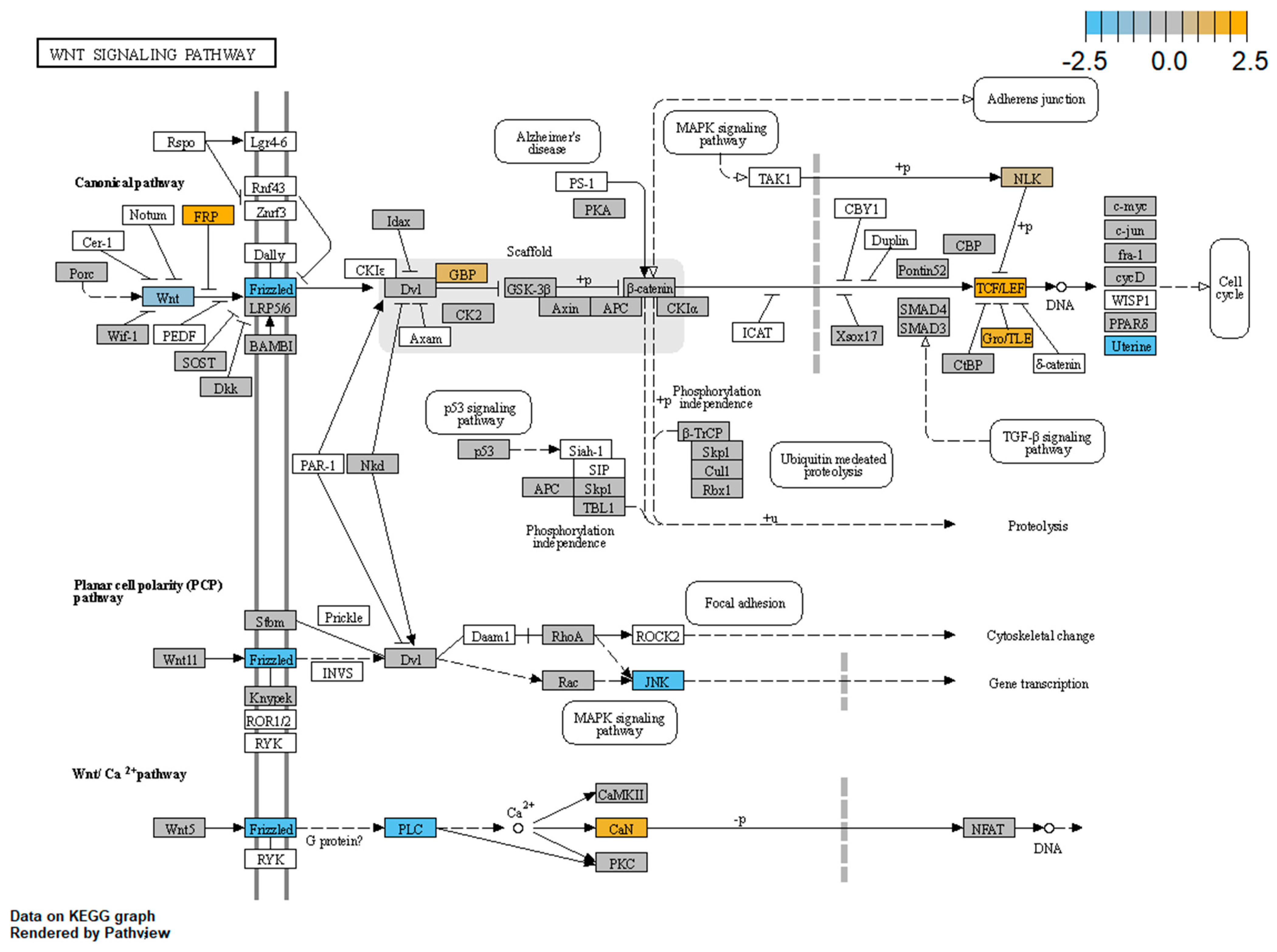
3.4. WNT Panel
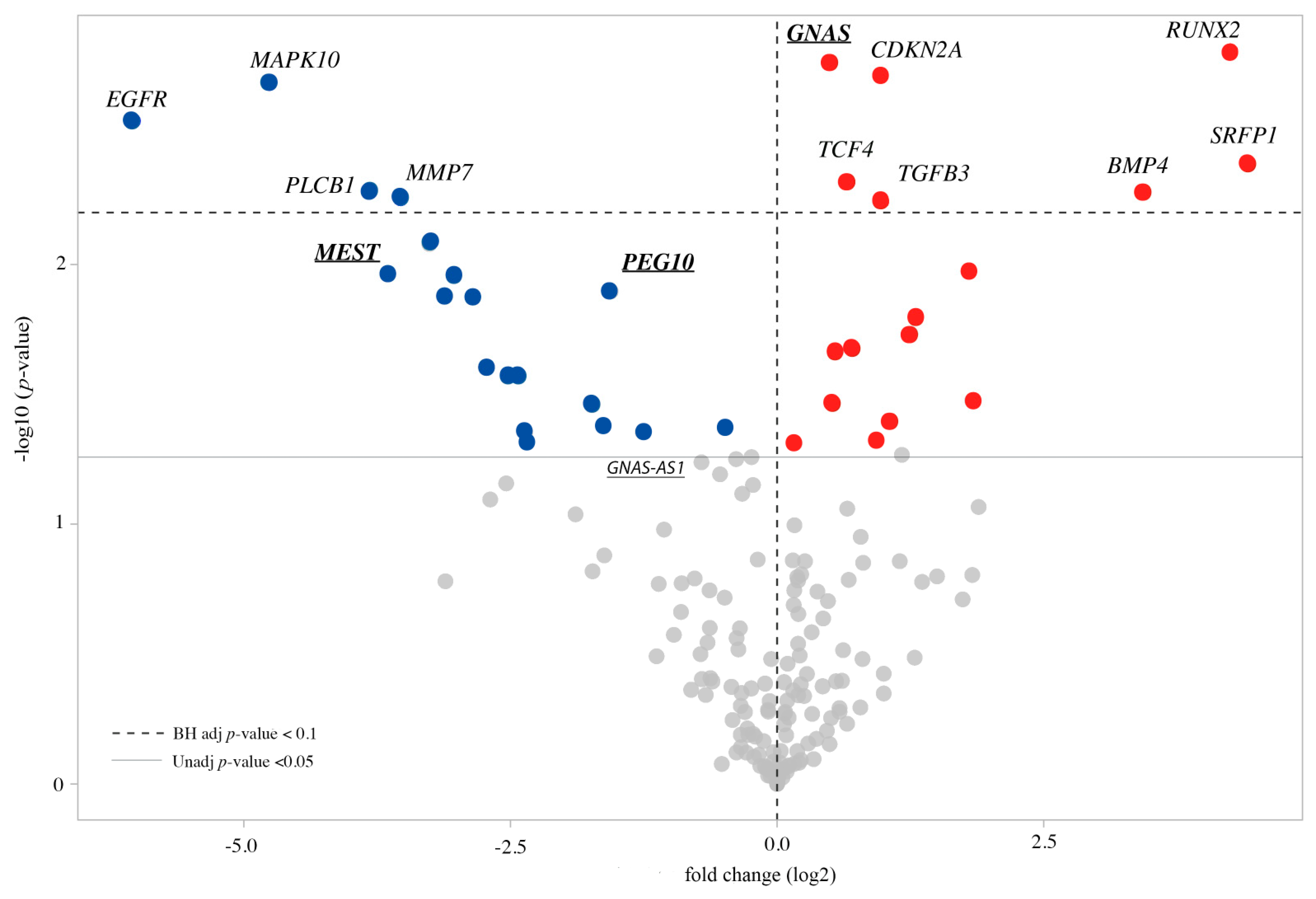
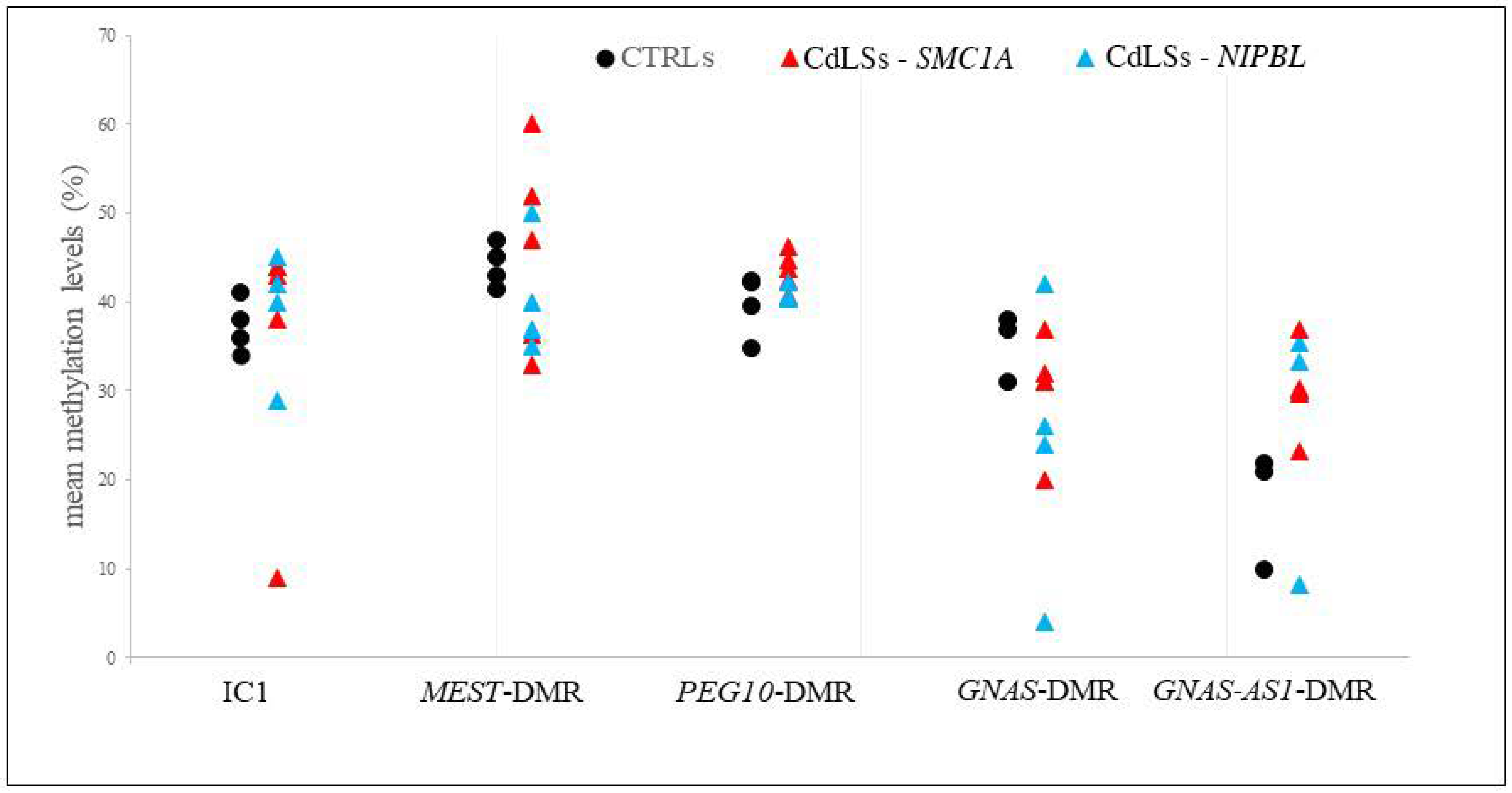
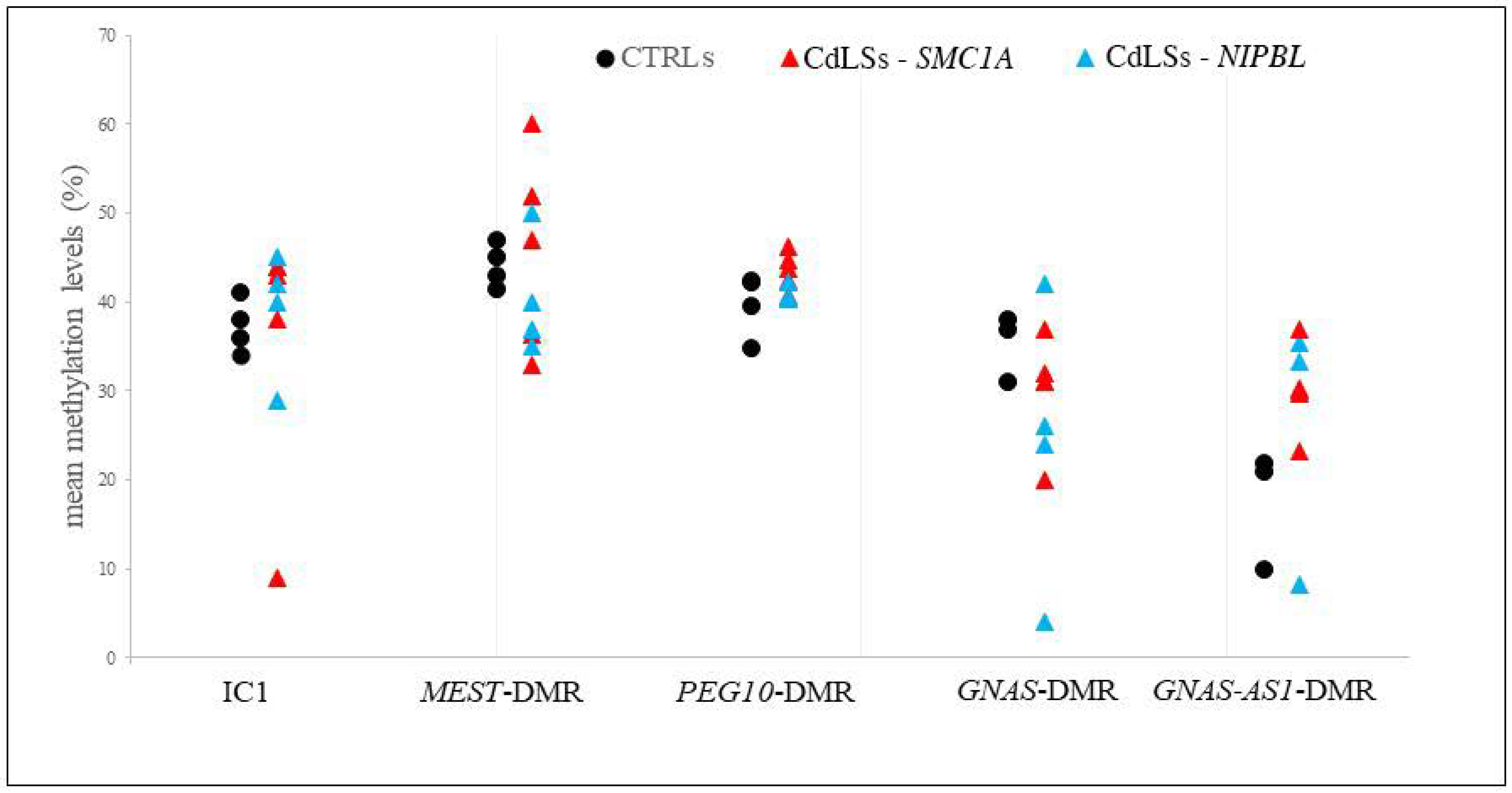
3.5. Methylation Analysis of the Imprinted DMRs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watrin, E.; Kaiser, F.J.; Wendt, K.S. Gene regulation and chromatin organization: Relevance of cohesin mutations to human disease. Curr. Opin. Genet. Dev. 2016, 37, 59–66. [Google Scholar] [CrossRef]
- Horsfield, J.A.; Print, C.G.; Mönnich, M. Diverse Developmental Disorders from the One Ring: Distinct Molecular Pathways Underlie the Cohesinopathies. Front. Genet. 2012, 3, 171. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.; Fernandez-Hernandez, R.; Cuadrado, A.; Coca, I.; Gomez, A.; Maqueda, M.; Latorre-Pellicer, A.; Puisac, B.; Ramos, F.J.; Sandoval, J.; et al. Disruption of NIPBL/Scc2 in Cornelia de Lange Syndrome provokes cohesin genome-wide redistribution with an impact in the transcriptome. Nat. Commun. 2021, 12, 4551. [Google Scholar] [CrossRef]
- Borrie, M.S.; Campor, J.S.; Joshi, H.; Gartenberg, M.R. Binding, sliding, and function of cohesin during transcriptional activation. Proc. Natl. Acad. Sci. USA 2017, 114, E1062–E1071. [Google Scholar] [CrossRef] [Green Version]
- Ciosk, R.; Shirayama, M.; Shevchenko, A.; Tanaka, T.; Toth, A.; Shevchenko, A.; Nasmyth, K. Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol. Cell 2000, 5, 243–254. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange Syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef]
- Krantz, I.D. Cohesin embraces new phenotypes. Nat. Genet. 2014, 46, 1157–1158. [Google Scholar] [CrossRef] [Green Version]
- Bose, T.; Gerton, J.L. Cohesinopathies, gene expression, and chromatin organization. J. Cell Biol. 2010, 189, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange Syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [Green Version]
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange Syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef]
- Tonkin, E.T.; Wang, T.J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange Syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Gillis, L.A.; McCallum, J.; Kaur, M.; DeScipio, C.; Yaeger, D.; Mariani, A.; Kline, A.D.; Li, H.H.; Devoto, M.; Jackson, L.G.; et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange Syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet. 2004, 75, 610–623. [Google Scholar] [CrossRef] [Green Version]
