
Structural and Functional Insights into α-Synuclein Fibril Polymorphism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical and Pathological Features of Synucleinopathies
2.1. Parkinson’s Disease (PD)
2.2. Multiple System Atrophy (MSA)
2.3. Dementia with Lewy Bodies (DLB)
3. Misfolding and Aggregation of α-Syn
4. Prion-like Strain Phenomena in α-Syn
4.1. Concept of Prion Strains
4.2. α-Syn Strains Generated In Vitro
4.3. α-Syn Strains in Human Synucleinopathy Samples
5. High-Resolution Structural Models of Existing α-Syn Fibril Strains
6. Familial Mutations of α-Syn Form Distinct Fibril Conformations
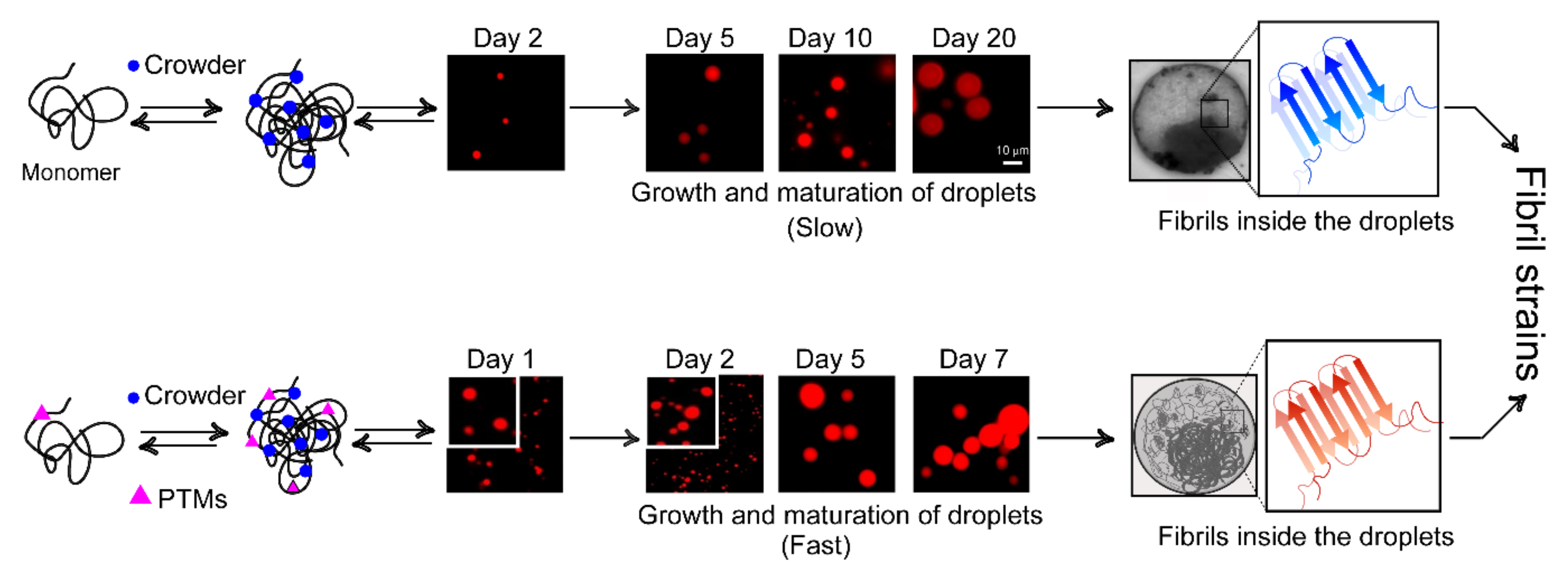
7. Phase Separation and Nucleation: Molecular Basis of Fibril Polymorphism
8. Clinical and Therapeutic Implications of Polymorphism
9. Concluding Remarks and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Spillantini, M.G.; Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. A-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Galvin, J.E.; Lee, V.M.; Trojanowski, J.Q. Synucleinopathies: Clinical and pathological implications. Arch. Neurol. 2001, 58, 186–190. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, M.B.; Jagannath, S.; Lashuel, H.A. Reverse engineering Lewy bodies: How far have we come and how far can we go? Nat. Rev. Neurosci. 2021, 22, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hsu, L.J.; Sisk, A.; Xia, Y.; Takeda, A.; Sundsmo, M.; Masliah, E. Human recombinant NACP/alpha-synuclein is aggregated and fibrillated in vitro: Relevance for Lewy body disease. Brain Res 1998, 799, 301–306. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Jakes, R.; Curran, M.D.; Wallace, A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of alpha-synuclein protein implicated in Parkinson’s disease. FEBS Lett. 1998, 440, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T., Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39, 2552–2563. [Google Scholar] [CrossRef]
- Cremades, N.; Chen, S.W.; Dobson, C.M. Structural Characteristics of alpha-Synuclein Oligomers. Int. Rev. Cell Mol. Biol. 2017, 329, 79–143. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell. Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [Green Version]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; McGarvey, N.H.; Rutherford, N.J.; Ceballos-Diaz, C.; Robertson, J.; Golde, T.E.; Giasson, B.I. Amyloidogenic alpha-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014, 127, 645–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helwig, M.; Klinkenberg, M.; Rusconi, R.; Musgrove, R.E.; Majbour, N.K.; El-Agnaf, O.M.A.; Ulusoy, A.; Di Monte, D.A. Brain propagation of transduced α-synuclein involves non-fibrillar protein species and is enhanced in α-synuclein null mice. Brain 2015, 139, 856–870. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illes-Toth, E.; Ramos, M.R.; Cappai, R.; Dalton, C.; Smith, D.P. Distinct higher-order α-synuclein oligomers induce intracellular aggregation. Biochem. J. 2015, 468, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Bockmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Lv, G.; Lee, J.S.; Jung, B.C.; Masuda-Suzukake, M.; Hong, C.S.; Valera, E.; Lee, H.J.; Paik, S.R.; Hasegawa, M.; et al. Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci. Rep. 2016, 6, 30891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; So, R.W.L.; Lau, H.H.C.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.C.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. alpha-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 2020, 23, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Lee, V.M. Distinct alpha-Synuclein strains and implications for heterogeneity among alpha-Synucleinopathies. Neurobiol Dis 2018, 109, 209–218. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Bousset, L.; Renner, M.; Redeker, V.; Savistchenko, J.; Triller, A.; Melki, R. Differential Membrane Binding and Seeding of Distinct alpha-Synuclein Fibrillar Polymorphs. Biophys. J. 2020, 118, 1301–1320. [Google Scholar] [CrossRef] [Green Version]
- De Giorgi, F.; Laferrière, F.; Zinghirino, F.; Faggiani, E.; Lends, A.; Bertoni, M.; Yu, X.; Grélard, A.; Morvan, E.; Habenstein, B.; et al. Novel self-replicating α-synuclein polymorphs that escape ThT monitoring can spontaneously emerge and acutely spread in neurons. Sci. Adv. 2020, 6, eabc4364. [Google Scholar] [CrossRef]
- Bhak, G.; Lee, J.; Kim, T.H.; Lee, S.; Lee, D.; Paik, S.R. Molecular inscription of environmental information into protein suprastructures: Temperature effects on unit assembly of α-synuclein oligomers into polymorphic amyloid fibrils. Biochem. J. 2014, 464, 259–269. [Google Scholar] [CrossRef]
- Meinhardt, J.; Sachse, C.; Hortschansky, P.; Grigorieff, N.; Fändrich, M. Abeta(1-40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J. Mol. Biol. 2009, 386, 869–877. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Ferreira, R.; Taylor, N.M.; Arteni, A.A.; Kumari, P.; Mona, D.; Ringler, P.; Britschgi, M.; Lauer, M.E.; Makky, A.; Verasdonck, J.; et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 2019, 8, e48907. [Google Scholar] [CrossRef]
- Birkett, C.R.; Hennion, R.M.; Bembridge, D.A.; Clarke, M.C.; Chree, A.; Bruce, M.E.; Bostock, C.J. Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. EMBO J. 2001, 20, 3351–3358. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef]
