
Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways


Abstract
1. Introduction
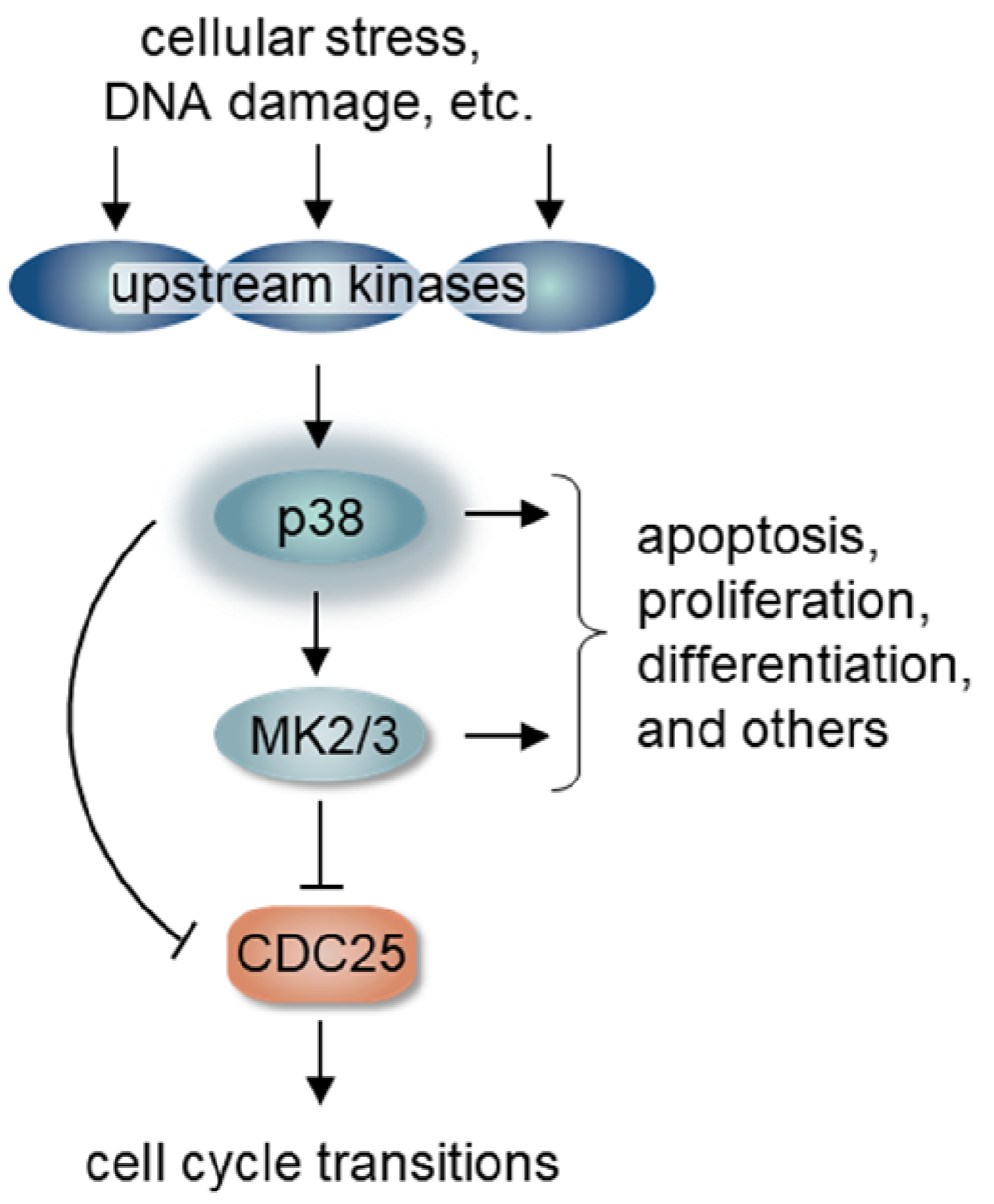
2. p38 Signaling and Cell Cycle Regulation
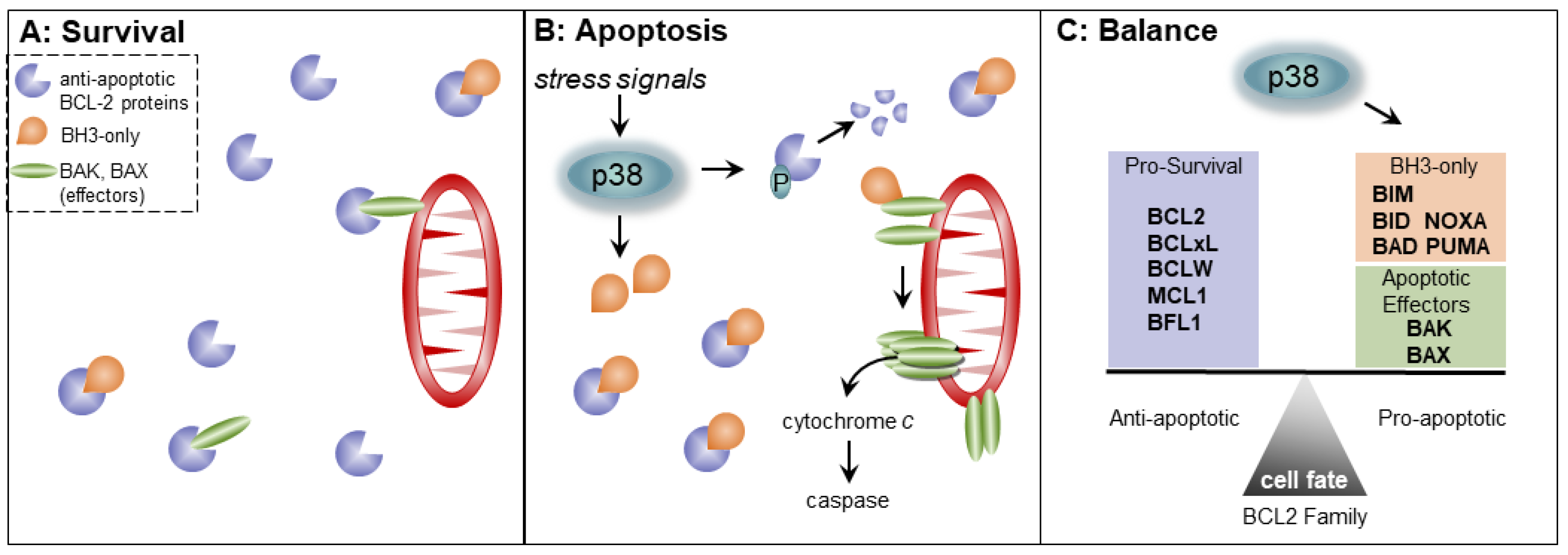
3. The BCL2 Family and the Cell Cycle
4. p38 MAPK Signaling and Regulation of Apoptosis by the BCL2 Family
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martinez-Limon, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci 2020, 21, 1913. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Johnson, G.L. MAPKs: Function, regulation, role in cancer and therapeutic targeting. Oncogene 2007, 26, 3097–3099. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Ray, L.B.; Sturgill, T.W. Rapid stimulation by insulin of a serine/threonine kinase in 3T3-L1 adipocytes that phosphorylates microtubule-associated protein 2 in vitro. Proc. Natl. Acad. Sci. USA 1987, 84, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Nishida, E.; Sakai, H. Activation of a Ca2+-inhibitable protein kinase that phosphorylates microtubule-associated protein 2 in vitro by growth factors, phorbol esters, and serum in quiescent cultured human fibroblasts. J. Biol. Chem. 1988, 263, 5396–5401. [Google Scholar] [CrossRef]
- Boulton, T.G.; Yancopoulos, G.D.; Gregory, J.S.; Slaughter, C.; Moomaw, C.; Hsu, J.; Cobb, M.H. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990, 249, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, W.E.; Kunisawa, R.; Thorner, J. A putative protein kinase overcomes pheromone-induced arrest of cell cycling in S. cerevisiae. Cell 1989, 58, 1107–1119. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Westfall, P.J.; Ballon, D.R.; Thorner, J. When the stress of your environment makes you go HOG wild. Science 2004, 306, 1511–1512. [Google Scholar] [CrossRef]
- Nebreda, A.R.; Porras, A. p38 MAP kinases: Beyond the stress response. Trends Biochem. Sci. 2000, 25, 257–260. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 2009, 21, 317–324. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Lu, S.; Wei, F.; Li, G. The evolution of the concept of stress and the framework of the stress system. Cell Stress 2021, 5, 76–85. [Google Scholar] [CrossRef]
- Thornton, T.M.; Rincon, M. Non-classical p38 map kinase functions: Cell cycle checkpoints and survival. Int. J. Biol. Sci. 2009, 5, 44–51. [Google Scholar] [CrossRef]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, S.P.; Hiebert, S.; Mudryj, M.; Horowitz, J.M.; Nevins, J.R. The E2F transcription factor is a cellular target for the RB protein. Cell 1991, 65, 1053–1061. [Google Scholar] [CrossRef]
- Frolov, M.V.; Dyson, N.J. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 2004, 117, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.D.; Phillips, R.A.; Gallie, B.L. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol. Cell Biol. 1999, 19, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Albanese, C.; Johnson, J.; Watanabe, G.; Eklund, N.; Vu, D.; Arnold, A.; Pestell, R.G. Transforming p21 Mutants and c-Ets-2 Activate the Cyclin D1 Promoter through Distinguishable Regions. J. Biol. Chem. 1995, 270, 23589–23597. [Google Scholar] [CrossRef] [PubMed]
