
Coordination of RNA Processing Regulation by Signal Transduction Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Regulation of Nuclear RNA Processing
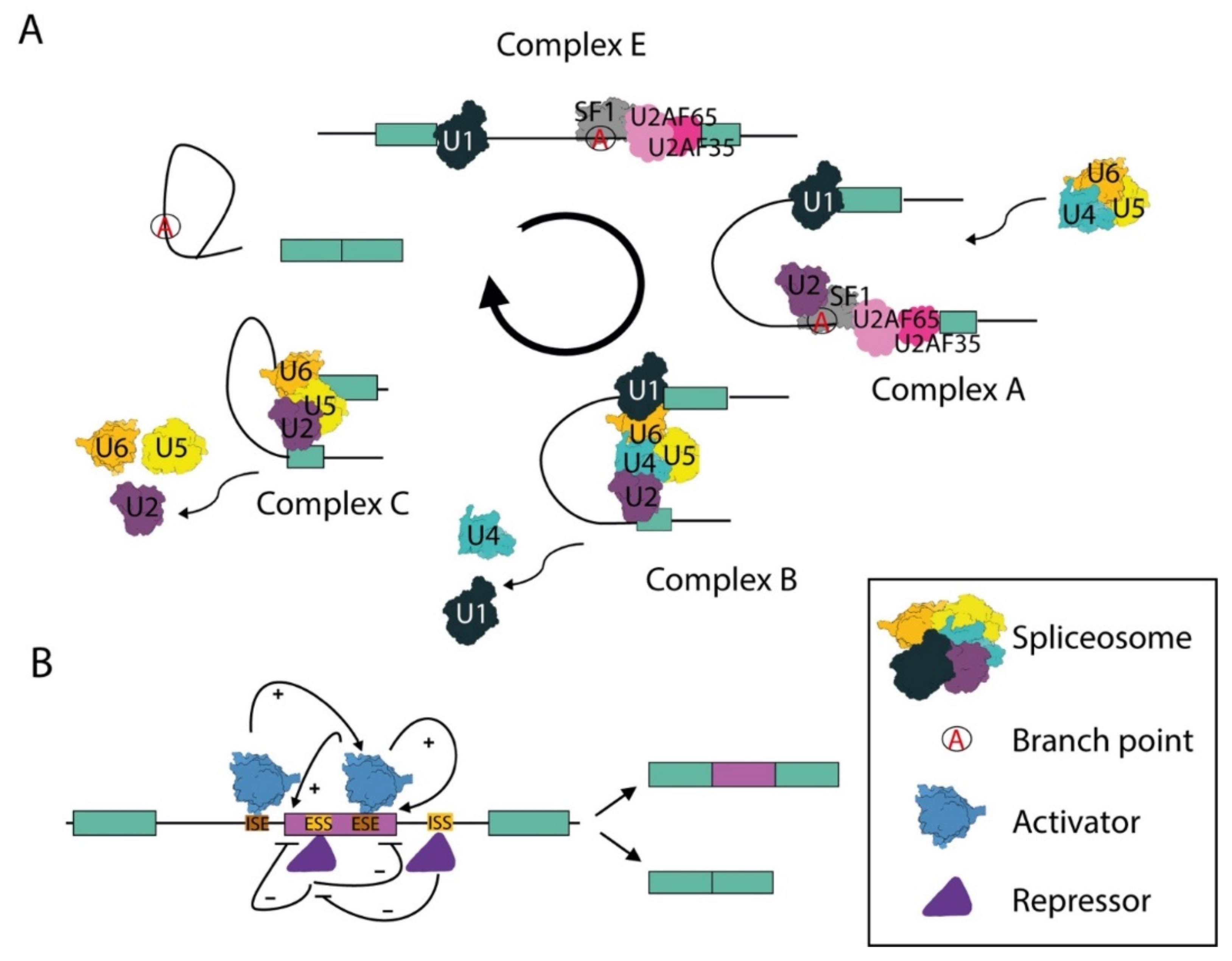
2.1. The Spliceosome and the Splicing Reaction
2.2. The Cleavage and Polyadenylation Complex and 3′ End Processing of Transcripts
2.3. Alternative Splicing and Alternative Polyadenylation: Evolutionary Devices That Amplify Genome Complexity and Plasticity
3. Post-Translational Modifications and RNA Processing
3.1. Sumoylation
3.2. Ubiquitination
3.3. Methylation
3.4. Acetylation
3.5. Phosphorylation
4. Post-Translational Modification of RNA-Binding Proteins
4.1. PTMs and Subcellular Localization of RBPs
4.2. PTMs and Activity of RBPs
5. Signaling Pathways That Regulate RNA Processing Machinery
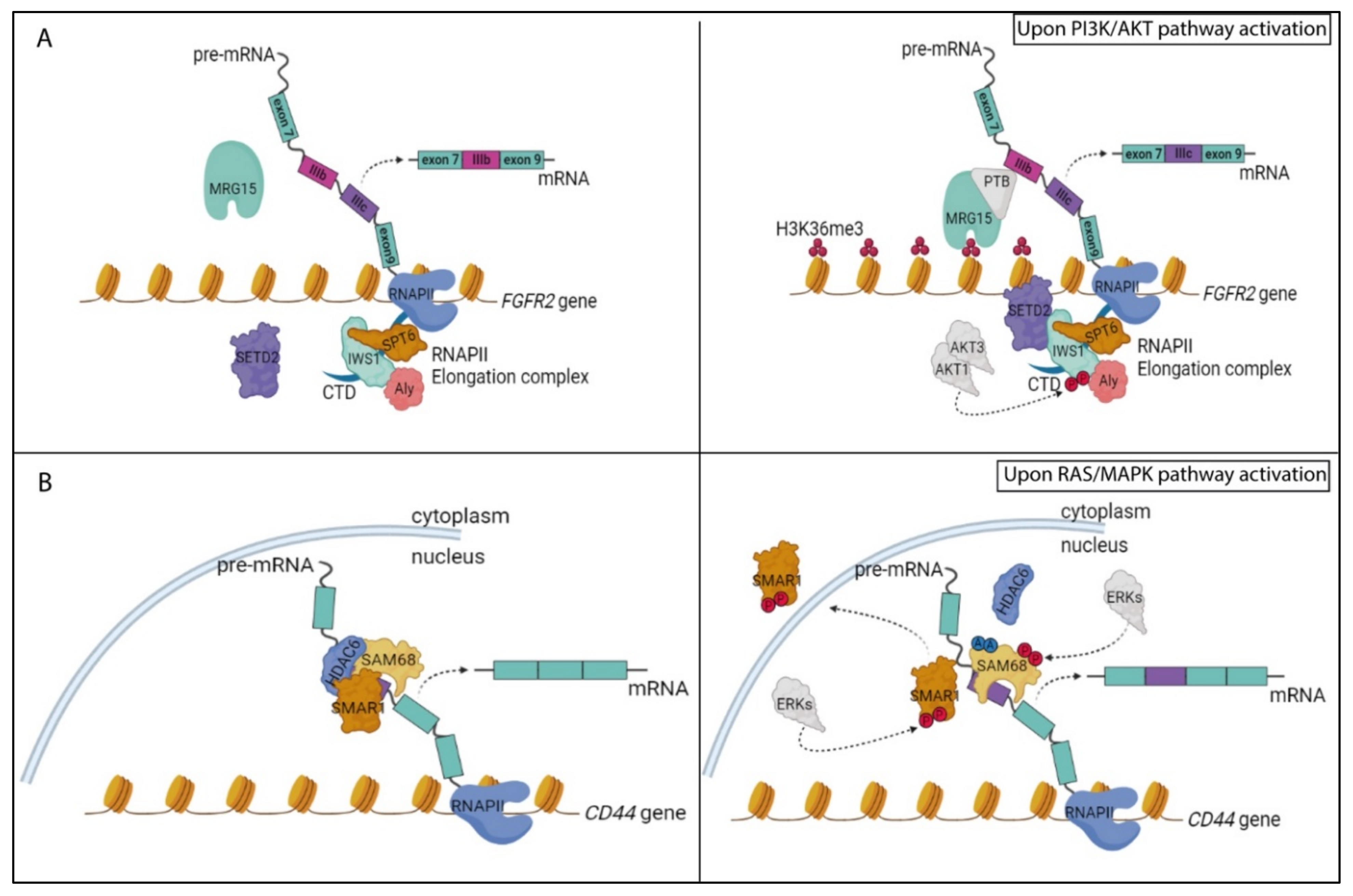
5.1. The PI3K/AKT Pathway and RNA Processing Regulation
5.2. The RAS/MAPK Pathway and RNA Processing Regulation
5.3. RNA Processing Regulation in Response to Heat Shock
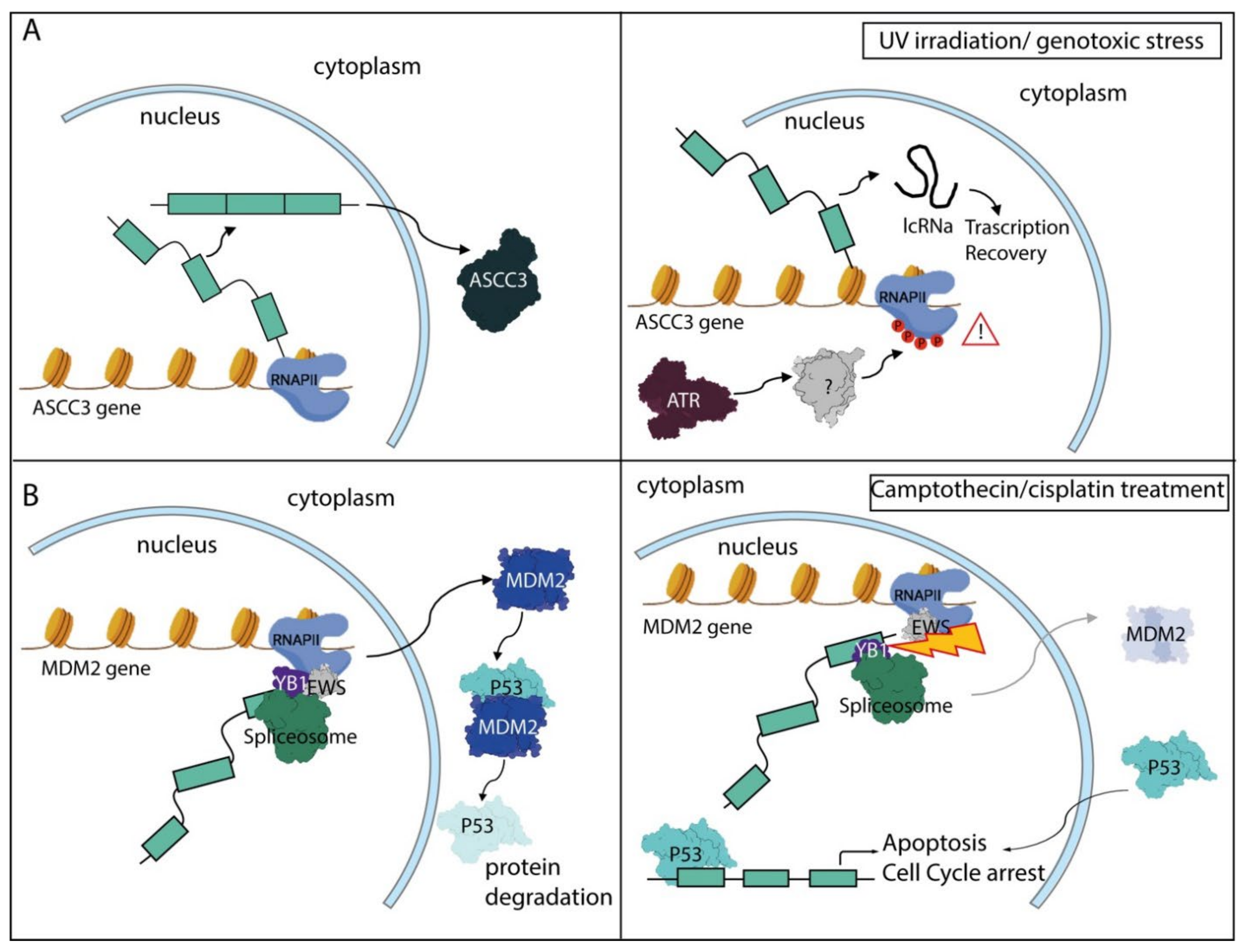
5.4. The DNA Damage Response Pathway and RNA Processing Regulation
5.5. The Circadian Clock Pathway and RNA Processing Regulation
6. RNA-Based Therapies as Tools to Correct Disease-Related Signal Transduction Pathways
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aDMA | ω-NG,NG-asymmetric di-methyl-arginine | mTORC1 | mTOR complex 1 |
| AKT/PKB | serine/threonine kinase | NER | nucleotide excision repair |
| ALS | amyotrophic lateral sclerosis | Noct | deadenylase nocturin |
| APA | alternative polyadenylation | pA | polyadenylation site |
| AS | alternative splicing | PABPN1 | nuclear poly(A) binding protein |
| ASCC3 | activating signal cointegrator 1 complex subunit 3 | PABPN1 | poly(A)-binding protein 1 |
| ASO | antisense oligonucleotide | PanNEN | pancreatic neuroendocrine neoplasms |
| ATM | Ataxia-telangiectasia mutated | PAP | poly(A) polymerase enzyme |
| ATR | ataxia telangiectasia and Rad3-related protein | PAS | polyadenylation signal |
| BMAIL1 | brain and muscle ARNT-like 1 | Pc2 | E3 ligase polycomb protein |
| C/P | cleavage and polyadenylation | PDAC | pancreatic ductal adenocarcinoma |
| CAMKIV | CA2+-calmodulin kinase IV | PER | period |
| cAMP | cyclin adenosine monophosphate | PI3K | phosphoinositide-3-kinase |
| CBP | CREB-binding protein | PKC | protein kinase C |
| CDK1 | cyclin dependent kinase 1 | Pre-mRNAs | precursor mRNAs |
| CDS | coding sequence | PRMT1-9 | arginine methyltransferases 1-9 |
| CFIm | cleavage factor I | Prp8 | pre-mRNA-processing-splicing-factor 8 |
| cGMP | cyclin guanosinemonophosphate | PTEN | phosphatase and tensin homolog |
| CHK1 | checkpoint kinase1 | PTM | post-translational modification |
| CHK2 | checkpoint kinase 2 | R15-LOX | reticulocyte-15-lipoxygenase |
| CIRBP | cold-inducible RNA-binding protein | RBM20 | RNA-binding motif 20 protein |
| CLK1-4 | CDC-like kinase | RBPs | RNA-binding proteins |
| CLOCK | circadian locomotor output cycles kaput | RGG | arginine–glycine–glycine-rich |
| CPSF | cleavage and polyadenylation specificity factor | RGG | glycine/arginine rich |
| CRY | cryptochrome | RNAPII | RNA polymerase II |
| CSTF | cleavage stimulation factor | RRE | receptor-binding element |
| CTD | carbossi-terminal domain | RRM | RNA recognition motif |
| DAZAP1 | DAZ-associated protein 1 | SCN | superchiasmatic nucleus of the hypothalamus |
| DDR | DNA damage response | sDMA | ω-NG, N’G-symmetric di-methyl-arginine |
| DESI1/2 | deSUMOylating isoptidase1/2 | SF1 | splicing factor 1 |
| DNA-PK | DNA-dependent protein kinase | SH3 | Src homology 3 |
| DSE | regulatory downstream sequence elements | SIMs | SUMO-interacting motifs |
| EDA | extra domain A | SIRT1 | stress response protein 1 |
| ERK | extracellular signal-regulated kinase | SMA | spinal muscular atrophy |
| FGFR2 | fibroblast growth factor receptor 2 | SMAR1 | scaffold/matrix associated region binding protein1 |
| FN | fibronectin | SMN | survival motor neuron protein |
| FUS | fused in sarcoma | snRNA | small nuclear RNA |
| FXR2P | fragile X-related protein 2 | snRNPs | small nuclear ribonucleoprotein particles |
| GCN5 | general control non-repressed 5 protein | SR | serine–arginine-rich proteins |
| H3K36me3 | trimethylation of lysine 36 of the histone 3 | SRPK2 | SR protein kinase 2 |
| HAT | histone acetyltransferase | SRSF1 | serine and arginine rich splicing factor 1 |
| HDACs | histone deacetylate enzymes | ssDNA | Single-stranded DNA |
| hnRNPs | heterogeneous nuclear ribonucleoproteins | STAGA | SAGA-like HAT complex |
| HSE | heat shock elements | STAR | signal transduction and activation of RNA |
| HSF1 | heat shock transcription factor 1 | TBK1-IRF3 | TANK-binding kinase1-interferon regulatory factor 3 |
| HsfA2 | heat shock transcription factor A2 | TRN1 | transportin 1 |
| HSPs | heat shock proteins | U2AF35 | U2 Auxiliary Factor of 35 kilodaltons |
| HSV-1 | herpes simplex virus 1 | U2AF65 | U2 Auxiliary Factor of 65 kilodaltons |
| HuR/ELAV1 | Hu antigen R/embryonic lethal, abnormal vision, Drosophila homolog-like 1 | UBDs | ubiquitin-binding domains |
| IPA | internal pA | USE | regulatory upstream sequence elements |
| ipRGCs | intrinsically photosensitive retinal ganglion cells | USPL1 | ubiquitin-specific protease-like 1 |
| IWS1 | interacts with SUPT6H, CTD assembly factor1 | UTR | untranslated region |
| MAPKs | Mitogen-activated protein kinases | UV | ultraviolet irradiation |
| MMA | ω-NG-mono-methyl-arginine | WTF9 | what’s this factor 9 |
| mTOR | target of rapamycin |
References
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ko, P.; Dixon, S.J. Protein palmitoylation and cancer. EMBO Rep. 2018, 19, e46666. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Singh, H.R.; Ostwal, Y.B. Post-Translational Modification, Phase Separation, and Robust Gene Transcription. Trends Genet. 2019, 35, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.G.; Rape, M. Ubiquitin-dependent regulation of transcription in development and disease. EMBO Rep. 2021, 22, e51078. [Google Scholar] [CrossRef] [PubMed]
- Filtz, T.M.; Vogel, W.; Leid, M. Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol. Sci. 2013, 35, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.D.; Li, H.; Ruthenburg, A.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.T.; Graf, T. The transcription factor code: A beacon for histone methyltransferase docking. Trends Cell Biol. 2021, 31, 792–800. [Google Scholar] [CrossRef]
- Papasaikas, P.; Valcárcel, J. The Spliceosome: The Ultimate RNA Chaperone and Sculptor. Trends Biochem. Sci. 2015, 41, 33–45. [Google Scholar] [CrossRef]