- Musio, A.; Selicorni, A.; Focarelli, M.L.; Gervasini, C.; Milani, D.; Russo, S.; Vezzoni, P.; Larizza, L. X linked Cornelia de Lange Syndrome owing to SMC1L1 mutations. Nat. Genet. 2006, 38, 528–530. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange Syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Gil-Rodríguez, M.C.; Deardorff, M.A.; Ansari, M.; Tan, C.A.; Parenti, I.; Baquero-Montoya, C.; Ousager, L.B.; Puisac, B.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E. De novo heterozygous mutations in SMC3 cause a range of Cornelia de Lange Syndrome-overlapping phenotypes. Hum. Mutat. 2015, 36, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Krantz, I.D. Cornelia de Lange Syndrome, cohesin, and beyond. Clin. Genet. 2009, 76, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Kline, A.D.; Krantz, I.D.; Bando, M.; Shirahige, K.; Chea, S.; Sakata, T.; Rao, S.; Dorsett, D.; Singh, V.P.; Gerton, J.L.; et al. Cornelia de Lange Syndrome, related disorders, and the Cohesin complex: Abstracts from the 8th biennial scientific and educational symposium 2018. Am. J. Med. Genet. A 2019, 179, 1080–1090. [Google Scholar] [CrossRef]
- Göndör, A.; Ohlsson, R. Chromatin insulators and cohesins. EMBO Rep. 2008, 9, 327–329. [Google Scholar] [CrossRef] [Green Version]
- Parelho, V.; Hadjur, S.; Spivakov, M.; Leleu, M.; Sauer, S.; Gregson, H.C.; Jarmuz, A.; Canzonetta, C.; Webster, Z.; Nesterova, T.; et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008, 132, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Rubio, E.D.; Reiss, D.J.; Welcsh, P.L.; Disteche, C.M.; Filippova, G.N.; Baliga, N.S.; Aebersold, R.; Ranish, J.A.; Krumm, A. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. USA 2008, 105, 8309–8314. [Google Scholar] [CrossRef] [Green Version]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef]
- Merkenschlager, M. Cohesin: A Global Player in Chromosome Biology with Local Ties to Gene Regulation. Curr. Opin. Genet. Dev. 2010, 20, 555–561. [Google Scholar] [CrossRef]
- Dorsett, D. Cohesin: Genomic Insights into Controlling Gene Transcription and Development. Curr. Opin. Genet. Dev. 2011, 21, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Weiss, F.D.; Calderon, L.; Wang, Y.F.; Georgieva, R.; Guo, Y.; Cvetesic, N.; Kaur, M.; Dharmalingam, G.; Krantz, I.D.; Lenhard, B.; et al. Neuronal genes deregulated in Cornelia de Lange Syndrome respond to removal and re-expression of cohesin. Nat. Commun. 2021, 12, 2919. [Google Scholar] [CrossRef]
- Murrell, A.; Heeson, S.; Reik, W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 2004, 36, 889–893. [Google Scholar] [CrossRef]