- Duronio, R.J.; Xiong, Y. Signaling Pathways that Control Cell Proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef] [PubMed]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000. [Google Scholar] [CrossRef]
- Zarkowska, T.; Mittnacht, S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem. 1997, 272, 12738–12746. [Google Scholar] [CrossRef]
- Johnson, D.G.; Ohtani, K.; Nevins, J.R. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994, 8, 1514–1525. [Google Scholar] [CrossRef]
- Slansky, J.E.; Farnham, P.J. Introduction to the E2F family: Protein structure and gene regulation. Curr. Top. Microbiol. Immunol. 1996, 208, 1–30. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef]
- Canepa, E.T.; Scassa, M.E.; Ceruti, J.M.; Marazita, M.C.; Carcagno, A.L.; Sirkin, P.F.; Ogara, M.F. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 2007, 59, 419–426. [Google Scholar] [CrossRef]
- Kishi, H.; Nakagawa, K.; Matsumoto, M.; Suga, M.; Ando, M.; Taya, Y.; Yamaizumi, M. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J. Biol. Chem. 2001, 276, 39115–39122. [Google Scholar] [CrossRef]
- Casanovas, O.; Miro, F.; Estanyol, J.M.; Itarte, E.; Agell, N.; Bachs, O. Osmotic stress regulates the stability of cyclin D1 in a p38SAPK2-dependent manner. J. Biol. Chem. 2000, 275, 35091–35097. [Google Scholar] [CrossRef]
- Faust, D.; Dolado, I.; Cuadrado, A.; Oesch, F.; Weiss, C.; Nebreda, A.R.; Dietrich, C. p38alpha MAPK is required for contact inhibition. Oncogene 2005, 24, 7941–7945. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Higashimoto, Y.; Popoff, I.J.; Gaarde, W.A.; Basrur, V.; Potapova, O.; Appella, E.; Fornace, A.J., Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001, 411, 102–107. [Google Scholar] [CrossRef]
- Kim, G.Y.; Mercer, S.E.; Ewton, D.Z.; Yan, Z.; Jin, K.; Friedman, E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J. Biol. Chem. 2002, 277, 29792–29802. [Google Scholar] [CrossRef]
- Gubern, A.; Joaquin, M.; Marques, M.; Maseres, P.; Garcia-Garcia, J.; Amat, R.; Gonzalez-Nunez, D.; Oliva, B.; Real, F.X.; de Nadal, E.; et al. The N-Terminal Phosphorylation of RB by p38 Bypasses Its Inactivation by CDKs and Prevents Proliferation in Cancer Cells. Mol. Cell 2016, 64, 25–36. [Google Scholar] [CrossRef]
- Tomas-Loba, A.; Manieri, E.; Gonzalez-Teran, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M.; Romero-Becerra, R.; Rodriguez, E.; Pintor-Chocano, A.; Feixas, F.; et al. p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature 2019, 568, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Saito, S.; Hollander, M.C.; Sakaguchi, K.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999, 18, 6845–6854. [Google Scholar] [CrossRef] [PubMed]
- Kopper, F.; Bierwirth, C.; Schon, M.; Kunze, M.; Elvers, I.; Kranz, D.; Saini, P.; Menon, M.B.; Walter, D.; Sorensen, C.S.; et al. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc. Natl. Acad. Sci. USA 2013, 110, 16856–16861. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Peterson, A.; Zinshteyn, B.; Regot, S.; Green, R. Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 2020, 182, 404–416. [Google Scholar] [CrossRef]