- Kastner, B.; Will, C.L.; Stark, H.; Lührmann, R. Structural Insights into Nuclear pre-mRNA Splicing in Higher Eukaryotes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032417. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 2016, 352. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2016, 18, 18–30. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Valcárcel, J. Building specificity with nonspecific RNA-binding proteins. Nat. Struct. Mol. Biol. 2005, 12, 645–653. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Paronetto, M.P.; Passacantilli, I.; Sette, C. Alternative splicing and cell survival: From tissue homeostasis to disease. Cell Death Differ. 2016, 23, 1919–1929. [Google Scholar] [CrossRef]
- Gruber, A.J.; Zavolan, M. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 2019, 20, 599–614. [Google Scholar] [CrossRef]
- Li, W.; You, B.; Hoque, M.; Zheng, D.; Luo, W.; Ji, Z.; Park, J.Y.; Gunderson, S.I.; Kalsotra, A.; Manley, J.; et al. Systematic Profiling of Poly(A)+ Transcripts Modulated by Core 3’ End Processing and Splicing Factors Reveals Regulatory Rules of Alternative Cleavage and Polyadenylation. PLoS Genet. 2015, 11, e1005166. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Bielli, P.; Compagnucci, C.; Cesari, E.; Volpe, E.; Vecchioli, S.F.; Sette, C. Sam68 promotes self-renewal and glycolytic metabolism in mouse neural progenitor cells by modulating Aldh1a3 pre-mRNA 3’-end processing. eLife 2016, 5, e20750. [Google Scholar] [CrossRef]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and Consequences of Alternative Polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef]
- Luco, R.F.; Allo, M.; Schor, I.E.; Kornblihtt, A.; Misteli, T. Epigenetics in Alternative Pre-mRNA Splicing. Cell 2011, 144, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of Alternative Splicing Through Coupling with Transcription and Chromatin Structure. Annu. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef] [PubMed]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Seeler, J.-S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Xu, H.; Wang, H.; Zhao, W.; Fu, S.; Li, Y.; Ni, W.; Xin, Y.; Li, W.; Yang, C.; Bai, Y.; et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics 2020, 10, 5671–5686. [Google Scholar] [CrossRef]
- Liang, Y.-C.; Lee, C.-C.; Yao, Y.-L.; Lai, C.-C.; Schmitz, L.; Yang, W.-M. SUMO5, a Novel Poly-SUMO Isoform, Regulates PML Nuclear Bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef]
- Hendriks, I.; Vertegaal, A. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef]
- Richard, P.; Vethantham, V.; Manley, J.L. Roles of Sumoylation in mRNA Processing and Metabolism. Adv. Exp. Med. Biol. 2017, 15–33. [Google Scholar] [CrossRef]
- Vethantham, V.; Rao, N.; Manley, J.L. Sumoylation Modulates the Assembly and Activity of the Pre-mRNA 3′ Processing Complex. Mol. Cell. Biol. 2007, 27, 8848–8858. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ryder, U.; Lamond, A.; Mann, M. Large-Scale Proteomic Analysis of the Human Spliceosome. Genome Res. 2002, 12, 1231–1245. [Google Scholar] [CrossRef]
- Hutten, S.; Chachami, G.; Winter, U.; Melchior, F.; Lamond, A.I. A role for the CB-associated SUMO isopeptidase USPL1 in RNAPII-mediated snRNA transcription. J. Cell Sci. 2014, 127, 1065–1078. [Google Scholar] [CrossRef]
- Navascues, J.; Bengoechea, R.; Tapia, O.; Casafont, I.; Berciano, M.; Lafarga, M. SUMO-1 transiently localizes to Cajal bodies in mammalian neurons. J. Struct. Biol. 2008, 163, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, B.; Bragado, L.; Will, C.L.; Mammi, P.; Risso, G.; Urlaub, H.; Lührmann, R.; Srebrow, A. SUMO conjugation to spliceosomal proteins is required for efficient pre-mRNA splicing. Nucleic Acids Res. 2017, 45, 6729–6745. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, B.; Mammi, P.; Bragado, L.; Giono, L.E.; Srebrow, A. When SUMO met splicing. RNA Biol. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, H.; Wang, X.; Ullah, K.; Xu, G. Proteomic approaches for the profiling of ubiquitylation events and their applications in drug discovery. J. Proteom. 2021, 231, 103996. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta Bioenerg. 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Wakatsuki, S.; Walters, K.J. Ubiquitin-binding domains—From structures to functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 659–671. [Google Scholar] [CrossRef]
- Williamson, A.; Wickliffe, K.E.; Mellone, B.; Song, L.; Karpen, G.H.; Rape, M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc. Natl. Acad. Sci. USA 2009, 106, 18213–18218. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of Proteins by Ubiquitin and Ubiquitin-Like Proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Williamson, A.; Banerjee, S.; Philipp, I.; Rape, M. Mechanism of Ubiquitin-Chain Formation by the Human Anaphase-Promoting Complex. Cell 2008, 133, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Sun, L.J. Nonproteolytic Functions of Ubiquitin in Cell Signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Bellare, P.; Kutach, A.K.; Rines, A.K.; Guthrie, C.; Sontheimer, E.J. Ubiquitin binding by a variant Jab1/MPN domain in the essential pre-mRNA splicing factor Prp8p. RNA 2005, 12, 292–302. [Google Scholar] [CrossRef][Green Version]
- Wilkinson, C.R.; Dittmar, G.; Ohi, M.D.; Uetz, P.; Jones, N.; Finley, D. Ubiquitin-like Protein Hub1 Is Required for Pre-mRNA Splicing and Localization of an Essential Splicing Factor in Fission Yeast. Curr. Biol. 2004, 14, 2283–2288. [Google Scholar] [CrossRef]
- Bellare, P.; Small, E.C.; Huang, X.; Wohlschlegel, J.A.; Staley, J.P.; Sontheimer, E.J. A role for ubiquitin in the spliceosome assembly pathway. Nat. Struct. Mol. Biol. 2008, 15, 444–451. [Google Scholar] [CrossRef]
- Song, E.J.; Werner, S.L.; Neubauer, J.; Stegmeier, F.; Aspden, J.; Rio, D.; Harper, J.; Elledge, S.J.; Kirschner, M.W.; Rape, M. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010, 24, 1434–1447. [Google Scholar] [CrossRef]
- Das, T.; Park, J.K.; Park, J.; Kim, E.; Rape, M.; Kim, E.E.; Song, E.J. USP15 regulates dynamic protein-protein interactions of the spliceosome through deubiquitination of PRP31. Nucleic Acids Res. 2017, 45, 4866–4880. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Wan, L.; Dreyfuss, G. Why do cells need an assembly machine for RNA–protein complexes? Trends Cell Biol. 2004, 14, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Eggert, C.; Bühler, D.; Brahms, H.; Kambach, C.; Fischer, U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001, 11, 1990–1994. [Google Scholar] [CrossRef]