- Eggermann, K.; Bliek, J.; Brioude, F.; Algar, E.; Buiting, K.; Russo, S.; Tümer, Z.; Monk, D.; Moore, G.; Antoniadi, T.; et al. EMQN best practice guidelines for the molecular genetic testing and reporting of chromosome 11p15 imprinting disorders: Silver-Russell and Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2016, 24, 1377–1387. [Google Scholar] [CrossRef]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef]
- Stedman, W.; Kang, H.; Lin, S.; Kissil, J.L.; Bartolomei, M.S.; Lieberman, P.M. Cohesins localize with CTCF at the KSHV latency control region and at cellular cmyc and H19/Igf2 insulators. EMBO J. 2008, 27, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Nativio, R.; Wendt, K.S.; Ito, Y.; Huddleston, J.E.; Uribe-Lewis, S.; Woodfine, K.; Krueger, C.; Reik, W.; Peters, J.M.; Murrell, A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009, 5, e1000739. [Google Scholar] [CrossRef]
- Nativio, R.; Sparago, A.; Ito, Y.; Weksberg, R.; Riccio, A.; Murrell, A. Disruption of genomic neighbourhood at the imprinted IGF2-H19 locus in Beckwith-Wiedemann syndrome and Silver-Russell syndrome. Hum. Mol. Genet. 2011, 20, 1363–1374. [Google Scholar] [CrossRef] [Green Version]
- Rovina, D.; La Vecchia, M.; Cortesi, A.; Fontana, L.; Pesant, M.; Maitz, S.; Tabano, S.; Bodega, B.; Miozzo, M.; Sirchia, S.M. Profound alterations of the chromatin architecture at chromosome 11p15.5 in cells from Beckwith-Wiedemann and Silver-Russell syndromes patients. Sci. Rep. 2020, 10, 8275. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Delgado, M.; Riccio, A.; Eggermann, T.; Maher, E.R.; Lapunzina, P.; Mackay, D.; Monk, D. Causes and Consequences of Multi-Locus Imprinting Disturbances in Humans. Trends Genet. 2016, 32, 444–455. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, D.; Feil, R. Differential 3D chromatin organization and gene activity in genomic imprinting. Curr. Opin. Genet. Dev. 2020, 61, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Selicorni, A.; Mariani, M.; Lettieri, A.; Massa, V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes (Basel) 2021, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, L.; Grazioli, P.; Mariani, M.; Bulfamante, G.P.; Selicorni, A.; Massa, V. Integrating molecular and structural findings: Wnt as a possible actor in shaping cognitive impairment in Cornelia de Lange Syndrome. Orphanet J. Rare Dis. 2017, 12, 174. [Google Scholar] [CrossRef] [Green Version]