- Pedraza-Alva, G.; Koulnis, M.; Charland, C.; Thornton, T.; Clements, J.L.; Schlissel, M.S.; Rincon, M. Activation of p38 MAP kinase by DNA double-strand breaks in V(D)J recombination induces a G2/M cell cycle checkpoint. EMBO J. 2006, 25, 763–773. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Amundson, S.A.; Fornace, A.J. p38 and Chk1 kinases: Different conductors for the G(2)/M checkpoint symphony. Curr. Opin. Genet. Dev. 2002, 12, 92–97. [Google Scholar] [CrossRef]
- Lemaire, M.; Froment, C.; Boutros, R.; Mondesert, O.; Nebreda, A.R.; Monsarrat, B.; Ducommun, B. CDC25B phosphorylation by p38 and MK-2. Cell Cycle 2006, 5, 1649–1653. [Google Scholar] [CrossRef][Green Version]
- Uchida, S.; Watanabe, N.; Kudo, Y.; Yoshioka, K.; Matsunaga, T.; Ishizaka, Y.; Nakagama, H.; Poon, R.Y.; Yamashita, K. SCFbeta(TrCP) mediates stress-activated MAPK-induced Cdc25B degradation. J. Cell Sci. 2011, 124, 2816–2825. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Hasskamp, P.; Schmedding, I.; Morandell, S.; van Vugt, M.A.; Wang, X.; Linding, R.; Ong, S.E.; Weaver, D.; Carr, S.A.; et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell 2010, 40, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kenny, A.E.; Rieder, C.L. P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol. Biol. Cell 2010, 21, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Phong, M.S.; Van Horn, R.D.; Li, S.; Tucker-Kellogg, G.; Surana, U.; Ye, X.S. p38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G(2) DNA damage checkpoint in human cancer cells. Mol. Cell Biol. 2010, 30, 3816–3826. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Nunez, G.; Clarke, M.F. The Bcl-2 family of proteins: Regulators of cell death and survival. Trends Cell Biol. 1994, 4, 399–403. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Reed, J.C.; Cuddy, M.; Slabiak, T.; Croce, C.M.; Nowell, P.C. Oncogenic potential of bcl-2 demonstrated by gene transfer. Nature 1988, 336, 259–261. [Google Scholar] [CrossRef]
- Yin, X.M.; Oltvai, Z.N.; Korsmeyer, S.J. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 1994, 369, 321–323. [Google Scholar] [CrossRef]
- Kozopas, K.M.; Yang, T.; Buchan, H.L.; Zhou, P.; Craig, R.W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 1993, 90, 3516–3520. [Google Scholar] [CrossRef] [PubMed]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Aouacheria, A.; Combet, C.; Tompa, P.; Hardwick, J.M. Redefining the BH3 Death Domain as a ‘Short Linear Motif’. Trends Biochem. Sci. 2015, 40, 736–748. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27 (Suppl. S1), S2–S19. [Google Scholar] [CrossRef]
- Du, H.; Wolf, J.; Schafer, B.; Moldoveanu, T.; Chipuk, J.E.; Kuwana, T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J. Biol. Chem. 2011, 286, 491–501. [Google Scholar] [CrossRef]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.H.; Placzek, W.J. Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents. Cells 2019, 8, 346. [Google Scholar] [CrossRef] [PubMed]
- Ruefli-Brasse, A.; Reed, J.C. Therapeutics targeting Bcl-2 in hematological malignancies. Biochem. J. 2017, 474, 3643–3657. [Google Scholar] [CrossRef] [PubMed]
- Crombie, J.; Davids, M.S. Venetoclax for the treatment of patients with chronic lymphocytic leukemia. Future Oncol. 2017, 13, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.W. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia 2002, 16, 444–454. [Google Scholar] [CrossRef]