- Boisvert, F.-M.; Côté, J.; Boulanger, M.-C.; Cléroux, P.; Bachand, F.; Autexier, C.; Richard, S. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J. Cell Biol. 2002, 159, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gu, L.; Liu, C.; Lu, T.; Lu, F.; Lu, Z.; Cui, P.; Pei, Y.; Wang, B.; Hu, S.; et al. Arginine methylation mediated by the Arabidopsis homolog of PRMT5 is essential for proper pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 2010, 107, 19114–19119. [Google Scholar] [CrossRef]
- Sanchez, S.E.; Petrillo, E.; Beckwith, E.J.; Zhang, X.; Rugnone, M.L.; Hernando, C.E.; Cuevas, J.C.; Herz, M.A.G.; Depetris-Chauvin, A.; Simpson, C.G.; et al. A methyl transferase links the circadian clock to the regulation of alternative splicing. Nature 2010, 468, 112–116. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Milliman, E.J.; Goulet, I.; Côté, J.; Jackson, C.; Vollbracht, J.A.; Yu, M.C. Protein Arginine Methylation Facilitates Cotranscriptional Recruitment of Pre-mRNA Splicing Factors. Mol. Cell. Biol. 2010, 30, 5245–5256. [Google Scholar] [CrossRef]
- Boisvert, F.-M.; Côté, J.; Boulanger, M.-C.; Richard, S. A Proteomic Analysis of Arginine-methylated Protein Complexes. Mol. Cell. Proteom. 2003, 2, 1319–1330. [Google Scholar] [CrossRef]
- Passos, D.O.; Quaresma, A.J.; Kobarg, J. The methylation of the C-terminal region of hnRNPQ (NSAP1) is important for its nuclear localization. Biochem. Biophys. Res. Commun. 2006, 346, 517–525. [Google Scholar] [CrossRef]
- Nichols, R.C.; Wang, X.W.; Tang, J.; Hamilton, B.; High, F.A.; Herschman, H.R.; Rigby, W.F. The RGG Domain in hnRNP A2 Affects Subcellular Localization. Exp. Cell Res. 2000, 256, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Allemand, E.; Zhang, Z.; Karni, R.; Myers, M.P.; Krainer, A.R. Arginine Methylation Controls the Subcellular Localization and Functions of the Oncoprotein Splicing Factor SF2/ASF. Mol. Cell. Biol. 2010, 30, 2762–2774. [Google Scholar] [CrossRef]
- Liu, Q.; Dreyfuss, G. In vivo and in vitro arginine methylation of RNA-binding proteins. Mol. Cell. Biol. 1995, 15, 2800–2808. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, R.; Chien, C.D.; Avantaggiati, M.L.; Muller, S. An Acetylation Switch Regulates SUMO-Dependent Protein Interaction Networks. Mol. Cell 2012, 46, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-Y.; Qi, Y.; Lu, J.-Y.; Pan, X.; Yuan, D.S.; Zhao, Y.; Bader, J.S.; Boeke, J.D. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev. 2008, 22, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Vogelauer, M.; Wu, J.; Suka, N.; Grunstein, M. Global histone acetylation and deacetylation in yeast. Nature 2000, 408, 495–498. [Google Scholar] [CrossRef]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C. Tetrahymena Histone Acetyltransferase A: A Homolog to Yeast Gcn5p Linking Histone Acetylation to Gene Activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Gunderson, F.Q.; Johnson, T.L. Acetylation by the Transcriptional Coactivator Gcn5 Plays a Novel Role in Co-Transcriptional Spliceosome Assembly. PLoS Genet. 2009, 5, e1000682. [Google Scholar] [CrossRef]
- Gunderson, F.Q.; Merkhofer, E.C.; Johnson, T.L. Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc. Natl. Acad. Sci. USA 2011, 108, 2004–2009. [Google Scholar] [CrossRef]
- Martinez, E.; Palhan, V.B.; Tjernberg, A.; Lymar, E.S.; Gamper, A.; Kundu, T.K.; Chait, B.T.; Roeder, R.G. Human STAGA Complex Is a Chromatin-Acetylating Transcription Coactivator That Interacts with Pre-mRNA Splicing and DNA Damage-Binding Factors In Vivo. Mol. Cell. Biol. 2001, 21, 6782–6795. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.N.; van Santen, M.A.; Schwienhorst, A.; Urlaub, H.; Lührmann, R. Stalling of spliceosome assembly at distinct stages by small-molecule inhibitors of protein acetylation and deacetylation. RNA 2008, 15, 153–175. [Google Scholar] [CrossRef] [PubMed]
- Matter, N.; Herrlich, P.; König, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Babic, I.; Jakymiw, A.; Fujita, D.J. The RNA binding protein Sam68 is acetylated in tumor cell lines, and its acetylation correlates with enhanced RNA binding activity. Oncogene 2004, 23, 3781–3789. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, V.; Tapia, O.; Sharma, S.; Bengoechea, R.; Stoecklin, G.; Lafarga, M.; Berciano, M.T. CBP-mediated SMN acetylation modulates Cajal body biogenesis and the cytoplasmic targeting of SMN. Cell. Mol. Life Sci. 2017, 75, 527–546. [Google Scholar] [CrossRef]
- Ubersax, J.A.; Farel, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Shi, Y.; Reddy, B.; Manley, J.L. PP1/PP2A Phosphatases Are Required for the Second Step of Pre-mRNA Splicing and Target Specific snRNP Proteins. Mol. Cell 2006, 23, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Hartmuth, K.; Möhlmann, S.; Urlaub, H.; Ficner, R.; Lührmann, R. Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 435–443. [Google Scholar] [CrossRef]
- Naro, C.; Sette, C. Phosphorylation-Mediated Regulation of Alternative Splicing in Cancer. Int. J. Cell Biol. 2013, 2013, 151839. [Google Scholar] [CrossRef]
- Naro, C.; Bielli, P.; Sette, C. Oncogenic dysregulation of pre-mRNA processing by protein kinases: Challenges and therapeutic opportunities. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.-D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2020, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Sette, C. Post-translational Regulation of STAR Proteins and Effects on Their Biological Functions. Adv. Exp. Med. Biol. 2010, 693, 54–66. [Google Scholar] [CrossRef]
- Lukong, K.E.; Larocque, D.; Tyner, A.; Richard, S. Tyrosine Phosphorylation of Sam68 by Breast Tumor Kinase Regulates Intranuclear Localization and Cell Cycle Progression. J. Biol. Chem. 2005, 280, 38639–38647. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Cappellari, M.; Busà, R.; Pedrotti, S.; Vitali, R.; Comstock, C.; Hyslop, T.; Knudsen, K.; Sette, C. Alternative Splicing of the Cyclin D1 Proto-Oncogene Is Regulated by the RNA-Binding Protein Sam68. Cancer Res. 2009, 70, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Pedrotti, S.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Stamm, S.; Manley, J.; Sette, C. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010, 29, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Tradewell, M.L.; Yu, Z.; Tibshirani, M.; Boulanger, M.-C.; Durham, H.D.; Richard, S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2011, 21, 136–149. [Google Scholar] [CrossRef]
- Arenas, A.; Chen, J.; Kuang, L.; Barnett, K.R.; Kasarskis, E.J.; Gal, J.; Zhu, H. Lysine acetylation regulates the RNA binding, subcellular localization and inclusion formation of FUS. Hum. Mol. Genet. 2020, 29, 2684–2697. [Google Scholar] [CrossRef] [PubMed]