- Bruce, M.; Chree, A.; McConnell, I.; Foster, J.; Pearson, G.; Fraser, H. Transmission of bovine spongiform encephalopathy and scrapie to mice: Strain variation and the species barrier. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1994, 343, 405–411. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, R.; Abid, K.; Soto, C. The prion strain phenomenon: Molecular basis and unprecedented features. Biochim. Biophys. Acta 2007, 1772, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Neuropathological spectrum of synucleinopathies. Mov. Disord. Off. J. Mov. Disord. Soc. 2003, 18 (Suppl. 6), S2–S12. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.R.; Holmes, B.B.; Furman, J.L.; Dhavale, D.D.; Su, B.W.; Song, E.S.; Cairns, N.J.; Kotzbauer, P.T.; Diamond, M.I. Parkinson’s disease and multiple system atrophy have distinct alpha-synuclein seed characteristics. J. Biol. Chem. 2019, 294, 1045–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peelaerts, W.; Bousset, L.; Baekelandt, V.; Melki, R. a-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: Similarities and differences. Cell Tissue Res. 2018, 373, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, G.; Imura, S.; Hosokawa, M.; Katsumata, R.; Nonaka, T.; Hisanaga, S.I.; Saeki, Y.; Hasegawa, M. alpha-Synuclein strains that cause distinct pathologies differentially inhibit proteasome. eLife 2020, 9, e56825. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Bousset, L.; George, S.; Madaj, Z.; Meyerdirk, L.; Schulz, E.; Steiner, J.A.; Melki, R.; Brundin, P. alpha-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol. Commun. 2019, 7, 221. [Google Scholar] [CrossRef] [Green Version]
- Cendrowska, U.; Silva, P.J.; Ait-Bouziad, N.; Müller, M.; Guven, Z.P.; Vieweg, S.; Chiki, A.; Radamaker, L.; Kumar, S.T.; Fändrich, M.; et al. Unraveling the complexity of amyloid polymorphism using gold nanoparticles and cryo-EM. Proc. Natl. Acad. Sci. USA 2020, 117, 6866–6874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Heise, H.; Hoyer, W.; Becker, S.; Andronesi, O.C.; Riedel, D.; Baldus, M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA 2005, 102, 15871–15876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fandrich, M. Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef]
- Gath, J.; Bousset, L.; Habenstein, B.; Melki, R.; Bockmann, A.; Meier, B.H. Unlike twins: An NMR comparison of two alpha-synuclein polymorphs featuring different toxicity. PLoS ONE 2014, 9, e90659. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of α-synuclein fibrils. eLife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Marti, M.J.; Tolosa, E.; Campdelacreu, J. Clinical overview of the synucleinopathies. Mov. Disord. Off. J. Mov. Disord. Soc. 2003, 18 (Suppl. 6), S21–S27. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Davie, C.A. A review of Parkinson’s disease. Br. Med Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef]
- Verbaan, D.; Marinus, J.; Visser, M.; van Rooden, S.M.; Stiggelbout, A.M.; van Hilten, J.J. Patient-reported autonomic symptoms in Parkinson disease. Neurology 2007, 69, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Colosimo, C. Nonmotor presentations of multiple system atrophy. Nat. Rev. Neurol. 2011, 7, 295–298. [Google Scholar] [CrossRef]
- Engen, P.A.; Dodiya, H.B.; Naqib, A.; Forsyth, C.B.; Green, S.J.; Voigt, R.M.; Kordower, J.H.; Mutlu, E.A.; Shannon, K.M.; Keshavarzian, A. The Potential Role of Gut-Derived Inflammation in Multiple System Atrophy. J. Parkinson’s Dis. 2017, 7, 331–346. [Google Scholar] [CrossRef]
- Mertsalmi, T.H.; Aho, V.T.E.; Pereira, P.A.B.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. More than constipation—Bowel symptoms in Parkinson’s disease and their connection to gut microbiota. Eur. J. Neurol. 2017, 24, 1375–1383. [Google Scholar] [CrossRef]
- Sakakibara, R.; Doi, H.; Fukudo, S. Lewy body constipation. J. Anus Rectum Colon 2019, 3, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Tursi, S.A.; Tükel, Ç. Curli-Containing Enteric Biofilms Inside and Out: Matrix Composition, Immune Recognition, and Disease Implications. Microbiol. Mol. Biol. Rev. 2018, 82, e00028-18. [Google Scholar] [CrossRef] [Green Version]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. eLife 2020, 9, e53111. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. A Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef]
- Yoshino, H.; Hirano, M.; Stoessl, A.J.; Imamichi, Y.; Ikeda, A.; Li, Y.; Funayama, M.; Yamada, I.; Nakamura, Y.; Sossi, V.; et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol. Aging 2017, 57, 248.e7–248.e12. [Google Scholar] [CrossRef]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure based aggregation studies reveal the presence of helix-rich intermediate during alpha-Synuclein aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates alpha-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Mondal, M.; Mohite, G.M.; Singh, P.K.; Ranjan, P.; Anoop, A.; Ghosh, S.; Jha, N.N.; Kumar, A.; Maji, S.K. The Parkinson’s disease-associated H50Q mutation accelerates alpha-Synuclein aggregation in vitro. Biochemistry 2013, 52, 6925–6927. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Mehra, S.; Sahay, S.; Singh, P.K.; Maji, S.K. alpha-synuclein aggregation and its modulation. Int. J. Biol. Macromol. 2017, 100, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Sahay, S.; Ghosh, D.; Dwivedi, S.; Anoop, A.; Mohite, G.M.; Kombrabail, M.; Krishnamoorthy, G.; Maji, S.K. Familial Parkinson disease-associated mutations alter the site-specific microenvironment and dynamics of alpha-synuclein. J. Biol. Chem. 2015, 290, 7804–7822. [Google Scholar] [CrossRef] [Green Version]
- Sahay, S.; Ghosh, D.; Singh, P.K.; Maji, S.K. Alteration of Structure and Aggregation of alpha-Synuclein by Familial Parkinson’s Disease Associated Mutations. Curr. Protein Pept. Sci. 2017, 18, 656–676. [Google Scholar] [CrossRef]
- Sahay, S.; Krishnamoorthy, G.; Maji, S.K. Site-specific structural dynamics of alpha-Synuclein revealed by time-resolved fluorescence spectroscopy: A review. Methods Appl. Fluoresc. 2016, 4, 042002. [Google Scholar] [CrossRef]
- Mohite, G.M.; Navalkar, A.; Kumar, R.; Mehra, S.; Das, S.; Gadhe, L.G.; Ghosh, D.; Alias, B.; Chandrawanshi, V.; Ramakrishnan, A.; et al. The Familial alpha-Synuclein A53E Mutation Enhances Cell Death in Response to Environmental Toxins Due to a Larger Population of Oligomers. Biochemistry 2018, 57, 5014–5028. [Google Scholar] [CrossRef]
