A Great Catch for Investigating Inborn Errors of Metabolism—Insights Obtained from Zebrafish


Abstract
1. Introduction
1.1. Rare Inherited Metabolic Disorders
1.2. Animal Models of IEM
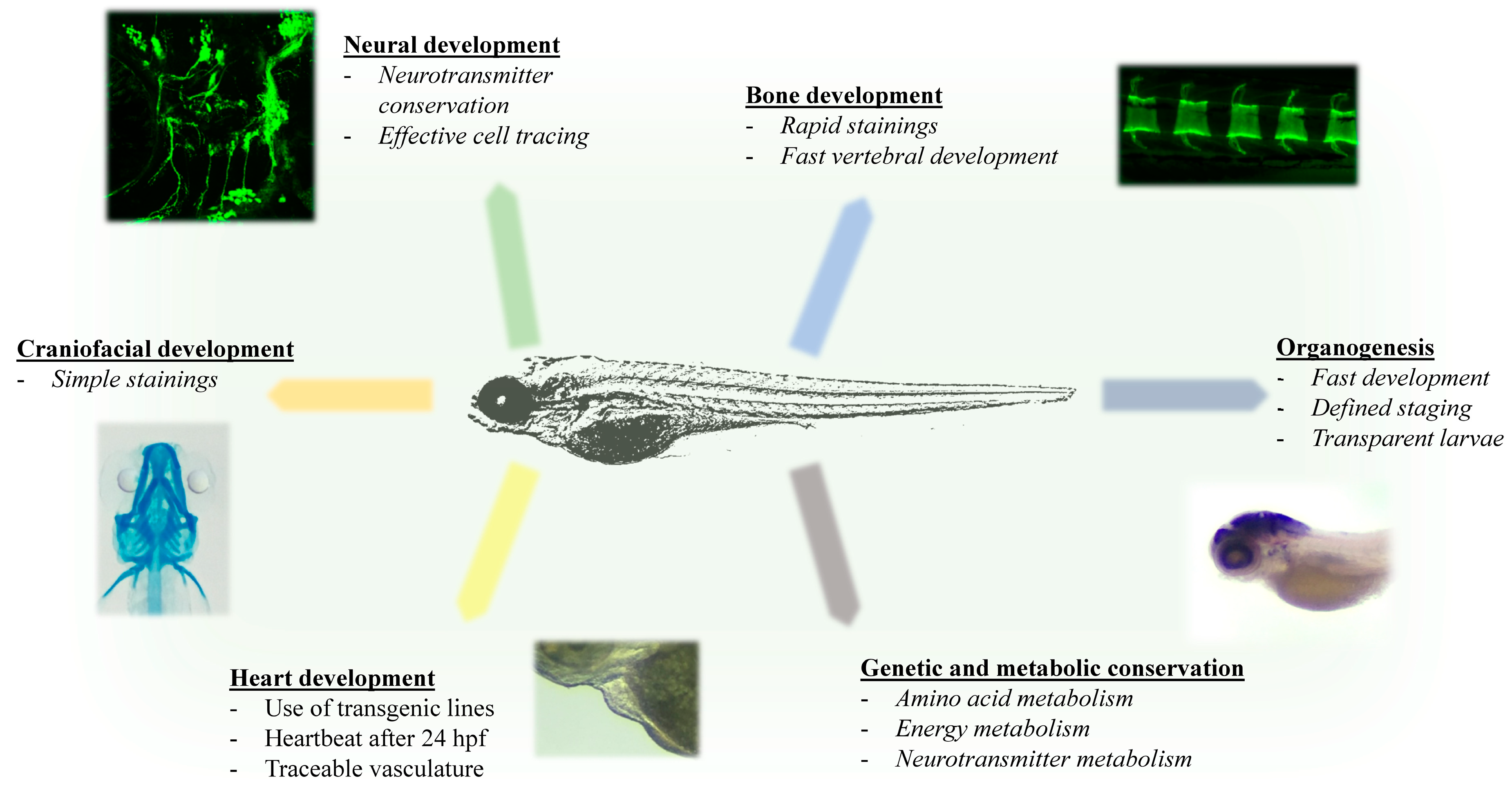
1.3. Advantages of Zebrafish for Investigating Rare Inherited Metabolic Diseases
2. Amino Acid and Peptide Metabolism
2.1. Maple Syrup Urine Disease (MSUD)
2.2. Methylmalonic Acidemia (MMA)
2.3. Multiple Acyl-CoA Dehydrogenase Deficiency/Glutaric Aciduria Type II
2.4. Other Amino Acid Disorders
3. Neurotransmission
3.1. Aromatic L-Amino Acid Decarboxylase (AADC) Deficiency
3.2. Atypical PKU/BH4 Deficiency
3.3. Hyperekplexia
4. Metabolism of Vitamins and Co-Factors
DHFR Deficiency
5. Urea Cycle Disorders
Hyperammonemia
6. Carbohydrate Metabolism
6.1. Glycogen Storage Disease Type II/Pompe Disease
6.2. Glycogen Storage Disease Type XI/Fanconi–Bickel Disease
6.3. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency
6.4. Galactosemia
6.5. GLUT1 Deficiency
7. Lipoprotein Metabolism
7.1. Hypercholesterolemia
7.2. Hypertriglyceridemia
8. Congenital Disorders of Glycosylation (CDG)
8.1. Pmm2-CDG
8.2. MPI-CDG
8.3. PGM3-CDG
8.4. TMEM165-CDG
8.5. TRAPPC11-CDG
9. Energy and Pyruvate Metabolism
Pyruvate Dehydrogenase (PDH) Complex
10. Lysosomal Storage Disorders
10.1. Gaucher’s Disease
10.2. Niemann-Pick Disease C (NPC)
10.3. Farber Disease
10.4. Mucopolysaccharidosis (MPS)
10.5. Mucopolysaccharidosis II (MPS II)/Hunter Syndrome
11. Novel Therapeutic Intervention for Metabolic Disorders
12. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Garrod, A.E. Inborn Errors of Metabolism, 2nd ed.; Oxford University Press: Oxford, UK, 1923. [Google Scholar]
- Martins, A.M. Inborn errors of metabolism: A clinical overview. Sao Paulo Med. J. 1999, 117, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Pampols, T. Inherited metabolic rare disease. Adv. Exp. Med. Biol. 2010, 686, 397–431. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.J.; Wei, N.; Li, M.; Xie, K.; Li, J.Q.; Huang, C.G.; Xiao, Y.S.; Liu, W.H.; Chen, X.G. Diagnosis and therapeutic monitoring of inborn errors of metabolism in 100,077 newborns from Jining city in China. BMC Pediatrics 2018, 18, 110. [Google Scholar] [CrossRef]
- Applegarth, D.A.; Toone, J.R.; Lowry, R.B. Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics 2000, 105, e10. [Google Scholar] [CrossRef] [PubMed]
- Hutchesson, A.C.; Bundey, S.; Preece, M.A.; Hall, S.K.; Green, A. A comparison of disease and gene frequencies of inborn errors of metabolism among different ethnic groups in the West Midlands, UK. J. Med. Genet. 1998, 35, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Keyfi, F.; Nasseri, M.; Nayerabadi, S.; Alaei, A.; Mokhtariye, A.; Varasteh, A. Frequency of Inborn Errors of Metabolism in a Northeastern Iranian Sample with High Consanguinity Rates. Hum. Hered. 2018, 83, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, S.; Green, A.; Preece, M.A.; Burton, H. The incidence of inherited metabolic disorders in the West Midlands, UK. Arch. Dis. Child. 2006, 91, 896–899. [Google Scholar] [CrossRef]