- Maraschi, A.; Gumina, V.; Dragotto, J.; Colombrita, C.; Mompeán, M.; Buratti, E.; Silani, V.; Feligioni, M.; Ratti, A. SUMOylation Regulates TDP-43 Splicing Activity and Nucleocytoplasmic Distribution. Mol. Neurobiol. 2021, 1–21. [Google Scholar] [CrossRef]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2014, 6, 93–110. [Google Scholar] [CrossRef]
- Aubol, B.E.; Wu, G.; Keshwani, M.M.; Movassat, M.; Fattet, L.; Hertel, K.; Fu, X.-D.; Adams, J.A. Release of SR Proteins from CLK1 by SRPK1: A Symbiotic Kinase System for Phosphorylation Control of Pre-mRNA Splicing. Mol. Cell 2016, 63, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Aubol, B.E.; Keshwani, M.M.; Fattet, L.; Adams, J.A. Mobilization of a splicing factor through a nuclear kinase–kinase complex. Biochem. J. 2018, 475, 677–690. [Google Scholar] [CrossRef]
- Lai, M.C.; Lin, R.I.; Huang, S.Y.; Tsai, C.W.; Tarn, W.Y. A human importin-β family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J. Biol. Chem. 2000, 275, 7950–7957. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.-C.; Lin, R.-I.; Tarn, W.-Y. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10154–10159. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, W.; Han, Q.; Wang, Y.; Li, C.; Zhang, P.; Xu, H. Post-translational modification control of RNA-binding protein hnRNPK function. Open Biol. 2019, 9, 180239. [Google Scholar] [CrossRef] [PubMed]
- Habelhah, H.; Shah, K.; Huang, L.; Ostareck-Lederer, A.; Burlingame, A.L.; Shokat, K.M.; Hentze, M.; Ronai, Z. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nature 2001, 3, 325–330. [Google Scholar] [CrossRef]
- Chang, Y.-I.; Hsu, S.-C.; Chau, G.-Y.; Huang, C.-Y.F.; Sung, J.-S.; Hua, W.-K.; Lin, W.-J. Identification of the methylation preference region in heterogeneous nuclear ribonucleoprotein K by protein arginine methyltransferase 1 and its implication in regulating nuclear/cytoplasmic distribution. Biochem. Biophys. Res. Commun. 2011, 404, 865–869. [Google Scholar] [CrossRef]
- Phoomak, C.; Park, D.; Silsirivanit, A.; Sawanyawisuth, K.; Vaeteewoottacharn, K.; Detarya, M.; Wongkham, C.; Lebrilla, C.B.; Wongkham, S. O-Glc NA c-induced nuclear translocation of hn RNP -K is associated with progression and metastasis of cholangiocarcinoma. Mol. Oncol. 2018, 13, 338–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wen, M.; Cao, X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science 2019, 365. [Google Scholar] [CrossRef]
- Fronz, K.; Guttinger, S.; Burkert, K.; Kühn, U.; Stöhr, N.; Schierhorn, A.; Wahle, E. Arginine Methylation of the Nuclear Poly(A) Binding Protein Weakens the Interaction with Its Nuclear Import Receptor, Transportin. J. Biol. Chem. 2011, 286, 32986–32994. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Hsieh, W.-Y.; Chen, L.-Y.; Li, C. Protein arginine methylation of SERBP1 by protein arginine methyltransferase 1 affects cytoplasmic/nuclear distribution. J. Cell. Biochem. 2012, 113, 2721–2728. [Google Scholar] [CrossRef] [PubMed]
- Côté, J.; Boisvert, F.-M.; Boulanger, M.-C.; Bedford, M.T.; Richard, S. Sam68 RNA Binding Protein Is an In Vivo Substrate for Protein ArginineN-Methyltransferase 1. Mol. Biol. Cell 2003, 14, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Du, A.; Gu, J.; Duan, G.; Wang, C.; Gui, X.; Ma, Z.; Qian, B.; Deng, X.; Zhang, K.; et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 2019, 29, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Stackpole, E.E.; Akins, M.; Fallon, J.R. N-myristoylation regulates the axonal distribution of the Fragile X-related protein FXR2P. Mol. Cell. Neurosci. 2014, 62, 42–50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhäusser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide Analysis of Regulatory Interactions of the RNA-Binding Protein HuR. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef]
- Masuda, K.; Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Lee, E.K.; Tominaga, K.; Selimyan, R.; Martindale, J.L.; Yang, X.; Lehrmann, E.; et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011, 30, 1040–1053. [Google Scholar] [CrossRef]
- Kim, H.H.; Yang, X.; Kuwano, Y.; Gorospe, M. Modification at HuR(S242) alters HuR localization and proliferative influence. Cell Cycle 2008, 7, 3371–3377. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Pullmann, R.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.; et al. Phosphorylation of HuR by Chk2 Regulates SIRT1 Expression. Mol. Cell 2007, 25, 543–557. [Google Scholar] [CrossRef]
- Yu, T.-X.; Wang, P.-Y.; Rao, J.N.; Zou, T.; Liu, L.; Xiao, L.; Gorospe, M.; Wang, J.-Y. Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res. 2011, 39, 8472–8487. [Google Scholar] [CrossRef]
- Frisone, P.; Pradella, D.; Di Matteo, A.; Belloni, E.; Ghigna, C.; Paronetto, M.P. SAM68: Signal Transduction and RNA Metabolism in Human Cancer. BioMed Res. Int. 2015, 2015, 528954. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Venables, J.P.; Elliott, D.J.; Geremia, R.; Rossi, P.; Sette, C. tr-kit promotes the formation of a multimolecular complex composed by Fyn, PLCγ1 and Sam68. Oncogene 2003, 22, 8707–8715. [Google Scholar] [CrossRef] [PubMed]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. SAM68 Regulates Neuronal Activity-Dependent Alternative Splicing of Neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Messina, V.; Bianchi, E.; Barchi, M.; Vogel, G.; Moretti, C.; Palombi, F.; Stefanini, M.; Geremia, R.; Richard, S.; et al. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. J. Cell Biol. 2009, 185, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Frankel, A.; Yaffe, M.B.; Clarke, S.; Leder, P.; Richard, S. Arginine Methylation Inhibits the Binding of Proline-rich Ligands to Src Homology 3, but Not WW, Domains. J. Biol. Chem. 2000, 275, 16030–16036. [Google Scholar] [CrossRef]
- Siam, A.; Baker, M.; Amit, L.; Regev, G.; Rabner, A.; Najar, R.A.; Bentata, M.; Dahan, S.; Cohen, K.; Araten, S.; et al. Regulation of alternative splicing by p300-mediated acetylation of splicing factors. RNA 2019, 25, 813–824. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, X.; Liu, N.; Wang, C.; Zhang, L.; Mo, W.; Hu, G. Arginine methylation of hnRNP K enhances p53 transcriptional activity. FEBS Lett. 2008, 582, 1761–1765. [Google Scholar] [CrossRef]
- Moumen, A.; Masterson, P.; O’Connor, M.J.; Jackson, S.P. hnRNP K: An HDM2 Target and Transcriptional Coactivator of p53 in Response to DNA Damage. Cell 2005, 123, 1065–1078. [Google Scholar] [CrossRef]