- Mohite, G.M.; Kumar, R.; Panigrahi, R.; Navalkar, A.; Singh, N.; Datta, D.; Mehra, S.; Ray, S.; Gadhe, L.G.; Das, S.; et al. Comparison of Kinetics, Toxicity, Oligomer Formation, and Membrane Binding Capacity of alpha-Synuclein Familial Mutations at the A53 Site, Including the Newly Discovered A53V Mutation. Biochemistry 2018, 57, 5183–5187. [Google Scholar] [CrossRef]
- Mohite, G.M.; Dwivedi, S.; Das, S.; Kumar, R.; Paluri, S.; Mehra, S.; Ruhela, N.; Jha, N.N.; Maji, S.K. Parkinson’s Disease Associated alpha-Synuclein Familial Mutants Promote Dopaminergic Neuronal Death in Drosophila melanogaster. ACS Chem. Neurosci. 2018, 9, 2628–2638. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Harper, J.D.; Williamson, R.E.; Lansbury, P.T., Jr. Accelerated oligomerization by Parkinson’s disease linked alpha-synuclein mutants. Ann. N. Y. Acad. Sci. 2000, 920, 42–45. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. alpha-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Antony, T.; Hoyer, W.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Cellular polyamines promote the aggregation of alpha-synuclein. J. Biol. Chem. 2003, 278, 3235–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Martin, J.L.; Klucken, J.; Outeiro, T.F.; Nguyen, P.; Keller-McGandy, C.; Cantuti-Castelvetri, I.; Grammatopoulos, T.N.; Standaert, D.G.; Hyman, B.T.; McLean, P.J. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J. Neurochem. 2007, 100, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Moloney, T.C.; Hyland, R.; O’Toole, D.; Paucard, A.; Kirik, D.; O’Doherty, A.; Gorman, A.M.; Dowd, E. Heat shock protein 70 reduces alpha-synuclein-induced predegenerative neuronal dystrophy in the alpha-synuclein viral gene transfer rat model of Parkinson’s disease. CNS Neurosci. Ther. 2014, 20, 50–58. [Google Scholar] [CrossRef]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef] [Green Version]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef]
- Cohlberg, J.A.; Li, J.; Uversky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Mehra, S.; Ghosh, D.; Kumar, R.; Mondal, M.; Gadhe, L.G.; Das, S.; Anoop, A.; Jha, N.N.; Jacob, R.S.; Chatterjee, D.; et al. Glycosaminoglycans have variable effects on alpha-synuclein aggregation and differentially affect the activities of the resulting amyloid fibrils. J. Biol. Chem. 2018, 293, 12975–12991. [Google Scholar] [CrossRef] [Green Version]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Lowe, R.; Pountney, D.L.; Jensen, P.H.; Gai, W.P.; Voelcker, N.H. Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. A Publ. Protein Soc. 2004, 13, 3245–3252. [Google Scholar] [CrossRef] [Green Version]
- Breydo, L.; Wu, J.W.; Uversky, V.N. Alpha-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2011, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, N. Multiple system atrophy--the nature of the beast. J. Neurol. Neurosurg. Psychiatry 1989, 52, 78–89. [Google Scholar] [CrossRef]
- Fanciulli, A.; Wenning, G.K. Multiple-System Atrophy. N. Engl. J. Med. 2015, 372, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Saito, Y.; Terao, S.; Ando, T.; Kachi, T.; Mukai, E.; Aiba, I.; Abe, Y.; Tamakoshi, A.; Doyu, M.; et al. Progression and prognosis in multiple system atrophy: An analysis of 230 Japanese patients. Brain 2002, 125, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, Y.; Wenning, G.K.; Tison, F.; Quinn, N.P. Survival of patients with pathologically proven multiple system atrophy: A meta-analysis. Neurology 1997, 48, 384–393. [Google Scholar] [CrossRef]
- Schrag, A.; Ben-Shlomo, Y.; Quinn, N.P. Prevalence of progressive supranuclear palsy and multiple system atrophy: A cross-sectional study. Lancet 1999, 354, 1771–1775. [Google Scholar] [CrossRef]
- Castellani, R. Multiple system atrophy: Clues from inclusions. Am. J. Pathol. 1998, 153, 671–676. [Google Scholar] [CrossRef]
- Lantos, P.L.; Papp, M.I. Cellular pathology of multiple system atrophy: A review. J. Neurol. Neurosurg. Psychiatry 1994, 57, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Wenning, G.K.; Ben Shlomo, Y.; Magalhães, M.; Daniel, S.E.; Quinn, N.P. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994, 117 Pt 4, 835–845. [Google Scholar] [CrossRef]
- Ozawa, T.; Paviour, D.; Quinn, N.P.; Josephs, K.A.; Sangha, H.; Kilford, L.; Healy, D.G.; Wood, N.W.; Lees, A.J.; Holton, J.L.; et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: Clinicopathological correlations. Brain 2004, 127, 2657–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, S.; Low, P.; Quinn, N.; Albanese, A.; Ben-Shlomo, Y.; Fowler, C.; Kaufmann, H.; Klockgether, T.; Lang, A.; Lantos, P.; et al. Consensus statement on the diagnosis of multiple system atrophy. Clin. Auton. Res. 1998, 8, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, S.; May, S.J.; Shults, C.W.; Tanner, C.M.; Kukull, W.; Lee, V.M.; Masliah, E.; Low, P.; Sandroni, P.; Trojanowski, J.Q.; et al. The North American Multiple System Atrophy Study Group. J. Neural Transm. 2005, 112, 1687–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Jeon, B.S.; Lee, J.Y.; Yun, J.Y. Survival of Korean patients with multiple system atrophy. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 909–912. [Google Scholar] [CrossRef]
- Köllensperger, M.; Geser, F.; Ndayisaba, J.P.; Boesch, S.; Seppi, K.; Ostergaard, K.; Dupont, E.; Cardozo, A.; Tolosa, E.; Abele, M.; et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: Final analysis of the European multiple system atrophy registry. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 2604–2612. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.H.; Galvin, J.E.; Baba, M.; Giasson, B.; Tomita, T.; Leight, S.; Nakajo, S.; Iwatsubo, T.; Trojanowski, J.Q.; Lee, V.M. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann. Neurol. 1998, 44, 415–422. [Google Scholar] [CrossRef]