- Waters, D.; Adeloye, D.; Woolham, D.; Wastnedge, E.; Patel, S.; Rudan, I. Global birth prevalence and mortality from inborn errors of metabolism: A systematic analysis of the evidence. J. Glob. Health 2018, 8, 021102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, Y.; Peng, W.; Chang, J.; Mei, Y.; Yan, L.; Chen, Y.; Wei, X.; Liu, Y.; Wang, Y.; et al. A 7-Year Report of Spectrum of Inborn Errors of Metabolism on Full-Term and Premature Infants in a Chinese Neonatal Intensive Care Unit. Front. Genet. 2019, 10, 1302. [Google Scholar] [CrossRef]
- Vernon, H.J. Inborn Errors of Metabolism: Advances in Diagnosis and Therapy. JAMA Pediatr. 2015, 169, 778–782. [Google Scholar] [CrossRef]
- Smith, L.D.; Willig, L.K.; Kingsmore, S.F. Whole-Exome Sequencing and Whole-Genome Sequencing in Critically Ill Neonates Suspected to Have Single-Gene Disorders. Cold Spring Harb. Perspect. Med. 2015, 6, a023168. [Google Scholar] [CrossRef] [PubMed]
- Jelin, A.C.; Vora, N. Whole Exome Sequencing: Applications in Prenatal Genetics. Obstet. Gynecol. Clin. North Am. 2018, 45, 69–81. [Google Scholar] [CrossRef] [PubMed]
- van Rijt, W.J.; Koolhaas, G.D.; Bekhof, J.; Heiner Fokkema, M.R.; de Koning, T.J.; Visser, G.; Schielen, P.C.; van Spronsen, F.J.; Derks, T.G. Inborn Errors of Metabolism That Cause Sudden Infant Death: A Systematic Review with Implications for Population Neonatal Screening Programmes. Neonatology 2016, 109, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.E.; Hummel, K.; Smith, A.G.; Longo, N. Acute Presentation and Management of the Encephalopathic Child With an Undiagnosed Inborn Error of Metabolism. J. Emerg. Med. 2019, 56, e5–e8. [Google Scholar] [CrossRef] [PubMed]
- Romao, A.; Simon, P.E.A.; Goes, J.E.C.; Pinto, L.L.C.; Giugliani, R.; Luca, G.R.; Carvalho, F.L.C. Initial Clinical Presentation in Cases of Inborn Errors of Metabolism in a Reference Children’s Hospital: Still a Diagnostic Challenge. Rev. Paul. Pediatr. 2017, 35, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.A.; Thomason, M.J.; Chalmers, R.A.; Addison, G.M.; Bain, M.D.; Cockburn, F.; Littlejohns, P.; Lord, J.; Wilcox, A.H. Newborn screening for inborn errors of metabolism: A systematic review. Health Technol. Assess 1997, 1, 1–95. [Google Scholar] [CrossRef]
- Camp, K.M.; Lloyd-Puryear, M.A.; Huntington, K.L. Nutritional treatment for inborn errors of metabolism: Indications, regulations, and availability of medical foods and dietary supplements using phenylketonuria as an example. Mol. Genet. Metab. 2012, 107, 3–9. [Google Scholar] [CrossRef]