- Ostareck-Lederer, A.; Ostareck, D.H.; Rucknagel, K.P.; Schierhorn, A.; Moritz, B.; Huttelmaier, S.; Flach, N.; Handoko, L.; Wahle, E. Asymmetric Arginine Dimethylation of Heterogeneous Nuclear Ribonucleoprotein K by Protein-arginine Methyltransferase 1 Inhibits Its Interaction with c-Src. J. Biol. Chem. 2006, 281, 11115–11125. [Google Scholar] [CrossRef]
- Ostareck-Lederer, A.; Ostareck, D.H.; Cans, C.; Neubauer, G.; Bomsztyk, K.; Superti-Furga, G.; Hentze, M.W. c-Src-Mediated Phosphorylation of hnRNP K Drives Translational Activation of Specifically Silenced mRNAs. Mol. Cell. Biol. 2002, 22, 4535–4543. [Google Scholar] [CrossRef]
- Ostareck-Lederer, A.; Ostareck, D.H. Control of mRNA translation and stability in haematopoietic cells: The function of hnRNPs K and E1/E2. Biol. Cell 2004, 96, 407–411. [Google Scholar] [CrossRef]
- Yang, J.-H.; Chiou, Y.-Y.; Fu, S.-L.; Shih, I.-Y.; Weng, T.-H.; Lin, W.-J.; Lin, C.-H. Arginine methylation of hnRNPK negatively modulates apoptosis upon DNA damage through local regulation of phosphorylation. Nucleic Acids Res. 2014, 42, 9908–9924. [Google Scholar] [CrossRef]
- Chu, P.-C.; Chuang, H.-C.; Kulp, S.K.; Chen, C.-S. The mRNA-stabilizing Factor HuR Protein Is Targeted by β-TrCP Protein for Degradation in Response to Glycolysis Inhibition. J. Biol. Chem. 2012, 287, 43639–43650. [Google Scholar] [CrossRef]
- Moumen, A.; Magill, C.; Dry, K.; Jackson, S.P. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle 2013, 12, 698–704. [Google Scholar] [CrossRef]
- Pelisch, F.; Pozzi, B.; Risso, G.; Muñoz, M.J.; Srebrow, A. DNA Damage-induced Heterogeneous Nuclear Ribonucleoprotein K SUMOylation Regulates p53 Transcriptional Activation*. J. Biol. Chem. 2012, 287, 30789–30799. [Google Scholar] [CrossRef]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef]
- Dummler, B.; Hemmings, B. Physiological roles of PKB/Akt isoforms in development and disease. Biochem. Soc. Trans. 2007, 35, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Sanidas, I.; Polytarchou, C.; Hatziapostolou, M.; Ezell, S.A.; Kottakis, F.; Hu, L.; Guo, A.; Xie, J.; Comb, M.J.; Iliopoulos, D.; et al. Phosphoproteomics Screen Reveals Akt Isoform-Specific Signals Linking RNA Processing to Lung Cancer. Mol. Cell 2014, 53, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of Alternative Splicing by Histone Modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.-L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.-C.; Chen, C.-T.; Ping, B.; Otte, A.P.; Hung, M.-C. Akt-Mediated Phosphorylation of EZH2 Suppresses Methylation of Lysine 27 in Histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U Enhances Caspase-9 Splicing and Is Modulated by AKT-dependent Phosphorylation of hnRNP L. J. Biol. Chem. 2013, 288, 8575–8584. [Google Scholar] [CrossRef]
- Shultz, J.C.; Goehe, R.W.; Wijesinghe, D.S.; Murudkar, C.; Hawkins, A.J.; Shay, J.W.; Minna, J.D.; Chalfant, C.E. Alternative Splicing of Caspase 9 Is Modulated by the Phosphoinositide 3-Kinase/Akt Pathway via Phosphorylation of SRp30a. Cancer Res. 2010, 70, 9185–9196. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.A.; Chalfant, C.E.; Watson, J.E.; Wyatt, J.R.; Dean, N.M.; Eichler, D.C.; Cooper, D.R. Insulin Regulates Alternative Splicing of Protein Kinase C βII through a Phosphatidylinositol 3-Kinase-dependent Pathway Involving the Nuclear Serine/Arginine-rich Splicing Factor, SRp40, in Skeletal Muscle Cells. J. Biol. Chem. 2001, 276, 22648–22654. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.A.; Kaneko, S.; Apostolatos, H.S.; Bae, S.S.; Watson, J.E.; Davidowitz, K.; Chappell, D.S.; Birnbaum, M.; Cheng, J.Q.; Cooper, D.R. Molecular and Genetic Studies Imply Akt-mediated Signaling Promotes Protein Kinase CβII Alternative Splicing via Phosphorylation of Serine/Arginine-rich Splicing Factor SRp40. J. Biol. Chem. 2005, 280, 14302–14309. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.; Pelisch, F.; Tanos, T.; Muñoz, M.J.; Wengier, D.; Quadrana, L.; Sanford, J.R.; Muschietti, J.; Kornblihtt, A.; Caceres, J.; et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 2005, 12, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Risso, G.; Pelisch, F.; Pozzi, B.; Mammi, P.; Blaustein, M.; Colman-Lerner, A.; Srebrow, A. Modification of Akt by SUMO conjugation regulates alternative splicing and cell cycle. Cell Cycle 2013, 12, 3165–3174. [Google Scholar] [CrossRef]
- Jiang, K.; Patel, N.A.; Watson, J.E.; Apostolatos, H.; Kleiman, E.; Hanson, O.; Hagiwara, M.; Cooper, D.R. Akt2 Regulation of Cdc2-Like Kinases (Clk/Sty), Serine/Arginine-Rich (SR) Protein Phosphorylation, and Insulin-Induced Alternative Splicing of PKCβII Messenger Ribonucleic Acid. Endocrinology 2008, 150, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.-W.; Liu, X.; Fu, H.; Rees, H.; Yepes, M.; Levey, A.; Ye, K. Interaction of Akt-phosphorylated SRPK2 with 14-3-3 Mediates Cell Cycle and Cell Death in Neurons. J. Biol. Chem. 2009, 284, 24512–24525. [Google Scholar] [CrossRef]
- Zhou, Z.; Qiu, J.; Liu, W.; Zhou, Y.; Plocinik, R.M.; Li, H.; Hu, Q.; Ghosh, G.; Adams, J.A.; Rosenfeld, M.G.; et al. The Akt-SRPK-SR Axis Constitutes a Major Pathway in Transducing EGF Signaling to Regulate Alternative Splicing in the Nucleus. Mol. Cell 2012, 47, 422–433. [Google Scholar] [CrossRef]
- White, E.S.; Sagana, R.L.; Booth, A.J.; Yan, M.; Cornett, A.M.; Bloomheart, C.A.; Tsui, J.L.; Wilke, C.A.; Moore, B.B.; Ritzenthaler, J.D.; et al. Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp. Cell Res. 2010, 316, 2644–2653. [Google Scholar] [CrossRef]
- Passacantilli, I.; Frisone, P.; de Paola, E.; Fidaleo, M.; Paronetto, M.P. hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 2017, 45, 12270–12284. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yin, Z.; Tan, B.; Guo, W. Insulin regulates titin pre-mRNA splicing through the PI3K-Akt-mTOR kinase axis in a RBM20-dependent manner. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schäfer, S.; Greaser, M.L.; Radke, M.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Dang, Y.; Zhang, W.; Zhao, X.; Zhang, C.; Hou, Z.; Jin, Y.; McNutt, M.A.; Marks, A.R.; Yin, Y. PTEN arginine methylation by PRMT6 suppresses PI3K–AKT signaling and modulates pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 2019, 116, 6868–6877. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Cheng, C.; Sharp, P.A. Regulation of CD44 Alternative Splicing by SRm160 and Its Potential Role in Tumor Cell Invasion. Mol. Cell. Biol. 2006, 26, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Batsché, E.; Yaniv, M.; Muchardt, C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat. Struct. Mol. Biol. 2005, 13, 22–29. [Google Scholar] [CrossRef]