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. alpha-Synucleinopathy phenotypes. Parkinsonism Relat. Disord. 2014, 20 (Suppl. 1), S62–S67. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.; Asi, Y.T.; Sailer, A.; Lees, A.J.; Houlden, H.; Revesz, T.; Holton, J.L. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol. Appl. Neurobiol. 2012, 38, 4–24. [Google Scholar] [CrossRef]
- Jin, H.; Ishikawa, K.; Tsunemi, T.; Ishiguro, T.; Amino, T.; Mizusawa, H. Analyses of copy number and mRNA expression level of the alpha-synuclein gene in multiple system atrophy. J. Med. Dent. Sci. 2008, 55, 145–153. [Google Scholar] [PubMed]
- Miller, D.W.; Johnson, J.M.; Solano, S.M.; Hollingsworth, Z.R.; Standaert, D.G.; Young, A.B. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J. Neural. Transm. 2005, 112, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Solano, S.M.; Miller, D.W.; Augood, S.J.; Young, A.B.; Penney, J.B., Jr. Expression of alpha-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: Genes associated with familial Parkinson’s disease. Ann. Neurol. 2000, 47, 201–210. [Google Scholar] [CrossRef]
- Yazawa, I.; Giasson, B.I.; Sasaki, R.; Zhang, B.; Joyce, S.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 2005, 45, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Kisos, H.; Pukaß, K.; Ben-Hur, T.; Richter-Landsberg, C.; Sharon, R. Increased neuronal α-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling α-synucleinopathies. PLoS ONE 2012, 7, e46817. [Google Scholar] [CrossRef] [Green Version]
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014, 62, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Vann Jones, S.A.; O’Brien, J.T. The prevalence and incidence of dementia with Lewy bodies: A systematic review of population and clinical studies. Psychol. Med. 2014, 44, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.P.M.; Surendranathan, A.; Bentley, A.; Barker, S.A.H.; Taylor, J.P.; Thomas, A.J.; Allan, L.M.; McNally, R.J.; James, P.W.; McKeith, I.G.; et al. Clinical prevalence of Lewy body dementia. Alzheimer’s Res. Ther. 2018, 10, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Levin, J.; Kurz, A.; Arzberger, T.; Giese, A.; Höglinger, G.U. The Differential Diagnosis and Treatment of Atypical Parkinsonism. Dtsch. Arztebl. Int. 2016, 113, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, D.B.; Fiest, K.M.; Roberts, J.I.; Maxwell, C.J.; Dykeman, J.; Pringsheim, T.; Steeves, T.; Smith, E.E.; Pearson, D.; Jetté, N. The Prevalence and Incidence of Dementia with Lewy Bodies: A Systematic Review. Can. J. Neurol. Sci. 2016, 43 (Suppl. 1), S83–S95. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Boeve, B.F.; Ahlskog, J.E.; Rocca, W.A. Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol. 2013, 70, 1396–1402. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front. Neurol. Neurosci. 2009, 24, 114–125. [Google Scholar] [CrossRef]
- Shimada, H.; Shinotoh, H.; Hirano, S.; Miyoshi, M.; Sato, K.; Tanaka, N.; Ota, T.; Fukushi, K.; Irie, T.; Ito, H.; et al. β-Amyloid in Lewy body disease is related to Alzheimer’s disease-like atrophy. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Flagmeier, P.; Habchi, J.; Lattanzi, V.; Linse, S.; Dobson, C.M.; Knowles, T.P.J.; Vendruscolo, M. Monomeric and fibrillar α-synuclein exert opposite effects on the catalytic cycle that promotes the proliferation of Aβ42 aggregates. Proc. Natl. Acad. Sci. USA 2017, 114, 8005–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K.; Pettegrew, J.W.; Masliah, E.; Hamilton, R.L.; Mandal, R. Interaction between Aβ Peptide and α Synuclein: Molecular Mechanisms in Overlapping Pathology of Alzheimer’s and Parkinson’s in Dementia with Lewy Body Disease. Neurochem. Res. 2006, 31, 1153–1162. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Desplats, P.A.; Overk, C.R.; Valera-Martin, E.; Rissman, R.A.; Wu, C.; Mante, M.; Adame, A.; Florio, J.; Rockenstein, E.; et al. Reducing Endogenous α-Synuclein Mitigates the Degeneration of Selective Neuronal Populations in an Alzheimer’s Disease Transgenic Mouse Model. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 7971–7984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsigelny, I.F.; Crews, L.; Desplats, P.; Shaked, G.M.; Sharikov, Y.; Mizuno, H.; Spencer, B.; Rockenstein, E.; Trejo, M.; Platoshyn, O.; et al. Mechanisms of Hybrid Oligomer Formation in the Pathogenesis of Combined Alzheimer’s and Parkinson’s Diseases. PLoS ONE 2008, 3, e3135. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Salmon, D.; Galasko, D.; Masliah, E.; Katzman, R.; DeTeresa, R.; Thal, L.; Pay, M.M.; Hofstetter, R.; Klauber, M.; et al. The Lewy body variant of Alzheimer’s disease: A clinical and pathologic entity. Neurology 1990, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Duda, J.E.; Xie, S.X.; Lee, E.B.; Van Deerlin, V.M.; Lopez, O.L.; Kofler, J.K.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-Beta (Aβ) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Aβ Pathology. Neuron 2020, 105, 260–275.e6. [Google Scholar] [CrossRef]