- Kabra, M. Dietary management of inborn errors of metabolism. Indian J. Pediatr. 2002, 69, 421–426. [Google Scholar] [CrossRef]
- Ris, M.D.; Williams, S.E.; Hunt, M.M.; Berry, H.K.; Leslie, N. Early-treated phenylketonuria: Adult neuropsychologic outcome. J. Pediatr. 1994, 124, 388–392. [Google Scholar] [CrossRef]
- McDonnell, G.V.; Esmonde, T.F.; Hadden, D.R.; Morrow, J.I. A neurological evaluation of adult phenylketonuria in Northern Ireland. Eur. Neurol. 1998, 39, 38–43. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Vaz, F.M.; Ferdinandusse, S.; van Kuilenburg, A.B.P.; Kemp, S.; van Karnebeek, C.D.; Waterham, H.R.; Houtkooper, R.H. Translational Metabolism: A multidisciplinary approach towards precision diagnosis of inborn errors of metabolism in the omics era. J. Inherit Metab. Dis. 2019, 42, 197–208. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.D. Production of mouse models for the study of human inborn errors of metabolism. Mol. Genet. Metab. 2000, 71, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Pronk, J.T.; van der Linden-Beuman, A.; Verduyn, C.; Scheffers, W.A.; van Dijken, J.P. Propionate metabolism in Saccharomyces cerevisiae: Implications for the metabolon hypothesis. Microbiology 1994, 140, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Struwe, W.B.; Hughes, B.L.; Osborn, D.W.; Boudreau, E.D.; Shaw, K.M.; Warren, C.E. Modeling a congenital disorder of glycosylation type I in C. elegans: A genome-wide RNAi screen for N-glycosylation-dependent loci. Glycobiology 2009, 19, 1554–1562. [Google Scholar] [CrossRef]
- Hindle, S.; Hebbar, S.; Sweeney, S.T. Invertebrate models of lysosomal storage disease: What have we learned so far? Invert. Neurosci. 2011, 11, 59–71. [Google Scholar] [CrossRef]
- de Voer, G.; Peters, D.; Taschner, P.E. Caenorhabditis elegans as a model for lysosomal storage disorders. Biochim. Biophys. Acta 2008, 1782, 433–446. [Google Scholar] [CrossRef][Green Version]
- Sellin, J.; Schulze, H.; Paradis, M.; Gosejacob, D.; Papan, C.; Shevchenko, A.; Psathaki, O.E.; Paululat, A.; Thielisch, M.; Sandhoff, K.; et al. Characterization of Drosophila Saposin-related mutants as a model for lysosomal sphingolipid storage diseases. Dis. Models Mech. 2017, 10, 737–750. [Google Scholar] [CrossRef]
- Daenzer, J.M.; Fridovich-Keil, J.L. Drosophila melanogaster Models of Galactosemia. Curr. Top Dev. Biol. 2017, 121, 377–395. [Google Scholar] [CrossRef]
- Vora, S.; Giger, U.; Turchen, S.; Harvey, J.W. Characterization of the enzymatic lesion in inherited phosphofructokinase deficiency in the dog: An animal analogue of human glycogen storage disease type VII. Proc. Natl. Acad. Sci. USA 1985, 82, 8109–8113. [Google Scholar] [CrossRef]
- Katz, M.L.; Rustad, E.; Robinson, G.O.; Whiting, R.E.H.; Student, J.T.; Coates, J.R.; Narfstrom, K. Canine neuronal ceroid lipofuscinoses: Promising models for preclinical testing of therapeutic interventions. Neurobiol. Dis. 2017, 108, 277–287. [Google Scholar] [CrossRef]