- Bottai, D.; Spreafico, M.; Pistocchi, A.; Fazio, G.; Adami, R.; Grazioli, P.; Canu, A.; Bragato, C.; Rigamonti, S.; Parodi, C.; et al. Modeling Cornelia de Lange Syndrome in vitro and in vivo reveals a role for cohesin complex in neuronal survival and differentiation. Hum. Mol. Genet. 2019, 28, 64–73. [Google Scholar] [CrossRef]
- Grazioli, P.; Parodi, C.; Mariani, M.; Bottai, D.; Di Fede, E.; Zulueta, A.; Avagliano, L.; Cereda, A.; Tenconi, R.; Wierzba, J.; et al. Lithium as a possible therapeutic strategy for Cornelia de Lange Syndrome. Cell Death Discov. 2021, 7, 34. [Google Scholar] [CrossRef]
- Gervasini, C.; Russo, S.; Cereda, A.; Parenti, I.; Masciadri, M.; Azzollini, J.; Melis, D.; Aravena, T.; Doray, B.; Ferrarini, A.; et al. Cornelia de Lange individuals with new and recurrent SMC1A mutations enhance delineation of mutation repertoire and phenotypic spectrum. Am. J. Med. Genet. 2013, 161A, 2909–2919. [Google Scholar] [CrossRef]
- Limongelli, G.; Russo, S.; Digilio, M.C.; Masciadri, M.; Pacileo, G.; Fratta, F.; Martone, F.; Maddaloni, V.; D’Alessandro, R.; Calabro, P.; et al. Hypertrophic cardiomyopathy in a girl with Cornelia de Lange Syndrome due to mutation in SMC1A. Am. J. Med. Genet. 2010, 152A, 2127–2129. [Google Scholar] [CrossRef]
- Selicorni, A.; Russo, S.; Gervasini, C.; Castronovo, P.; Milani, D.; Cavalleri, F.; Bentivegna, A.; Masciadri, M.; Domi, A.; Divizia, M.T.; et al. Clinical score of 62 Italian patients with Cornelia de Lange Syndrome and correlations with the presence and type of NIPBL mutation. Clin. Genet. 2007, 72, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Masciadri, M.; Gervasini, C.; Azzollini, J.; Cereda, A.; Zampino, G.; Haas, O.; Scarano, G.; Di Rocco, M.; Finelli, P.; et al. Intragenic and large NIPBL rearrangements revealed by MLPA in Cornelia de Lange patients. Eur. J. Hum. Genet. 2012, 20, 734–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holwerda, S.; de Laat, W. Chromatin loops, gene positioning, and gene expression. Front. Genet. 2012, 3, 217. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, J.; Heard, E. Structural and functional diversity of Topologically Associating Domains. FEBS Lett. 2015, 589, 2877–2884. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Higashimoto, K.; Jozaki, K.; Yatsuki, H.; Nakabayashi, K.; Makita, Y.; Tonoki, H.; Okamoto, N.; Takada, F.; Ohashi, H.; et al. Comprehensive and quantitative multilocus methylation analysis reveals the susceptibility of specific imprinted differentially methylated regions to aberrant methylation in Beckwith-Wiedemann syndrome with epimutations. Genet. Med. 2014, 16, 903–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potabattula, R.; Dittrich, M.; Schorsch, M.; Hahn, T.; Haaf, T.; El Hajj, N. Male obesity effects on sperm and next-generation cord blood DNA methylation. PLoS ONE 2019, 14, e0218615. [Google Scholar] [CrossRef]