- Kraybill, M.L.; Larson, E.B.; Tsuang, D.W.; Teri, L.; McCormick, W.C.; Bowen, J.D.; Kukull, W.A.; Leverenz, J.B.; Cherrier, M.M. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 2005, 64, 2069–2073. [Google Scholar] [CrossRef] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [Green Version]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- George, J.M. The synucleins. Genome Biol. 2002, 3, 3002.1–3002.6. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-bound Human α-Synuclein *. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, E.A.; Mazzulli, J.R.; Giasson, B.I. Characterization of hydrophobic residue requirements for alpha-synuclein fibrillization. Biochemistry 2009, 48, 9427–9436. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zaer, S.; Drori, P.; Zamel, J.; Joron, K.; Kalisman, N.; Lerner, E.; Dokholyan, N.V. The structural heterogeneity of α-synuclein is governed by several distinct subpopulations with interconversion times slower than milliseconds. Structure 2021, 29, 1048–1064.e6. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- Lautenschlager, J.; Stephens, A.D.; Fusco, G.; Strohl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, S.R.; Shin, H.J.; Lee, J.H. Metal-catalyzed oxidation of alpha-synuclein in the presence of Copper(II) and hydrogen peroxide. Arch. Biochem. Biophys. 2000, 378, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry 2004, 43, 16233–16242. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. beta-Synuclein inhibits alpha-synuclein aggregation: A possible role as an anti-parkinsonian factor. Neuron 2001, 32, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Fink, A.L. Amino acid determinants of alpha-synuclein aggregation: Putting together pieces of the puzzle. FEBS Lett. 2002, 522, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, H.; Limprasert, P.; Fan, Y.; Onodera, O.; Kakita, A.; Takahashi, H.; Bonner, L.T.; Tsuang, D.W.; Murray, I.V.; Lee, V.M.; et al. Beta-synuclein gene alterations in dementia with Lewy bodies. Neurology 2004, 63, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Watabe, K.; Akatsu, H.; Rockenstein, E.; Masliah, E.; Hashimoto, M. Enhanced lysosomal pathology caused by beta-synuclein mutants linked to dementia with Lewy bodies. J. Biol. Chem. 2007, 282, 28904–28914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, M.; Sugama, S.; Sekiyama, K.; Sekigawa, A.; Tsukui, T.; Nakai, M.; Waragai, M.; Takenouchi, T.; Takamatsu, Y.; Wei, J.; et al. A beta-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat. Commun. 2010, 1, 110. [Google Scholar] [CrossRef]
- Sharma, K.; Mehra, S.; Sawner, A.S.; Markam, P.S.; Panigrahi, R.; Navalkar, A.; Chatterjee, D.; Kumar, R.; Kadu, P.; Patel, K.; et al. Effect of Disease-Associated P123H and V70M Mutations on β-Synuclein Fibrillation. ACS Chem. Neurosci. 2020, 11, 2836–2848. [Google Scholar] [CrossRef]
- Ji, H.; Liu, Y.E.; Jia, T.; Wang, M.; Liu, J.; Xiao, G.; Joseph, B.K.; Rosen, C.; Shi, Y.E. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res. 1997, 57, 759–764. [Google Scholar] [PubMed]
- Akopian, A.N.; Wood, J.N. Peripheral Nervous System-specific Genes Identified by Subtractive cDNA Cloning (∗). J. Biol. Chem. 1995, 270, 21264–21270. [Google Scholar] [CrossRef] [Green Version]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, A.L. The aggregation and fibrillation of alpha-synuclein. Acc. Chem. Res. 2006, 39, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H., 3rd. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar]
- Knowles, T.P.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Arosio, P.; Knowles, T.P.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: A critical analysis of alpha-synuclein staging. Neuropathol. Appl. Neurobiol. 2008, 34, 284–295. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2008, 34, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Parkkinen, L.; Pirttilä, T.; Alafuzoff, I. Applicability of current staging/categorization of α-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008, 115, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Alpha-synuclein pathology in Parkinson’s and Alzheimer’s disease brain: Incidence and topographic distribution—A pilot study. Acta Neuropathol. 2003, 106, 191–201. [Google Scholar] [CrossRef]
- Zaccai, J.; Brayne, C.; McKeith, I.; Matthews, F.; Ince, P.G. Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology 2008, 70, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Arizona Parkinson’s Disease, Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rietdijk, C.D.; Perez-Pardo, P.; Garssen, J.; van Wezel, R.J.A.; Kraneveld, A.D. Exploring Braak’s Hypothesis of Parkinson’s Disease. Front. Neurol. 2017, 8, 37. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinson’s Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longhena, F.; Faustini, G.; Missale, C.; Pizzi, M.; Spano, P.; Bellucci, A. The Contribution of α-Synuclein Spreading to Parkinson’s Disease Synaptopathy. Neural Plast. 2017, 2017, 5012129. [Google Scholar] [CrossRef] [Green Version]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.-N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Barker, R.A.; Williams-Gray, C.H. Review: The spectrum of clinical features seen with alpha synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef]
- Jellinger, K.A. A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 2009, 1792, 730–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Vilar, M.; Chou, H.T.; Luhrs, T.; Maji, S.K.; Riek-Loher, D.; Verel, R.; Manning, G.; Stahlberg, H.; Riek, R. The fold of alpha-synuclein fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 8637–8642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv. Protein Chem. 1997, 50, 123–159. [Google Scholar]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.O.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.C.; Citron, M.; Biere, A.L. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 1999, 274, 19509–19512. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Wang, L.Q.; Zhao, K.; Yuan, H.Y.; Wang, Q.; Guan, Z.; Tao, J.; Li, X.N.; Sun, Y.; Yi, C.W.; Chen, J.; et al. Cryo-EM structure of an amyloid fibril formed by full-length human prion protein. Nat. Struct. Mol. Biol. 2020, 27, 598–602. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Porta, S.; Xu, Y.; Lehr, T.; Zhang, B.; Meymand, E.; Olufemi, M.; Stieber, A.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Distinct brain-derived TDP-43 strains from FTLD-TDP subtypes induce diverse morphological TDP-43 aggregates and spreading patterns in vitro and in vivo. Neuropathol. Appl. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Collins, S.R.; Toyama, B.H.; Weissman, J.S. The physical basis of how prion conformations determine strain phenotypes. Nature 2006, 442, 585–589. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Mahmood, S. An overview of animal prion diseases. Virol. J. 2011, 8, 493. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Montrasio, F.; Kaeser, P.S. Prions: Health scare and biological challenge. Nat. Rev. Mol. Cell Biol. 2001, 2, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 1961, 71, 101–109. [Google Scholar] [CrossRef]