- Vairo, G.; Soos, T.J.; Upton, T.M.; Zalvide, J.; DeCaprio, J.A.; Ewen, M.E.; Koff, A.; Adams, J.M. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol. Cell Biol. 2000, 20, 4745–4753. [Google Scholar] [CrossRef]
- Greider, C.; Chattopadhyay, A.; Parkhurst, C.; Yang, E. BCL-x(L) and BCL2 delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene 2002, 21, 7765–7775. [Google Scholar] [CrossRef]
- Janumyan, Y.M.; Sansam, C.G.; Chattopadhyay, A.; Cheng, N.; Soucie, E.L.; Penn, L.Z.; Andrews, D.; Knudson, C.M.; Yang, E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003, 22, 5459–5470. [Google Scholar] [CrossRef]
- Huang, D.C.; O’Reilly, L.A.; Strasser, A.; Cory, S. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 1997, 16, 4628–4638. [Google Scholar] [CrossRef]
- Deng, X.; Gao, F.; May, W.S., Jr. Bcl2 retards G1/S cell cycle transition by regulating intracellular ROS. Blood 2003, 102, 3179–3185. [Google Scholar] [CrossRef] [PubMed]
- Viant, C.; Guia, S.; Hennessy, R.J.; Rautela, J.; Pham, K.; Bernat, C.; Goh, W.; Jiao, Y.; Delconte, R.; Roger, M.; et al. Cell cycle progression dictates the requirement for BCL2 in natural killer cell survival. J. Exp. Med. 2017, 214, 491–510. [Google Scholar] [CrossRef]
- Whitaker, R.H.; Placzek, W.J. MCL1 binding to the reverse BH3 motif of P18INK4C couples cell survival to cell proliferation. Cell Death Dis. 2020, 11, 156. [Google Scholar] [CrossRef]
- Placzek, W.J.; Sturlese, M.; Wu, B.; Cellitti, J.F.; Wei, J.; Pellecchia, M. Identification of a novel Mcl-1 protein binding motif. J. Biol. Chem. 2011, 286, 39829–39835. [Google Scholar] [CrossRef]
- Fujise, K.; Zhang, D.; Liu, J.; Yeh, E.T. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J. Biol. Chem. 2000, 275, 39458–39465. [Google Scholar] [CrossRef]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; He, X.; Hsu, J.M.; Xia, W.; Chen, C.T.; Li, L.Y.; Lee, D.F.; Liu, J.C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell Biol. 2007, 27, 4006–4017. [Google Scholar] [CrossRef]
- Allan, L.A.; Skowyra, A.; Rogers, K.I.; Zeller, D.; Clarke, P.R. Atypical APC/C-dependent degradation of Mcl-1 provides an apoptotic timer during mitotic arrest. EMBO J. 2018, 37, e96831. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Li, G.; Cao, B.; Liu, L.; Cheng, Q.; Kong, H.; Shan, C.; Huang, X.; Chen, J.; Gao, N. Downregulation of Mcl-1 through inhibition of translation contributes to benzyl isothiocyanate-induced cell cycle arrest and apoptosis in human leukemia cells. Cell Death Dis. 2013, 4, e515. [Google Scholar] [CrossRef]
- Hasan, S.M.; Sheen, A.D.; Power, A.M.; Langevin, L.M.; Xiong, J.; Furlong, M.; Day, K.; Schuurmans, C.; Opferman, J.T.; Vanderluit, J.L. Mcl1 regulates the terminal mitosis of neural precursor cells in the mammalian brain through p27Kip1. Development 2013, 140, 3118–3127. [Google Scholar] [CrossRef][Green Version]
- Maryanovich, M.; Oberkovitz, G.; Niv, H.; Vorobiyov, L.; Zaltsman, Y.; Brenner, O.; Lapidot, T.; Jung, S.; Gross, A. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 2012, 14, 535–541. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zaltsman, Y.; Ruggiero, A.; Goldman, A.; Shachnai, L.; Zaidman, S.L.; Porat, Z.; Golan, K.; Lapidot, T.; Gross, A. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun. 2015, 6, 7901. [Google Scholar] [CrossRef]