- Cheng, C.; Yaffe, M.B.; Sharp, P.A. A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev. 2006, 20, 1715–1720. [Google Scholar] [CrossRef]
- Valacca, C.; Bonomi, S.; Buratti, E.; Pedrotti, S.; Baralle, F.E.; Sette, C.; Ghigna, C.; Biamonti, G. Sam68 regulates EMT through alternative splicing–activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 2010, 191, 87–99. [Google Scholar] [CrossRef]
- Al-Ayoubi, A.M.; Zheng, H.; Liu, Y.; Bai, T.; Eblen, S.T. Mitogen-Activated Protein Kinase Phosphorylation of Splicing Factor 45 (SPF45) Regulates SPF45 Alternative Splicing Site Utilization, Proliferation, and Cell Adhesion. Mol. Cell. Biol. 2012, 32, 2880–2893. [Google Scholar] [CrossRef]
- Choudhury, R.; Roy, S.G.; Tsai, Y.S.; Tripathy, A.; Graves, L.M.; Wang, Z. The splicing activator DAZAP1 integrates splicing control into MEK/Erk regulated cell proliferation and migration. Nat. Commun. 2014, 5, 3078. [Google Scholar] [CrossRef]
- Nakka, K.K.; Chaudhary, N.; Joshi, S.; Bhat, J.; Singh, K.; Chatterjee, S.; Malhotra, R.; De, A.; Santra, M.K.; Dilworth, F.J.; et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc. Natl. Acad. Sci. USA 2015, 112, E3374–E3383. [Google Scholar] [CrossRef]
- Naro, C.; Pellegrini, L.; Jolly, A.; Farini, D.; Cesari, E.; Bielli, P.; de la Grange, P.; Sette, C. Functional Interaction between U1snRNP and Sam68 Insures Proper 3′ End Pre-mRNA Processing during Germ Cell Differentiation. Cell Rep. 2019, 26, 2929–2941. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Shalgi, R.; Hurt, J.A.; Lindquist, S.; Burge, C.B. Widespread Inhibition of Posttranscriptional Splicing Shapes the Cellular Transcriptome following Heat Shock. Cell Rep. 2014, 7, 1362–1370. [Google Scholar] [CrossRef]
- Shi, Y.; Manley, J. A Complex Signaling Pathway Regulates SRp38 Phosphorylation and Pre-mRNA Splicing in Response to Heat Shock. Mol. Cell 2007, 28, 79–90. [Google Scholar] [CrossRef]
- Bond, U.; James, T.C. Dynamic changes in small nuclear ribonucleoproteins of heat-stressed and thermotolerant HeLa cells. Int. J. Biochem. Cell Biol. 2000, 32, 643–656. [Google Scholar] [CrossRef]
- Metz, A.; Soret, J.; Vourc’H, C.; Tazi, J.; Jolly, C. A key role for stress-induced satellite III transcripts in the relocalization of splicing factors into nuclear stress granules. J. Cell Sci. 2004, 117, 4551–4558. [Google Scholar] [CrossRef]
- Bracken, A.P.; Bond, U. Reassembly and protection of small nuclear ribonucleoprotein particles by heat shock proteins in yeast cells. RNA 1999, 5, 1586–1596. [Google Scholar] [CrossRef]
- Bond, U. Heat shock but not other stress inducers leads to the disruption of a sub-set of snRNPs and inhibition of in vitro splicing in HeLa cells. EMBO J. 1988, 7, 3509–3518. [Google Scholar] [CrossRef]
- Jolly, C.; Vourc’H, C.; Robert-Nicoud, M.; Morimoto, R.I. Intron-independent Association of Splicing Factors with Active Genes. J. Cell Biol. 1999, 145, 1133–1143. [Google Scholar] [CrossRef]
- Marin-Vinader, L.; Shin, C.; Onnekink, C.; Manley, J.L.; Lubsen, N.H. Hsp27 Enhances Recovery of Splicing as well as Rephosphorylation of SRp38 after Heat Shock. Mol. Biol. Cell 2006, 17, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Katsogiannou, M.; Andrieu, C.; Baylot, V.; Baudot, A.; Dusetti, N.; Gayet, O.; Finetti, P.; Garrido, C.; Birnbaum, D.; Bertucci, F.; et al. The Functional Landscape of Hsp27 Reveals New Cellular Processes such as DNA Repair and Alternative Splicing and Proposes Novel Anticancer Targets. Mol. Cell. Proteom. 2014, 13, 3585–3601. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, A.; Ferrandino, G.; Credendino, S.C.; Moccia, C.; D’Angelo, F.; Miranda, B.; D’Ambrosio, C.; Bielli, P.; Spadaro, O.; Ceccarelli, M.; et al. DNAJC17 is localized in nuclear speckles and interacts with splicing machinery components. Sci. Rep. 2018, 8, 7794. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Howell, S. Heat Stress Responses and Thermotolerance in Maize. Int. J. Mol. Sci. 2021, 22, 948. [Google Scholar] [CrossRef]
- Hsu, Y.-W.; Juan, C.-T.; Wang, C.-M.; Jauh, G.-Y. Mitochondrial Heat Shock Protein 60s Interact with What’s This Factor 9 to Regulate RNA Splicing ofccmFCandrpl2. Plant Cell Physiol. 2018, 60, 116–125. [Google Scholar] [CrossRef]
- Liu, J.; Sun, N.; Liu, M.; Liu, J.C.; Du, B.; Wang, X.; Qi, X. An Autoregulatory Loop Controlling Arabidopsis HsfA2 Expression: Role of Heat Shock-Induced Alternative Splicing. Plant Physiol. 2013, 162, 512–521. [Google Scholar] [CrossRef]
- Lans, H.; Hoeijmakers, J.H.J.; Vermeulen, W.; Marteijn, J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019, 20, 766–784. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Carey, K.T.; Wickramasinghe, V.O. Regulatory Potential of the RNA Processing Machinery: Implications for Human Disease. Trends Genet. 2018, 34, 279–290. [Google Scholar] [CrossRef]
- Mikolaskova, B.; Jurcik, M.; Cipakova, I.; Kretova, M.; Chovanec, M.; Cipak, L. Maintenance of genome stability: The unifying role of interconnections between the DNA damage response and RNA-processing pathways. Curr. Genet. 2018, 64, 971–983. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Muñoz, M.J.; Perez-Santangelo, M.S.; Paronetto, M.P.; de la Mata, M.; Pelisch, F.; Boireau, S.; Glover-Cutter, K.; Ben-Dov, C.; Blaustein, M.; Lozano, J.J.; et al. DNA Damage Regulates Alternative Splicing through Inhibition of RNA Polymerase II Elongation. Cell 2009, 137, 708–720. [Google Scholar] [CrossRef]
- Hsin, J.-P.; Manley, J. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef]
- Muñoz, M.J.; Moreno, N.N.; Giono, L.E.; Botto, A.E.C.; Dujardin, G.; Bastianello, G.; Lavore, S.; Torres-Méndez, A.; Menck, C.F.; Blencowe, B.J.; et al. Major Roles for Pyrimidine Dimers, Nucleotide Excision Repair, and ATR in the Alternative Splicing Response to UV Irradiation. Cell Rep. 2017, 18, 2868–2879. [Google Scholar] [CrossRef] [PubMed]
- Williamson, L.; Saponaro, M.; Boeing, S.; East, P.; Mitter, R.; Kantidakis, T.; Kelly, G.; Lobley, A.; Walker, J.; Spencer-Dene, B.; et al. UV Irradiation Induces a Non-coding RNA that Functionally Opposes the Protein Encoded by the Same Gene. Cell 2017, 168, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.A.; van Ijcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.J.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Bielli, P.; Pagliarini, V.; Sette, C. The interplay between DNA damage response and RNA processing: The unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015, 6, 142. [Google Scholar] [CrossRef]