- Veldman-Jones, M.H.; Brant, R.; Rooney, C.; Geh, C.; Emery, H.; Harbron, C.G.; Wappett, M.; Sharpe, A.; Dymond, M.; Barrett, J.C.; et al. Evaluating Robustness and Sensitivity of the NanoString Technologies nCounter Platform to Enable Multiplexed Gene Expression Analysis of Clinical Samples. Cancer Res. 2015, 75, 2587–2593. [Google Scholar] [CrossRef] [Green Version]
- Gentien, D.; Piqueret-Stephan, L.; Henry, E.; Albaud, B.; Rapinat, A.; Koscielny, S.; Scoazec, J.Y.; Vielh, P. Digital Multiplexed Gene Expression Analysis of mRNA and miRNA from Routinely Processed and Stained Cytological Smears: A Proof-of-Principle Study. Acta Cytol. 2021, 65, 88–98. [Google Scholar] [CrossRef]
- Prokopec, S.D.; Watson, J.D.; Waggott, D.M.; Smith, A.B.; Wu, A.H.; Okey, A.B.; Pohjanvirta, R.; Boutros, P.C. Systematic evaluation of medium-throughput mRNA abundance platforms. RNA 2013, 19, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Pistocchi, A.; Fazio, G.; Cereda, A.; Ferrari, L.; Bettini, L.R.; Messina, G.; Cotelli, F.; Biondi, A.; Selicorni, A.; Massa, V. Cornelia de Lange Syndrome: NIPBL haploinsufficiency downregulates canonical Wnt pathway in zebrafish embryos and patients fibroblasts. Cell Death Dis. 2013, 4, e866. [Google Scholar] [CrossRef] [Green Version]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef] [PubMed]
- Boudaoud, I.; Fournier, É.; Baguette, A.; Vallée, M.; Lamaze, F.C.; Droit, A.; Bilodeau, S. Connected Gene Communities Underlie Transcriptional Changes in Cornelia de Lange Syndrome. Genetics 2017, 207, 139–151. [Google Scholar] [CrossRef]
- Avagliano, L.; Parenti, I.; Grazioli, P.; Di Fede, E.; Parodi, C.; Mariani, M.; Kaiser, F.J.; Selicorni, A.; Gervasini, C.; Massa, V. Chromatinopathies: A focus on Cornelia de Lange Syndrome. Clin. Genet. 2020, 97, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.H.; Maher, E.R. Human imprinting syndromes. Epigenomics 2009, 1, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Tabano, S.; Maitz, S.; Colapietro, P.; Garzia, E.; Gerli, A.G.; Sirchia, S.M.; Miozzo, M. Clinical and Molecular Diagnosis of Beckwith-Wiedemann Syndrome with Single- or Multi-Locus Imprinting Disturbance. Int. J. Mol. Sci. 2021, 22, 3445. [Google Scholar] [CrossRef]