- Wang, P.; Lindsay, J.; Owens, T.W.; Mularczyk, E.J.; Warwood, S.; Foster, F.; Streuli, C.H.; Brennan, K.; Gilmore, A.P. Phosphorylation of the proapoptotic BH3-only protein bid primes mitochondria for apoptosis during mitotic arrest. Cell Rep. 2014, 7, 661–671. [Google Scholar] [CrossRef]
- Kamer, I.; Sarig, R.; Zaltsman, Y.; Niv, H.; Oberkovitz, G.; Regev, L.; Haimovich, G.; Lerenthal, Y.; Marcellus, R.C.; Gross, A. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 2005, 122, 593–603. [Google Scholar] [CrossRef]
- Zinkel, S.S.; Hurov, K.E.; Ong, C.; Abtahi, F.M.; Gross, A.; Korsmeyer, S.J. A role for proapoptotic BID in the DNA-damage response. Cell 2005, 122, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Hershko, T.; Ginsberg, D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 2004, 279, 8627–8634. [Google Scholar] [CrossRef] [PubMed]
- Brady, H.J.; Gil-Gomez, G.; Kirberg, J.; Berns, A.J. Bax alpha perturbs T cell development and affects cell cycle entry of T cells. EMBO J. 1996, 15, 6991–7001. [Google Scholar] [CrossRef]
- Knudson, C.M.; Johnson, G.M.; Lin, Y.; Korsmeyer, S.J. Bax accelerates tumorigenesis in p53-deficient mice. Cancer Res. 2001, 61, 659–665. [Google Scholar]
- Zinkel, S.; Gross, A.; Yang, E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006, 13, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Ianari, A.; Natale, T.; Calo, E.; Ferretti, E.; Alesse, E.; Screpanti, I.; Haigis, K.; Gulino, A.; Lees, J.A. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 2009, 15, 184–194. [Google Scholar] [CrossRef]
- Gordon, G.M.; Du, W. Conserved RB functions in development and tumor suppression. Protein Cell 2011, 2, 864–878. [Google Scholar] [CrossRef]
- Croxton, R.; Ma, Y.; Song, L.; Haura, E.B.; Cress, W.D. Direct repression of the Mcl-1 promoter by E2F1. Oncogene 2002, 21, 1359–1369. [Google Scholar] [CrossRef]
- Eischen, C.M.; Packham, G.; Nip, J.; Fee, B.E.; Hiebert, S.W.; Zambetti, G.P.; Cleveland, J.L. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene 2001, 20, 6983–6993. [Google Scholar] [CrossRef]
- Hilgendorf, K.I.; Leshchiner, E.S.; Nedelcu, S.; Maynard, M.A.; Calo, E.; Ianari, A.; Walensky, L.D.; Lees, J.A. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 2013, 27, 1003–1015. [Google Scholar] [CrossRef]
- Antonucci, L.A.; Egger, J.V.; Krucher, N.A. Phosphorylation of the Retinoblastoma protein (Rb) on serine-807 is required for association with Bax. Cell Cycle 2014, 13, 3611–3617. [Google Scholar] [CrossRef]
- Krishna, M.; Narang, H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol. Life Sci. 2008, 65, 3525–3544. [Google Scholar] [CrossRef]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lyle, C.S.; Obey, T.B.; Gaarde, W.A.; Muir, J.A.; Bennett, B.L.; Chambers, T.C. Inhibition of cell proliferation and cell cycle progression by specific inhibition of basal JNK activity: Evidence that mitotic Bcl-2 phosphorylation is JNK-independent. J. Biol. Chem. 2004, 279, 11957–11966. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, M.; Saucourt, C.; Duval, D.; Gauthereau, X.; Thibault, C.; Dembele, D.; Feraud, O.; Menager, J.; Rallu, M.; Pradier, L.; et al. Bcl2, a transcriptional target of p38alpha, is critical for neuronal commitment of mouse embryonic stem cells. Cell Death Differ. 