- Akaike, Y.; Masuda, K.; Kuwano, Y.; Nishida, K.; Kajita, K.; Kurokawa, K.; Satake, Y.; Shoda, K.; Imoto, I.; Rokutan, K. HuR Regulates Alternative Splicing of the TRA2 β Gene in Human Colon Cancer Cells under Oxidative Stress. Mol. Cell. Biol. 2014, 34, 2857–2873. [Google Scholar] [CrossRef]
- Zhang, S. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res. 2004, 32, 1–10. [Google Scholar] [CrossRef]
- Liu, S.; Shao, Y.; Wang, Q.; Zhai, Y.; Li, X. Genotoxic stress causes the accumulation of DNA-dependent protein kinase catalytic subunit phosphorylated at serine 2056 at nuclear speckles and alters pre- mRNA alternative splicing. FEBS Open Bio 2018, 9, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Comiskey, D.F.; Jacob, A.G.; Singh, R.K.; Tapia-Santos, A.S.; Chandler, D.S. Splicing factor SRSF1 negatively regulates alternative splicing of MDM2 under damage. Nucleic Acids Res. 2015, 43, 4202–4218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Trépanier, V.; Beaujois, R.; Viranaicken, W.; Drobetsky, E.; DesGroseillers, L. The downregulation of the RNA-binding protein Staufen2 in response to DNA damage promotes apoptosis. Nucleic Acids Res. 2016, 44, 3695–3712. [Google Scholar] [CrossRef] [PubMed]
- Adesso, L.; Calabretta, S.; Barbagallo, F.; Capurso, G.; Pilozzi, E.; Geremia, R.; Fave, G.D.; Sette, C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene 2012, 32, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- Busà, R.; Geremia, R.; Sette, C. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 2010, 38, 3005–3018. [Google Scholar] [CrossRef]
- Paronetto, M.P.; Miñana, B.; Valcárcel, J. The Ewing Sarcoma Protein Regulates DNA Damage-Induced Alternative Splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Leva, V.; Giuliano, S.; Bardoni, A.; Camerini, S.; Crescenzi, M.; Lisa, A.; Biamonti, G.; Montecucco, A. Phosphorylation of SRSF1 is modulated by replicational stress. Nucleic Acids Res. 2011, 40, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Sanchez, G.; De Cian, M.-C.; Barbier, J.; Dardenne, E.; Gratadou, L.; Dujardin, G.; Le Jossic-Corcos, C.; Corcos, L.; Auboeuf, D. Cotranscriptional exon skipping in the genotoxic stress response. Nat. Struct. Mol. Biol. 2010, 17, 1358–1366. [Google Scholar] [CrossRef]
- Shkreta, L.; Toutant, J.; Durand, M.; Manley, J.; Chabot, B. SRSF10 Connects DNA Damage to the Alternative Splicing of Transcripts Encoding Apoptosis, Cell-Cycle Control, and DNA Repair Factors. Cell Rep. 2016, 17, 1990–2003. [Google Scholar] [CrossRef]
- Shkreta, L.; Michelle, L.; Toutant, J.; Tremblay, M.L.; Chabot, B. The DNA Damage Response Pathway Regulates the Alternative Splicing of the Apoptotic Mediator Bcl-x. J. Biol. Chem. 2011, 286, 331–340. [Google Scholar] [CrossRef]
- Dutertre, M.; Sfaxi, R.; Vagner, S. Reciprocal Links between Pre-messenger RNA 3′-End Processing and Genome Stability. Trends Biochem. Sci. 2021, 46, 579–594. [Google Scholar] [CrossRef]
- Decorsière, A.; Cayrel, A.; Vagner, S.; Millevoi, S. Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3’-end processing and function during DNA damage. Genes Dev. 2011, 25, 220–225. [Google Scholar] [CrossRef]
- Newman, M.; Sfaxi, R.; Saha, A.; Monchaud, D.; Teulade-Fichou, M.-P.; Vagner, S. The G-Quadruplex-Specific RNA Helicase DHX36 Regulates p53 Pre-mRNA 3′-End Processing Following UV-Induced DNA Damage. J. Mol. Biol. 2017, 429, 3121–3131. [Google Scholar] [CrossRef]
- Gavish-Izakson, M.; Velpula, B.B.; Elkon, R.; Prados, R.; Barnabas, G.D.; Ugalde, A.P.; Agami, R.; Geiger, T.; Huertas, P.; Ziv, Y.; et al. Nuclear poly(A)-binding protein 1 is an ATM target and essential for DNA double-strand break repair. Nucleic Acids Res. 2017, 46, 730–747. [Google Scholar] [CrossRef]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; Hoon, D.; Anelli, V.V.; Mione, M.C.; Carninci, P.; Di Fagagna, F.D. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef]
- Fonseca, D.; Baquero, J.; Murphy, M.R.; Aruggoda, G.; Varriano, S.; Sapienza, C.; Mashadova, O.; Rahman, S.; Kleiman, F.E. mRNA Processing Factor CstF-50 and Ubiquitin Escort Factor p97 Are BRCA1/BARD1 Cofactors Involved in Chromatin Remodeling during the DNA Damage Response. Mol. Cell. Biol. 2018, 38, e00364-17. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; Heuvel, D.V.D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M.; et al. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180, 1228–1244.e24. [Google Scholar] [CrossRef] [PubMed]
- Patke, A.; Young, M.W.; Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 2019, 21, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Kinouchi, K.; Sassone-Corsi, P. Metabolic rivalry: Circadian homeostasis and tumorigenesis. Nat. Rev. Cancer 2020, 20, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Reinke, H.; Asher, G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 2019, 20, 227–241. [Google Scholar] [CrossRef]
- Hastings, M.H.; Maywood, E.S.; Brancaccio, M. Generation of circadian rhythms in the suprachiasmatic nucleus. Nat. Rev. Neurosci. 2018, 19, 453–469. [Google Scholar] [CrossRef]
- Menaker, M.; Murphy, Z.C.; Sellix, M.T. Central control of peripheral circadian oscillators. Curr. Opin. Neurobiol. 2020, 23, 741–746. [Google Scholar] [CrossRef]
- Cox, K.H.; Takahashi, J.S. Circadian clock genes and the transcriptional architecture of the clock mechanism. J. Mol. Endocrinol. 2019, 63, R93–R102. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Kinouchi, K.; Welz, P.-S.; Smith, J.G.; Zinna, V.M.; Shi, J.; Samad, M.; Chen, S.; Magnan, C.N.; Kinchen, J.M.; et al. Defining the Independence of the Liver Circadian Clock. Cell 2019, 177, 1448–1462.e14. [Google Scholar] [CrossRef]
- Bell-Pedersen, D.; Cassone, V.M.; Earnest, D.J.; Golden, S.S.; Hardin, P.E.; Thomas, T.L.; Zoran, M.J. Circadian rhythms from multiple oscillators: Lessons from diverse organisms. Nat. Rev. Genet. 2005, 6, 544–556. [Google Scholar] [CrossRef]
- Hirano, A.; Fu, Y.-H.; Ptáček, A.H.Y.-H.F.L.J. The intricate dance of post-translational modifications in the rhythm of life. Nat. Struct. Mol. Biol. 2016, 23, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Etchegaray, J.-P.; Cagampang, F.R.; Loudon, A.; Reppert, S.M. Posttranslational Mechanisms Regulate the Mammalian Circadian Clock. Cell 2001, 107, 855–867. [Google Scholar] [CrossRef]
- Koronowski, K.B.; Sassone-Corsi, P. Communicating clocks shape circadian homeostasis. Science 2021, 371, eabd0951. [Google Scholar] [CrossRef] [PubMed]
- Brenna, A.; Albrecht, U. Phosphorylation and Circadian Molecular Timing. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Parnell, A.A.; de Nobrega, A.K.; Lyons, L.C. Translating around the clock: Multi-level regulation of post-transcriptional processes by the circadian clock. Cell. Signal. 2020, 80, 109904. [Google Scholar] [CrossRef]