- Turan, S.; Bastepe, M. GNAS Spectrum of Disorders. Curr. Osteoporos. Rep. 2015, 13, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koukoura, O.; Sifakis, S.; Spandidos, D.A. DNA methylation in the human placenta and fetal growth (review). Mol. Med. Rep. 2012, 5, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Bedeschi, M.F.; Maitz, S.; Cereda, A.; Faré, C.; Motta, S.; Seresini, A.; D’Ursi, P.; Orro, A.; Pecile, V.; et al. Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15.5 related imprinting disorders. Epigenetics 2018, 13, 897–909. [Google Scholar] [CrossRef]
- Mehta, G.D.; Kumar, R.; Srivastava, S.; Ghosh, S.K. Cohesin: Functions beyond sister chromatid cohesion. FEBS Lett. 2013, 587, 2299–2312. [Google Scholar] [CrossRef] [Green Version]
- Zuin, J.; Franke, V.; van Ijcken, W.F.; van der Sloot, A.; Krantz, I.D.; van der Reijden, M.I.; Nakato, R.; Lenhard, B.; Wendt, K.S. A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet. 2014, 10, e1004153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, J.A.; Herrera, P.S.; Kaur, M.; Leo, L.; McEldrew, D.; Tintos-Hernandez, J.A.; Rajagopalan, R.; Gagne, A.; Zhang, Z.; Ortiz-Gonzalez, X.R.; et al. NIPBL+/− haploinsufficiency reveals a constellation of transcriptome disruptions in the pluripotent and cardiac states. Sci. Rep. 2018, 8, 1056. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [Green Version]
- Miyabayashi, T.; Teo, J.L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef] [Green Version]
- Ooki, A.; Onodera, S.; Saito, A.; Oguchi, A.; Murakawa, Y.; Sakamoto, T.; Sueishi, K.; Nishii, Y.; Azuma, T. CAGE-seq analysis of osteoblast derived from cleidocranial dysplasia human induced pluripotent stem cells. Bone 2020, 141, 115582. [Google Scholar] [CrossRef]
- Motaei, J.; Salmaninejad, A.; Jamali, E.; Khorsand, I.; Ahmadvand, M.; Shabani, S.; Karimi, F.; Nazari, M.S.; Ketabchi, G.; Naqipour, F. Molecular Genetics of Cleidocranial Dysplasia. Fetal Pediatr. Pathol. 2021, 40, 442–454. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Causative Genetic Alteration | Dmr Methylation Analysis | 3C Assay | Ncounter Analysis | ||
|---|---|---|---|---|---|---|
| IC1 | MEST-DMR | PEG10-DMR | ||||
| CTRL1 | + | + | + | + | + | |
| CTRL2 | + | + | + | + | + | |
| CTRL3 | + | + | + | − | + | |
| CTRL4 | + | + | + | − | + | |
| CdLS1 | SMC1A c.2351T > C | + | + | + | + | + |
| CdLS2 | SMC1A c.173del16 | + | + | + | + | + |
| CdLS3 | SMC1A c.2351T > C | + | + | + | − | + |