2008, 15, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Bradham, C.; McClay, D.R. p38 MAPK in development and cancer. Cell Cycle 2006, 5, 824–828. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Son, J.K.; Varadarajan, S.; Bratton, S.B. TRAIL-activated stress kinases suppress apoptosis through transcriptional upregulation of MCL-1. Cell Death Differ. 2010, 17, 1288–1301. [Google Scholar] [CrossRef]
- Azijli, K.; Yuvaraj, S.; van Roosmalen, I.; Flach, K.; Giovannetti, E.; Peters, G.J.; de Jong, S.; Kruyt, F.A. MAPK p38 and JNK have opposing activities on TRAIL-induced apoptosis activation in NSCLC H460 cells that involves RIP1 and caspase-8 and is mediated by Mcl-1. Apoptosis 2013, 18, 851–860. [Google Scholar] [CrossRef]
- Nijhawan, D.; Fang, M.; Traer, E.; Zhong, Q.; Gao, W.; Du, F.; Wang, X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003, 17, 1475–1486. [Google Scholar] [CrossRef]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Z.; Westberg, J.A.; Holtta, E.; Andersson, L.C. BCL2 regulates neural differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 4504–4508. [Google Scholar] [CrossRef]
- Opferman, J.T.; Kothari, A. Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 2018, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Oeztuerk-Winder, F.; Ventura, J.J. The many faces of p38 mitogen-activated protein kinase in progenitor/stem cell differentiation. Biochem. J. 2012, 445, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Ghatnekar, G.S.; Ghatnekar, A.V.; You, X.; Bu, M.; Guo, X.; Bu, S.; Shen, B.; Huang, Q. 2-Methoxyestradiol induced Bax phosphorylation and apoptosis in human retinoblastoma cells via p38 MAPK activation. Mol. Carcinog. 2012, 51, 576–585. [Google Scholar] [CrossRef]
- Cai, B.; Chang, S.H.; Becker, E.B.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef]
- Lu, J.; Quearry, B.; Harada, H. p38-MAP kinase activation followed by BIM induction is essential for glucocorticoid-induced apoptosis in lymphoblastic leukemia cells. FEBS Lett. 2006, 580, 3539–3544. [Google Scholar] [CrossRef]
- Cai, B.; Xia, Z. p38 MAP kinase mediates arsenite-induced apoptosis through FOXO3a activation and induction of Bim transcription. Apoptosis 2008, 13, 803–810. [Google Scholar] [CrossRef]
- Sridevi, P.; Nhiayi, M.K.; Setten, R.L.; Wang, J.Y. Persistent inhibition of ABL tyrosine kinase causes enhanced apoptotic response to TRAIL and disrupts the pro-apoptotic effect of chloroquine. PLoS ONE 2013, 8, e77495. [Google Scholar] [CrossRef]
- Tonino, S.H.; van Laar, J.; van Oers, M.H.; Wang, J.Y.; Eldering, E.; Kater, A.P. ROS-mediated upregulation of Noxa overcomes chemoresistance in chronic lymphocytic leukemia. Oncogene 2011, 30, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Ambroise, G.; Portier, A.; Roders, N.; Arnoult, D.; Vazquez, A. Subcellular localization of PUMA regulates its pro-apoptotic activity in Burkitt’s lymphoma B cells. Oncotarget 2015, 6, 38181–38194. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle 2009, 8, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Alaoui, A.; Feyen, O.; Mirmohammadsadegh, A.; Essmann, F.; Tannapfel, A.; Gulbins, E.; Schulze-Osthoff, K.; Hengge, U.R. The BH3-only member Noxa causes apoptosis in melanoma cells by multiple pathways. Oncogene 2008, 27, 4557–4568. [Google Scholar] [CrossRef]