- Fraser, H.; Dickinson, A.G. Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J. Comp. Pathol. 1973, 83, 29–40. [Google Scholar] [CrossRef]
- Haldiman, T.; Kim, C.; Cohen, Y.; Chen, W.; Blevins, J.; Qing, L.; Cohen, M.L.; Langeveld, J.; Telling, G.C.; Kong, Q.; et al. Co-existence of distinct prion types enables conformational evolution of human PrPSc by competitive selection. J. Biol. Chem. 2013, 288, 29846–29861. [Google Scholar] [CrossRef] [Green Version]
- Makarava, N.; Baskakov, I.V. The same primary structure of the prion protein yields two distinct self-propagating states. J. Biol. Chem. 2008, 283, 15988–15996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccardo, P.; Liepnieks, J.J.; William, A.; Dlouhy, S.R.; Farlow, M.R.; Young, K.; Nochlin, D.; Bird, T.D.; Nixon, R.R.; Ball, M.J.; et al. Prion Proteins with Different Conformations Accumulate in Gerstmann-Sträussler-Scheinker Disease Caused by A117V and F198S Mutations. Am. J. Pathol. 2001, 158, 2201–2207. [Google Scholar] [CrossRef] [Green Version]
- Collinge, J.; Sidle, K.C.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Stockel, J.; Mehlhorn, I.; Groth, D.; Baldwin, M.A.; Prusiner, S.B.; James, T.L.; Cohen, F.E. Physical studies of conformational plasticity in a recombinant prion protein. Biochemistry 1997, 36, 3543–3553. [Google Scholar] [CrossRef]
- Zou, W.Q.; Gambetti, P. Prion: The chameleon protein. Cell. Mol. Life Sci. 2007, 64, 3266–3270. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Heikenwalder, M.; Polymenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell. Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef]
- Muramoto, T.; Kitamoto, T.; Tateishi, J.; Goto, I. The sequential development of abnormal prion protein accumulation in mice with Creutzfeldt-Jakob disease. Am. J. Pathol. 1992, 140, 1411–1420. [Google Scholar]
- Hill, A.F.; Collinge, J. Prion strains and species barriers. Contrib. Microbiol. 2004, 11, 33–49. [Google Scholar] [CrossRef]
- Puoti, G.; Giaccone, G.; Rossi, G.; Canciani, B.; Bugiani, O.; Tagliavini, F. Sporadic Creutzfeldt-Jakob disease: Co-occurrence of different types of PrP(Sc) in the same brain. Neurology 1999, 53, 2173–2176. [Google Scholar] [CrossRef]
- Schoch, G.; Seeger, H.; Bogousslavsky, J.; Tolnay, M.; Janzer, R.C.; Aguzzi, A.; Glatzel, M. Analysis of Prion Strains by PrPSc Profiling in Sporadic Creutzfeldt–Jakob Disease. PLOS Med. 2005, 3, e14. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Fraser, H.; McConnell, I.; Outram, G.W.; Sales, D.I.; Taylor, D.M. Extraneural competition between different scrapie agents leading to loss of infectivity. Nature 1975, 253, 556. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology 1985, 23, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Aiken, J.M.; Bessen, R.A. Delay in onset of prion disease for the HY strain of transmissible mink encephalopathy as a result of prior peripheral inoculation with the replication-deficient DY strain. J. Gen. Virol. 2004, 85, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melki, R. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J. Parkinson’s Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.; Ma, B.; Nussinov, R. Polymorphism in Alzheimer Aβ Amyloid Organization Reflects Conformational Selection in a Rugged Energy Landscape. Chem. Rev. 2010, 110, 4820–4838. [Google Scholar] [CrossRef]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of alpha-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Ahlawat, S.; Kumar, H.; Singh, N.; Navalkar, A.; Patel, K.; Kadu, P.; Kumar, R.; Jha, N.N.; Udgaonkar, J.B.; et al. α-Synuclein aggregation intermediates form fibril polymorphs with distinct prion-like properties. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jiménez, J.L.; Nettleton, E.J.; Bouchard, M.; Robinson, C.V.; Dobson, C.M.; Saibil, H.R. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 9196–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.W.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klement, K.; Wieligmann, K.; Meinhardt, J.; Hortschansky, P.; Richter, W.; Fändrich, M. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer’s abeta(1-40) amyloid fibrils. J. Mol. Biol. 2007, 373, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 2019, 8, e43584. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, K.; Liberta, F.; Vielberg, M.T.; Close, W.; Lilie, H.; Gührs, K.H.; Schierhorn, A.; Koehler, R.; Schmidt, A.; Haupt, C.; et al. Common Fibril Structures Imply Systemically Conserved Protein Misfolding Pathways In Vivo. Angew. Chem. 2017, 56, 7510–7514. [Google Scholar] [CrossRef]
- Annamalai, K.; Gührs, K.H.; Koehler, R.; Schmidt, M.; Michel, H.; Loos, C.; Gaffney, P.M.; Sigurdson, C.J.; Hegenbart, U.; Schönland, S.; et al. Polymorphism of Amyloid Fibrils In Vivo. Angew. Chem. 2016, 55, 4822–4825. [Google Scholar] [CrossRef]