- McGlincy, N.J.; Valomon, A.; Chesham, J.E.; Maywood, E.S.; Hastings, M.H.; Ule, J. Regulation of alternative splicing by the circadian clock and food related cues. Genome Biol. 2012, 13, R54. [Google Scholar] [CrossRef] [PubMed]
- Preußner, M.; Wilhelmi, I.; Schultz, A.-S.; Finkernagel, F.; Michel, M.; Möröy, T.; Heyd, F. Rhythmic U2af26 Alternative Splicing Controls PERIOD1 Stability and the Circadian Clock in Mice. Mol. Cell 2014, 54, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Preußner, M.; Goldammer, G.; Neumann, A.; Haltenhof, T.; Rautenstrauch, P.; Müller-McNicoll, M.; Heyd, F. Body Temperature Cycles Control Rhythmic Alternative Splicing in Mammals. Mol. Cell 2017, 67, 433–446.e4. [Google Scholar] [CrossRef] [PubMed]
- Pico, E.G.; Niu, S.; Rollag, M.D.; Strayer, C.A.; Besharse, J.C.; Green, C.B. Immediate early response of the circadian polyA ribonuclease nocturnin to two extracellular stimuli. RNA 2007, 13, 745–755. [Google Scholar] [CrossRef]
- Wang, Y.; Osterbur, D.L.; Megaw, P.L.; Tosini, G.; Fukuhara, C.; Green, C.B.; Besharse, J.C. Rhythmic expression of Nocturnin mRNA in multiple tissues of the mouse. BMC Dev. Biol. 2001, 1, 9. [Google Scholar] [CrossRef]
- Kojima, S.; Sher-Chen, E.L.; Green, C.B. Circadian control of mRNA polyadenylation dynamics regulates rhythmic protein expression. Genes Dev. 2012, 26, 2724–2736. [Google Scholar] [CrossRef]
- Torres, M.; Becquet, D.; Franc, J.-L.; François-Bellan, A.-M. Circadian processes in the RNA life cycle. Wiley Interdiscip. Rev. RNA 2018, 9, e1467. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, W.; Murakawa, Y.; Yin, J.; Wang, G.; Landthaler, M.; Yan, J. Cold-induced RNA-binding proteins regulate circadian gene expression by controlling alternative polyadenylation. Sci. Rep. 2013, 3, 2054. [Google Scholar] [CrossRef]
- Bendix, C.; Marshall, C.M.; Harmon, F.G. Circadian Clock Genes Universally Control Key Agricultural Traits. Mol. Plant 2015, 8, 1135–1152. [Google Scholar] [CrossRef]
- Blümel, M.; Dally, N.; Jung, C. Flowering time regulation in crops—what did we learn from Arabidopsis? Curr. Opin. Biotechnol. 2014, 32, 121–129. [Google Scholar] [CrossRef]
- Filichkin, S.A.; Priest, H.D.; Givan, S.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.-K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2009, 20, 45–58. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Y.; Sancar, A.; Oztas, O. The circadian clock shapes the Arabidopsis transcriptome by regulating alternative splicing and alternative polyadenylation. J. Biol. Chem. 2020, 295, 7608–7619. [Google Scholar] [CrossRef]
- Riegler, S.; Servi, L.; Scarpin, M.R.; Herz, M.A.G.; Kubaczka, M.G.; Venhuizen, P.; Meyer, C.; Brunkard, J.O.; Kalyna, M.; Barta, A.; et al. Light regulates alternative splicing outcomes via the TOR kinase pathway. Cell Rep. 2021, 36. [Google Scholar] [CrossRef]
- Black, A.; Gamarra, J.; Giudice, J. More than a messenger: Alternative splicing as a therapeutic target. Biochim. Biophys. Acta Bioenerg. 2019, 1862, 194395. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.; Bak-Gordon, P.; Carmo-Fonseca, M. Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov. 2019, 19, 112–129. [Google Scholar] [CrossRef]
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-C.; To, M.D.; Westcott, P.M.K.; Delrosario, R.; Kim, I.-J.; Philips, M.; Tran, Q.; Bollam, S.R.; Goodarzi, H.; Bayani, N.; et al. Targeting KRAS4A splicing through the RBM39/DCAF15 pathway inhibits cancer stem cells. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Salmón, M.; Paniagua, G.; Lechuga, C.G.; Fernández-García, F.; Zarzuela, E.; Álvarez-Díaz, R.; Musteanu, M.; Guerra, C.; Caleiras, E.; Muñoz, J.; et al. KRAS4A induces metastatic lung adenocarcinomas in vivo in the absence of the KRAS4B isoform. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.-H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020, 38, 198–211. [Google Scholar] [CrossRef]
- Hofmann, Y.; Lorson, C.L.; Stamm, S.; Androphy, E.J.; Wirth, B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc. Natl. Acad. Sci. USA 2000, 97, 9618–9623. [Google Scholar] [CrossRef] [PubMed]
- Novoyatleva, T.; Heinrich, B.; Tang, Y.; Benderska, N.; Butchbach, M.E.; Lorson, C.L.; Lorson, M.A.; Ben-Dov, C.; Fehlbaum, P.; Bracco, L.; et al. Protein phosphatase 1 binds to the RNA recognition motif of several splicing factors and regulates alternative pre-mRNA processing. Hum. Mol. Genet. 2007, 17, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, F.; Capone, A.; Montori, A.; Rinzivillo, M.; Partelli, S.; Panzuto, F.; Pilozzi, E.; Arcidiacono, P.G.; Sette, C.; Capurso, G. MYC Upregulation Confers Resistance to Everolimus and Establishes Vulnerability to Cyclin-Dependent Kinase Inhibitors in Pancreatic Neuroendocrine Neoplasm Cells. Neuroendocrinology 2020, 111, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.-Y.; Cody, N.A.L.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.-J.; Wu, C.-S.; Chen, C.-Y.; Hung, L.-Y.; Chiang, T.-W.; Yang, M.-Y. NCLscan: Accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res. 2015, 44, e29. [Google Scholar] [CrossRef]
- Chuang, T.-J.; Chen, Y.-J.; Chen, C.-Y.; Mai, T.-L.; Wang, Y.-D.; Yeh, C.-S.; Yang, M.-Y.; Hsiao, Y.-T.; Chang, T.-H.; Kuo, T.-C.; et al. Integrative transcriptome sequencing reveals extensive alternative trans-splicing and cis-backsplicing in human cells. Nucleic Acids Res. 2018, 46, 3671–3691. [Google Scholar] [CrossRef]
- Chen, L.-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- Siegfried, Z.; Bonomi, S.; Ghigna, C.; Karni, R. Regulation of the Ras-MAPK and PI3K-mTOR Signalling Pathways by Alternative Splicing in Cancer. Int. J. Cell Biol. 2013, 2013, 568931. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruta, V.; Pagliarini, V.; Sette, C. Coordination of RNA Processing Regulation by Signal Transduction Pathways. Biomolecules 2021, 11, 1475. https://doi.org/10.3390/biom11101475
Ruta V, Pagliarini V, Sette C. Coordination of RNA Processing Regulation by Signal Transduction Pathways. Biomolecules. 2021; 11(10):1475. https://doi.org/10.3390/biom11101475
Chicago/Turabian StyleRuta, Veronica, Vittoria Pagliarini, and Claudio Sette. 2021. "Coordination of RNA Processing Regulation by Signal Transduction Pathways" Biomolecules 11, no. 10: 1475. https://doi.org/10.3390/biom11101475
APA StyleRuta, V., Pagliarini, V., & Sette, C. (2021). Coordination of RNA Processing Regulation by Signal Transduction Pathways. Biomolecules, 11(10), 1475. https://doi.org/10.3390/biom11101475