- Wu, F.; Wang, Z.; Gu, J.H.; Ge, J.B.; Liang, Z.Q.; Qin, Z.H. p38(MAPK)/p53-Mediated Bax induction contributes to neurons degeneration in rotenone-induced cellular and rat models of Parkinson’s disease. Neurochem. Int. 2013, 63, 133–140. [Google Scholar] [CrossRef]
- Gascon, S.; Murenu, E.; Masserdotti, G.; Ortega, F.; Russo, G.L.; Petrik, D.; Deshpande, A.; Heinrich, C.; Karow, M.; Robertson, S.P.; et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell 2016, 18, 396–409. [Google Scholar] [CrossRef]
- Zhang, C.L.; Song, F.; Zhang, J.; Song, Q.H. Hypoxia-induced Bcl-2 expression in endothelial cells via p38 MAPK pathway. Biochem. Biophys. Res. Commun. 2010, 394, 976–980. [Google Scholar] [CrossRef]
- Nelyudova, A.; Aksenov, N.; Pospelov, V.; Pospelova, T. By blocking apoptosis, Bcl-2 in p38-dependent manner promotes cell cycle arrest and accelerated senescence after DNA damage and serum withdrawal. Cell Cycle 2007, 6, 2171–2177. [Google Scholar] [CrossRef]
- Gupta, J.; del Barco Barrantes, I.; Igea, A.; Sakellariou, S.; Pateras, I.S.; Gorgoulis, V.G.; Nebreda, A.R. Dual function of p38alpha MAPK in colon cancer: Suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell 2014, 25, 484–500. [Google Scholar] [CrossRef]
- Scheiblecker, L.; Kollmann, K.; Sexl, V. CDK4/6 and MAPK-Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals 2020, 13, 418. [Google Scholar] [CrossRef]
- Takenaka, K.; Moriguchi, T.; Nishida, E. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science 1998, 280, 599–602. [Google Scholar] [CrossRef]
- Deacon, K.; Mistry, P.; Chernoff, J.; Blank, J.L.; Patel, R. p38 Mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeutic drug-induced mitotic arrest. Mol. Biol. Cell 2003, 14, 2071–2087. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Igea, A.; Sartori, A.A.; Gomis, R.R.; Paull, T.T.; Isoda, M.; Perez-Montoyo, H.; Serra, V.; Gonzalez-Suarez, E.; Stracker, T.H.; et al. Targeting p38alpha Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells. Cancer Cell 2018, 33, 1094–1110. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Wang, J.; Xu, H.; Yang, X. Resistance to docetaxel-induced apoptosis in prostate cancer cells by p38/p53/p21 signaling. Prostate 2011, 71, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Ma, N.; Wang, J.; Song, J.; Bu, X.; Cheng, Y.; Sun, K.; Xiong, H.; Jiang, G.; Zhang, B.; et al. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008, 8, 375. [Google Scholar] [CrossRef]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef]
- Fang, S.; Qiu, J.; Wu, Z.; Bai, T.; Guo, W. Down-regulation of UBC9 increases the sensitivity of hepatocellular carcinoma to doxorubicin. Oncotarget 2017, 8, 49783–49795. [Google Scholar] [CrossRef]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; Wicky John, S.; Liccardi, G.; et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol. Cell 2017, 66, 698–710. [Google Scholar] [CrossRef]