| CdLS4 | SMC1A c.3497A > C | + | + | + | − | + |
| CdLS5 | SMC1A c.2078G > A | + | + | + | − | + |
| CdLS6 | NIPBL c.-75_ + 65del | + | + | + | + | + |
| CdLS7 | NIPBL c.4253G > A | + | + | + | − | + |
| CdLS8 | NIPBL c.231-1_231-2del | + | + | + | − | + |
| CdLS9 | NIPBL t(5;15) | + | + | + | + | + |
| Gene | Accession | Log2 Fold Change | p-Value | BH p-Value |
|---|---|---|---|---|
| GNAS * | NM_080425.1 | 0.494 | 0.00166 | 0.0887 |
| MEST * | NM_177525.1 | −3.65 | 0.0109 | 0.13 |
| EG10 * | NM_001040152.1 | −1.56 | 0.0127 | 0.131 |
| GNAS-AS1 | NR_002785.2:1026 | −0.709 | 0.0578 | 0.256 |
| KCNQ1OT1 | NR_002728.2:31875 | −0.535 | 0.0643 | 0.277 |
| FAM50B | NM_012135.1:1272 | 0.197 | 0.289 | 0.594 |
| PLAGL1 | NM_006718.3:1872 | 0.222 | 0.414 | 0.698 |
| Gene | Accession | Log2 Fold Change | p-Value | BH p-Value | Pathway Annotation |
|---|---|---|---|---|---|
| RUNX2 | NM_004348.3 | 4.25 | 0.00151 | 0.0887 | Development & Differentiation, Transcription Factors |
| CDKN2A | NM_000077.3 | 0.971 | 0.00187 | 0.0887 | Cell Cycle, Development & Differentiation, Transcription Factors |
| MAPK10 | NM_002753.2 | −4.76 | 0.002 | 0.0887 | KEGG WNT Annotation |
| EGFR | NM_201282.1 | −6.04 | 0.0028 | 0.0919 | Adhesion, Calcium Binding and Signaling, Cell Cycle, Development & Differentiation, Migration |
| SFRP1 | NM_003012.3 | 4.41 | 0.00411 | 0.0919 | Canonical Wnt Pathway, KEGG WNT Annotation, WNT Signaling Negative Regulation |
| TCF4 | NM_003199.1 | 0.661 | 0.00481 | 0.0919 | Transcription Factors |
| BMP4 | NM_001202.3 | 3.43 | 0.00524 | 0.0919 | Development & Differentiation |
| PLCB1 | NM_182734.1 | −3.82 | 0.00524 | 0.0919 | KEGG WNT Annotation |
| MMP7 | NM_002423.3 | −3.53 | 0.00556 | 0.0919 | Calcium Binding and Signaling, KEGG WNT Annotation, Proteolysis, WNT Signaling Target Genes |
| TGFB3 | NM_003239.2 | 0.978 | 0.00571 | 0.0919 | Development & Differentiation |
| PTGS2 | NM_000963.1 | −3.26 | 0.00834 | 0.123 | Calcium Binding and Signaling, Cell Cycle |
| TLE1 | NM_005077.3 | 1.8 | 0.0106 | 0.13 | WNT Signaling Negative Regulation |
| FZD5 | NM_003468.2 | −3.03 | 0.011 | 0.13 | Canonical Wnt Pathway, KEGG WNT Annotation |
| CXCL12 | NM_000609.5 | −3.11 | 0.0132 | 0.131 | EMTMetastasis |
| GDNF | NM_000514.2 | −2.85 | 0.0133 | 0.131 | Development & Differentiation, Migration |
| IRS1 | NM_005544.2 | 1.3 | 0.016 | 0.149 | Migration |
| FRAT1 | NM_005479.3 | 1.25 | 0.0185 | 0.164 | Canonical Wnt Pathway, KEGG WNT Annotation |
| FZD3 | NM_017412.2 | 0.699 | 0.0211 | 0.173 | Canonical Wnt Pathway, KEGG WNT Annotation |