- Qualman, S.J.; Haupt, H.M.; Yang, P.; Hamilton, S.R. Esophageal Lewy bodies associated with ganglion cell loss in achalasia: Similarity to Parkinson’s disease. Gastroenterology 1984, 87, 848–856. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Takahashi, H.; Takeda, S.; Ohama, E.; Ikuta, F. Parkinson’s disease: The presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 1988, 76, 217–221. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Ikuta, F. Parkinson’s disease: An immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 1990, 79, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Chen, Y.; Li, M.; Wang, Y.; Zhang, W.; Chen, X.; Ye, Q. Gastrointestinal nervous system α-synuclein as a potential biomarker of Parkinson disease. Medicine 2018, 97, e11337. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kwok, S.K.; Choe, J.Y.; Park, S.H. Recent Advances in Our Understanding of the Link between the Intestinal Microbiota and Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2019, 20, 4871. [Google Scholar] [CrossRef] [Green Version]
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.A.; Gradinaru, V. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 2020, 23, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.; Kumar, R.; Mehra, S.; Ghosh, A.; Maji, S.K.; Bhunia, A. Multitude NMR studies of alpha-synuclein familial mutants: Probing their differential aggregation propensities. Chem. Commun. 2018, 54, 3605–3608. [Google Scholar] [CrossRef]
- Watson, M.D.; Lee, J.C. N-Terminal Acetylation Affects α-Synuclein Fibril Polymorphism. Biochemistry 2019, 58, 3630–3633. [Google Scholar] [CrossRef]
- Yang, X.; Wang, B.; Hoop, C.L.; Williams, J.K.; Baum, J. NMR unveils an N-terminal interaction interface on acetylated-α-synuclein monomers for recruitment to fibrils. Proc. Natl. Acad. Sci. USA 2021, 118, e2017452118. [Google Scholar] [CrossRef]
- Ma, M.R.; Hu, Z.W.; Zhao, Y.F.; Chen, Y.X.; Li, Y.M. Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 2016, 6, 37130. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [Green Version]
- Candelise, N.; Schmitz, M.; Thüne, K.; Cramm, M.; Rabano, A.; Zafar, S.; Stoops, E.; Vanderstichele, H.; Villar-Pique, A.; Llorens, F.; et al. Effect of the micro-environment on α-synuclein conversion and implication in seeded conversion assays. Transl. Neurodegener. 2020, 9, 5. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, T.; Qureshi, M.M.; Ardah, M.T.; Varghese, S.; Shehab, S.A.; Kasai, T.; Ishigami, N.; Tamaoka, A.; Nakagawa, M.; El-Agnaf, O.M. Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 2010, 75, 1766–1772. [Google Scholar] [CrossRef] [PubMed]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Tanudjojo, B.; Shaikh, S.S.; Fenyi, A.; Bousset, L.; Agarwal, D.; Marsh, J.; Zois, C.; Heman-Ackah, S.; Fischer, R.; Sims, D.; et al. Phenotypic manifestation of α-synuclein strains derived from Parkinson’s disease and multiple system atrophy in human dopaminergic neurons. Nat. Commun. 2021, 12, 3817. [Google Scholar] [CrossRef]
- Strohaker, T.; Jung, B.C.; Liou, S.H.; Fernandez, C.O.; Riedel, D.; Becker, S.; Halliday, G.M.; Bennati, M.; Kim, W.S.; Lee, S.J.; et al. Structural heterogeneity of alpha-synuclein fibrils amplified from patient brain extracts. Nat. Commun. 2019, 10, 5535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerez, J.A.; Riek, R. Neurodegenerative diseases distinguished through protein-structure analysis. Nature 2020, 578, 223–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.; Falcon, B.; Kotecha, A.; van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-based Classification of Tauopathies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Gremer, L.; Scholzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-beta(1-42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational behavior and aggregation of alpha-synuclein in organic solvents: Modeling the effects of membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef]
- Uversky, V.N. Alpha-synuclein misfolding and neurodegenerative diseases. Curr. Protein Pept. Sci. 2008, 9, 507–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Choi, C.; Lee, S.J. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, P.; Kumar, A. Perturbation in Long-Range Contacts Modulates the Kinetics of Amyloid Formation in alpha-Synuclein Familial Mutants. ACS Chem. Neurosci. 2017, 8, 2235–2246. [Google Scholar] [CrossRef]
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovicic, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef]
- Jao, C.C.; Der-Sarkissian, A.; Chen, J.; Langen, R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA 2004, 101, 8331–8336. [Google Scholar] [CrossRef] [Green Version]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. alpha-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell. Dev. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, M.R. The biochemistry of Parkinson’s disease. Annu. Rev. Biochem. 2005, 74, 29–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Anthony Crowther, R.; Goedert, M. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 2004, 576, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Krohnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef]
- Sahay, S.; Anoop, A.; Krishnamoorthy, G.; Maji, S.K. Site-specific fluorescence dynamics of alpha-synuclein fibrils using time-resolved fluorescence studies: Effect of familial Parkinson’s disease-associated mutations. Biochemistry 2014, 53, 807–809. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Flagmeier, P.; Kumita, J.R.; Meisl, G.; Chirgadze, D.Y.; Bongiovanni, M.N.; Knowles, T.P.J.; Dobson, C.M. The Influence of Pathogenic Mutations in alpha-Synuclein on Biophysical and Structural Characteristics of Amyloid Fibrils. ACS Nano 2020, 14, 5213–5222. [Google Scholar] [CrossRef]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Zhou, K.; Hughes, M.P.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. The alpha-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc. Natl. Acad. Sci. USA 2020, 117, 3592–3602. [Google Scholar] [CrossRef]