- Menon, M.B.; Gropengiesser, J.; Fischer, J.; Novikova, L.; Deuretzbacher, A.; Lafera, J.; Schimmeck, H.; Czymmeck, N.; Ronkina, N.; Kotlyarov, A.; et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 2017, 19, 1248–1259. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Delanghe, T.; Rojas-Rivera, D.; Priem, D.; Delvaeye, T.; Bruggeman, I.; Van Herreweghe, F.; Vandenabeele, P.; Bertrand, M.J.M. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 2017, 19, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.L.; Wu, Z.; Zhang, P.; Wood, L.D.; Bhakta, K.S.; Han, J.; Feramisco, J.R.; Karin, M.; Wang, J.Y. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000, 14, 574–584. [Google Scholar] [PubMed]
- Haq, R.; Brenton, J.D.; Takahashi, M.; Finan, D.; Finkielsztein, A.; Damaraju, S.; Rottapel, R.; Zanke, B. Constitutive p38HOG mitogen-activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 2002, 62, 5076–5082. [Google Scholar] [PubMed]
- Faust, D.; Schmitt, C.; Oesch, F.; Oesch-Bartlomowicz, B.; Schreck, I.; Weiss, C.; Dietrich, C. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 2012, 10, 6. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein Family/Category | Protein | Cell Cycle and Survival Roles | p38 Integration/Effect |
|---|---|---|---|
| BCL2 Anti-Apoptotic Proteins | BCL2 | G1 phase ↑; survival | +P degradation; transcription ↓ |
| MCL1 | G1/S trans. ↑, M phase timer; survival | +P degradation; transcription ↓ | |
| BCLxL | G1 phase ↑; survival | +P degradation | |
| BCL2 Apoptotic Effectors | BAX | RB interaction; apoptosis | +P apoptosis ↑ |
| BAK | S phase ↑; apoptosis | - | |
| BCL2 BH3-Only Proteins | BIM | transcription ↑ by E2F; apoptosis | +P, apoptosis ↑; transcription ↑ |
| BID | G0/G1, M; apoptosis | - | |
| NOXA | transcription ↑ by E2F; apoptosis | transcription ↑ | |
| PUMA | transcription ↑ by E2F; apoptosis | transcription ↑ | |
| cyclin D | CDK4/6 activation | +P degradation | |
| CDK4/6 | RB inhibition | inhibits | |
| Cell Cycle: G1/S Transition | RB | E2F inhibition; (BAX interaction) | p38α activates RB; γ inhibits |
| E2F | S transcription; (BCL2/MCL1 transcription ↓) | - | |
| cyclin E | CDK2 activation | - | |
| CDC25 | CDK dephosphorylation and activation | inhibition | |
| cyclin B | CDK1 activation | - | |
| Cell Cycle: G2/M Transition | CDK1 | M entry and progression | - |
| CDC25 | CDK dephosphorylation and activation | inhibition | |
| p27 | CDK2 inhibitor; (MCL1 & terminal mitosis) | ||
| Cell Cycle Inhibitors | p18 | CDK4/6 inhibitor; (destabilized by MCL1) | - |
| p38 | Cell cycle arrest in response to stress | - | |
| p38 MAPK Pathway | MK2 | CDC25 inhibition and cycle arrest | +P activated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitaker, R.H.; Cook, J.G. Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways. Biomolecules 2021, 11, 1444. https://doi.org/10.3390/biom11101444
Whitaker RH, Cook JG. Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways. Biomolecules. 2021; 11(10):1444. https://doi.org/10.3390/biom11101444
Chicago/Turabian StyleWhitaker, Robert H., and Jeanette Gowen Cook. 2021. "Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways" Biomolecules 11, no. 10: 1444. https://doi.org/10.3390/biom11101444
APA StyleWhitaker, R. H., & Cook, J. G. (2021). Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways. Biomolecules, 11(10), 1444. https://doi.org/10.3390/biom11101444