| PPP3CC | NM_005605.4 | 0.552 | 0.0216 | 0.173 | KEGG WNT Annotation |
| SNAI2 | NM_003068.3 | −2.73 | 0.0249 | 0.191 | EMTMetastasis |
| KREMEN1 | NM_001039570.1 | −2.52 | 0.0268 | 0.191 | WNT Signaling Negative Regulation |
| FZD10 | NM_007197.2 | −2.43 | 0.027 | 0.191 | KEGG WNT Annotation |
| LEF1 | NM_016269.3 | 1.83 | 0.0334 | 0.22 | Canonical Wnt Pathway, KEGG WNT Annotation, Transcription Factors |
| NLK | NM_016231.2 | 0.519 | 0.0342 | 0.22 | KEGG WNT Annotation, WNT Signaling Negative Regulation |
| SOX2 | NM_003106.2 | −1.74 | 0.0348 | 0.22 | Cell Cycle, Development & Differentiation, Transcription Factors |
| PPP3CA | NM_000944.4 | 1.06 | 0.0401 | 0.237 | KEGG WNT Annotation |
| CXCR4 | NM_003467.2 | −1.63 | 0.0418 | 0.237 | EMTMetastasis |
| BIRC5 | NM_001168.2 | −0.491 | 0.0427 | 0.237 | Cell Cycle |
| WNT10B | NM_003394.2 | −1.26 | 0.0442 | 0.237 | KEGG WNT Annotation |
| SERPINE1 | NM_001165413.1 | −2.36 | 0.0442 | 0.237 | EMTMetastasis |
| TCF7L1 | NM_031283.1 | 0.931 | 0.0477 | 0.24 | Canonical Wnt Pathway, KEGG WNT Annotation, Transcription Factors |
| FZD8 | NM_031866.1 | −2.35 | 0.0479 | 0.24 | Canonical Wnt Pathway, KEGG WNT Annotation |
| SMAD2 | NM_005901.5 | 0.165 | 0.0488 | 0.24 | EMTMetastasis, KEGG WNT Annotation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pileggi, S.; La Vecchia, M.; Colombo, E.A.; Fontana, L.; Colapietro, P.; Rovina, D.; Morotti, A.; Tabano, S.; Porta, G.; Alcalay, M.; et al. Cohesin Mutations Induce Chromatin Conformation Perturbation of the H19/IGF2 Imprinted Region and Gene Expression Dysregulation in Cornelia de Lange Syndrome Cell Lines. Biomolecules 2021, 11, 1622. https://doi.org/10.3390/biom11111622
Pileggi S, La Vecchia M, Colombo EA, Fontana L, Colapietro P, Rovina D, Morotti A, Tabano S, Porta G, Alcalay M, et al. Cohesin Mutations Induce Chromatin Conformation Perturbation of the H19/IGF2 Imprinted Region and Gene Expression Dysregulation in Cornelia de Lange Syndrome Cell Lines. Biomolecules. 2021; 11(11):1622. https://doi.org/10.3390/biom11111622
Chicago/Turabian StylePileggi, Silvana, Marta La Vecchia, Elisa Adele Colombo, Laura Fontana, Patrizia Colapietro, Davide Rovina, Annamaria Morotti, Silvia Tabano, Giovanni Porta, Myriam Alcalay, and et al. 2021. "Cohesin Mutations Induce Chromatin Conformation Perturbation of the H19/IGF2 Imprinted Region and Gene Expression Dysregulation in Cornelia de Lange Syndrome Cell Lines" Biomolecules 11, no. 11: 1622. https://doi.org/10.3390/biom11111622