- Sun, Y.; Hou, S.; Zhao, K.; Long, H.; Liu, Z.; Gao, J.; Zhang, Y.; Su, X.D.; Li, D.; Liu, C. Cryo-EM structure of full-length alpha-synuclein amyloid fibril with Parkinson’s disease familial A53T mutation. Cell Res. 2020, 30, 360–362. [Google Scholar] [CrossRef]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. Structures of fibrils formed by alpha-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat. Struct. Mol. Biol. 2019, 26, 1044–1052. [Google Scholar] [CrossRef]
- Zhao, K.; Li, Y.; Liu, Z.; Long, H.; Zhao, C.; Luo, F.; Sun, Y.; Tao, Y.; Su, X.-D.; Li, D.; et al. Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nat. Commun. 2020, 11, 2643. [Google Scholar] [CrossRef]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M. Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Paslawski, W.; Mysling, S.; Thomsen, K.; Jorgensen, T.J.; Otzen, D.E. Co-existence of two different alpha-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry. Angew. Chem. 2014, 53, 7560–7563. [Google Scholar] [CrossRef] [PubMed]
- Antonschmidt, L.; Dervisoglu, R.; Sant, V.; Tekwani Movellan, K.; Mey, I.; Riedel, D.; Steinem, C.; Becker, S.; Andreas, L.B.; Griesinger, C. Insights into the molecular mechanism of amyloid filament formation: Segmental folding of alpha-synuclein on lipid membranes. Sci. Adv. 2021, 7, eabg2174. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Maderna, E.; Ghidoni, R.; Benussi, L.; Tonoli, E.; Giaccone, G.; Moda, F.; Paterlini, A.; Campagnani, I.; et al. Molecular subtypes of Alzheimer’s disease. Sci. Rep. 2018, 8, 3269. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Martin, S.; Jeffrey, M. Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: Implications for differential cell targeting and PrP processing. J. Gen. Virol. 2003, 84, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [Green Version]
- Hyman, A.A.; Weber, C.A.; Julicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [Green Version]
- Bergeron-Sandoval, L.P.; Safaee, N.; Michnick, S.W. Mechanisms and Consequences of Macromolecular Phase Separation. Cell 2016, 165, 1067–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, aaf4382. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Alberti, S.; Saha, S.; Woodruff, J.B.; Franzmann, T.M.; Wang, J.; Hyman, A.A. A User’s Guide for Phase Separation Assays with Purified Proteins. J. Mol. Biol. 2018, 430, 4806–4820. [Google Scholar] [CrossRef]
- Sawner, A.S.; Ray, S.; Yadav, P.; Mukherjee, S.; Panigrahi, R.; Poudyal, M.; Patel, K.; Ghosh, D.; Kummerant, E.; Kumar, A.; et al. Modulating α-Synuclein Liquid–Liquid Phase Separation. Biochemistry 2021. [Google Scholar] [CrossRef] [PubMed]
- Malinovska, L.; Kroschwald, S.; Alberti, S. Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim. Biophys. Acta 2013, 1834, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Das, S.; Mohite, G.M.; Rout, S.K.; Halder, S.; Jha, N.N.; Ray, S.; Mehra, S.; Agarwal, V.; Maji, S.K. Cytotoxic Oligomers and Fibrils Trapped in a Gel-like State of alpha-Synuclein Assemblies. Angew. Chem. 2018, 57, 5262–5266. [Google Scholar] [CrossRef]
- Volles, M.J.; Lansbury, P.T., Jr. Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry 2003, 42, 7871–7878. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Lansbury, P.T., Jr. Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat. Cell. Biol. 2000, 2, E115–E119. [Google Scholar] [CrossRef]
- Mathieu, C.; Pappu, R.V.; Taylor, J.P. Beyond aggregation: Pathological phase transitions in neurodegenerative disease. Science 2020, 370, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Pandey, S.; Kumar, R.; Gadhe, L.; Datta, D.; Patel, K.; Mahato, J.; Navalkar, A.; Panigrahi, R.; et al. Liquid-liquid phase separation and liquid-to-solid transition mediate α-synuclein amyloid fibril containing hydrogel formation. bioRxiv 2019. [Google Scholar] [CrossRef]
- Thompson, M.J.; Sievers, S.A.; Karanicolas, J.; Ivanova, M.I.; Baker, D.; Eisenberg, D. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4074–4078. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [Green Version]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef] [Green Version]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017, 74, 163–172. [Google Scholar] [CrossRef]
- Groveman, B.R.; Orrù, C.D.; Hughson, A.G.; Raymond, L.D.; Zanusso, G.; Ghetti, B.; Campbell, K.J.; Safar, J.; Galasko, D.; Caughey, B. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol. Commun. 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, K.; Ikenaka, K.; Araki, K.; So, M.; Aguirre, C.; Kajiyama, Y.; Konaka, K.; Noi, K.; Baba, K.; Tsuda, H.; et al. Ultrasonication-based rapid amplification of α-synuclein aggregates in cerebrospinal fluid. Sci. Rep. 2019, 9, 6001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelise, N.; Schmitz, M.; Llorens, F.; Villar-Piqué, A.; Cramm, M.; Thom, T.; da Silva Correia, S.M.; da Cunha, J.E.G.; Möbius, W.; Outeiro, T.F.; et al. Seeding variability of different alpha synuclein strains in synucleinopathies. Ann. Neurol. 2019, 85, 691–703. [Google Scholar] [CrossRef] [PubMed]
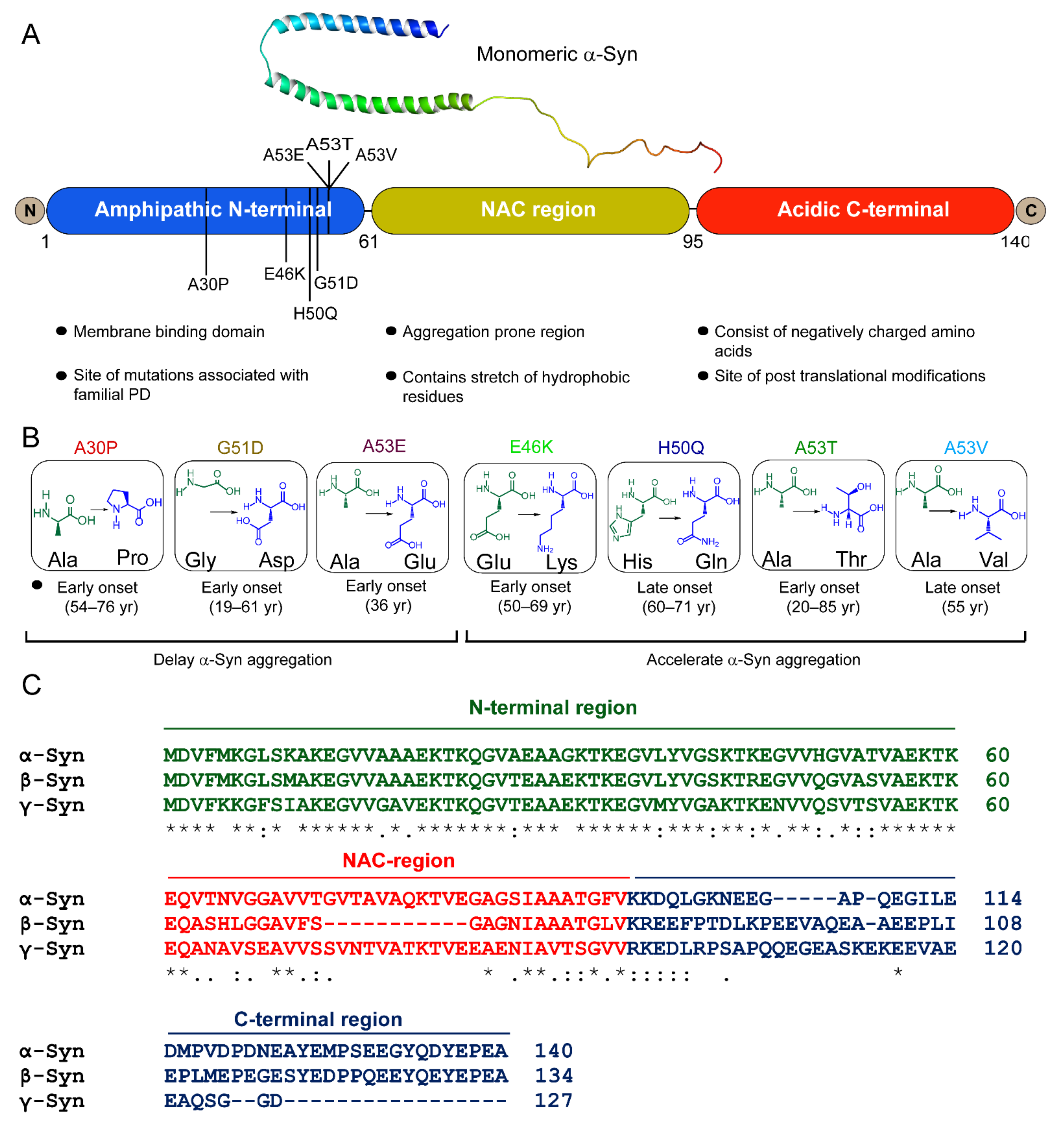



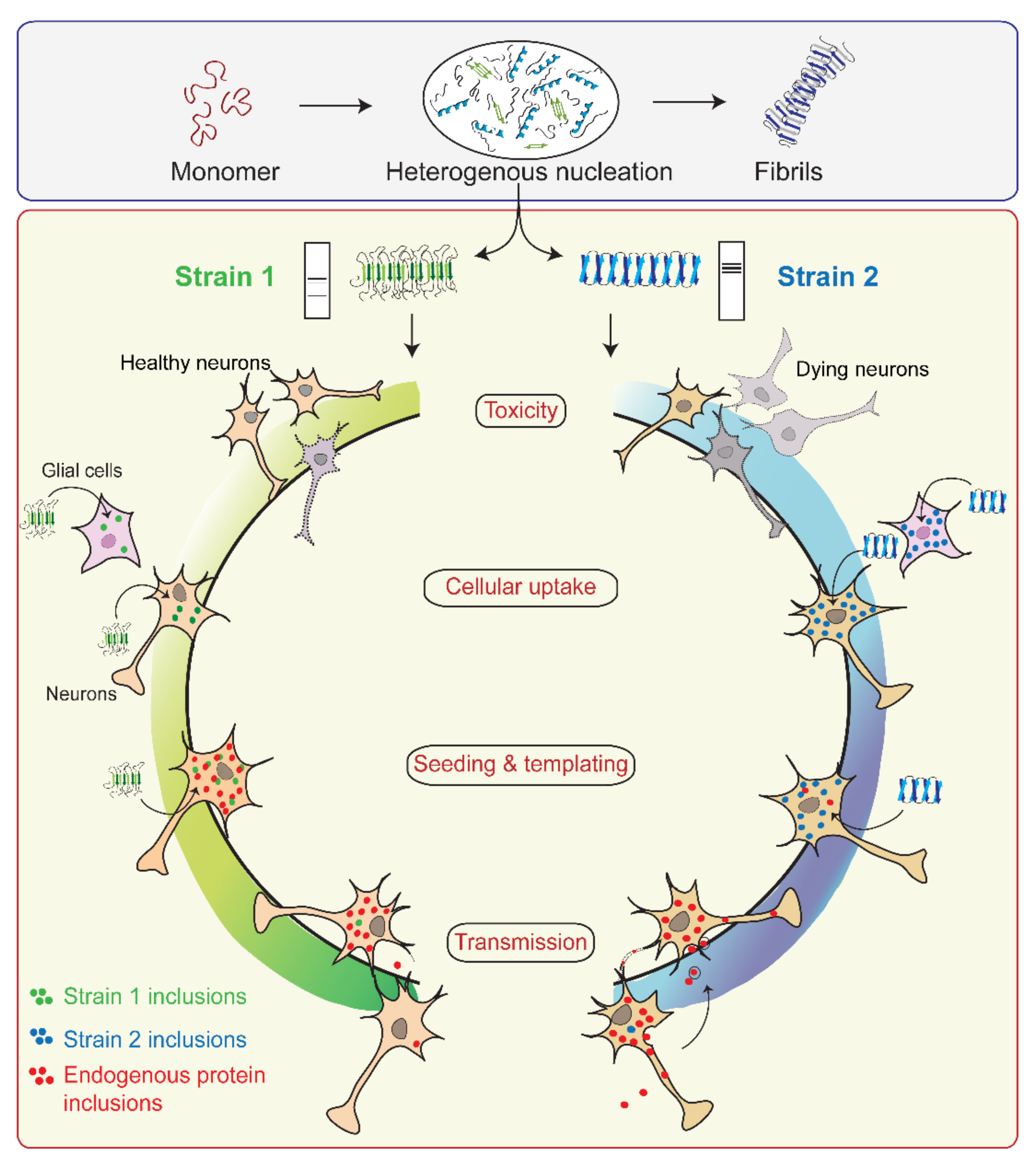

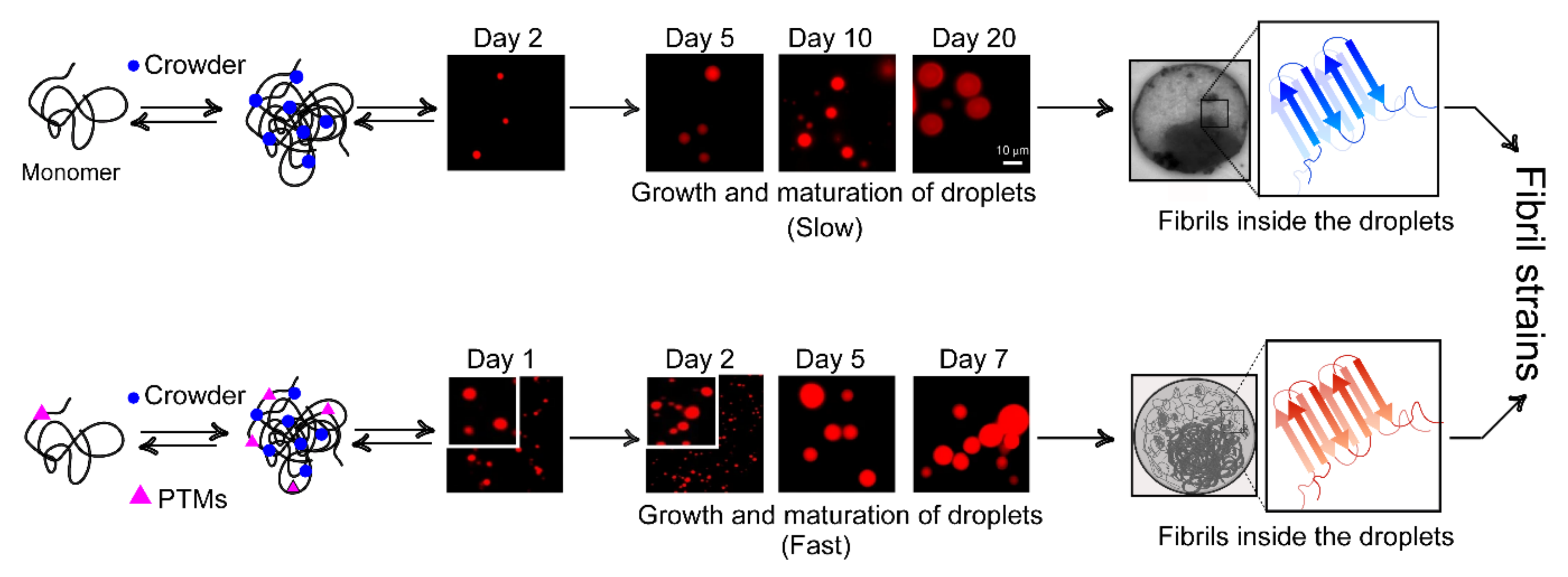
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehra, S.; Gadhe, L.; Bera, R.; Sawner, A.S.; Maji, S.K. Structural and Functional Insights into α-Synuclein Fibril Polymorphism. Biomolecules 2021, 11, 1419. https://doi.org/10.3390/biom11101419
Mehra S, Gadhe L, Bera R, Sawner AS, Maji SK. Structural and Functional Insights into α-Synuclein Fibril Polymorphism. Biomolecules. 2021; 11(10):1419. https://doi.org/10.3390/biom11101419
Chicago/Turabian StyleMehra, Surabhi, Laxmikant Gadhe, Riya Bera, Ajay Singh Sawner, and Samir K. Maji. 2021. "Structural and Functional Insights into α-Synuclein Fibril Polymorphism" Biomolecules 11, no. 10: 1419. https://doi.org/10.3390/biom11101419
APA StyleMehra, S., Gadhe, L., Bera, R., Sawner, A. S., & Maji, S. K. (2021). Structural and Functional Insights into α-Synuclein Fibril Polymorphism. Biomolecules, 11(10), 1419. https://doi.org/10.3390/biom11101419