- Sewell, A.C.; Haskins, M.E.; Giger, U. Inherited metabolic disease in companion animals: Searching for nature’s mistakes. Vet. J. (London, England: 1997) 2007, 174, 252–259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schlegel, A.; Gut, P. Metabolic insights from zebrafish genetics, physiology, and chemical biology. Cell Mol. Life Sci. 2015, 72, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Akhmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef]
- Ata, H.; Ekstrom, T.L.; Martinez-Galvez, G.; Mann, C.M.; Dvornikov, A.V.; Schaefbauer, K.J.; Ma, A.C.; Dobbs, D.; Clark, K.J.; Ekker, S.C. Robust activation of microhomology-mediated end joining for precision gene editing applications. PLoS Genet. 2018, 14, e1007652. [Google Scholar] [CrossRef]
- Wierson, W.A.; Simone, B.W.; WareJoncas, Z.; Mann, C.; Welker, J.M.; Kar, B.; Emch, M.J.; Friedberg, I.; Gendron, W.A.C.; Barry, M.A.; et al. Expanding the CRISPR Toolbox with ErCas12a in Zebrafish and Human Cells. CRISPR J. 2019, 2, 417–433. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Parichy, D.M.; Elizondo, M.R.; Mills, M.G.; Gordon, T.N.; Engeszer, R.E. Normal table of postembryonic zebrafish development: Staging by externally visible anatomy of the living fish. Dev. Dyn. 2009, 238, 2975–3015. [Google Scholar] [CrossRef]
- Randlett, O.; Wee, C.L.; Naumann, E.A.; Nnaemeka, O.; Schoppik, D.; Fitzgerald, J.E.; Portugues, R.; Lacoste, A.M.B.; Riegler, C.; Engert, F.; et al. Whole-brain activity mapping onto a zebrafish brain atlas. Nat. Methods 2015, 12, 1039–1046. [Google Scholar] [CrossRef]
- Rocha, M.; Singh, N.; Ahsan, K.; Beiriger, A.; Prince, V.E. Neural crest development: Insights from the zebrafish. Dev. Dyn. 2020, 249, 88–111. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Wullimann, M. Atlas of Early Zebrafish Brain Development: A Tool for Molecular Neurogenetics; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.S.; Braasch, I.; Frickey, T.; Meyer, A.; Van de Peer, Y. Genome duplication, a trait shared by 22000 species of ray-finned fish. Genome Res. 2003, 13, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Schauerte, H.E.; van Eeden, F.J.; Fricke, C.; Odenthal, J.; Strahle, U.; Haffter, P. Sonic hedgehog is not required for the induction of medial floor plate cells in the zebrafish. Development 1998, 125, 2983–2993. [Google Scholar] [PubMed]
- Benchoula, K.; Khatib, A.; Jaffar, A.; Ahmed, Q.U.; Sulaiman, W.; Wahab, R.A.; El-Seedi, H.R. The promise of zebrafish as a model of metabolic syndrome. Exp. Anim. 2019, 68, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Salmi, T.M.; Tan, V.W.T.; Cox, A.G. Dissecting metabolism using zebrafish models of disease. Biochem. Soc. Trans. 2019, 47, 305–315. [Google Scholar] [CrossRef]
- Zhang, Y.; Qin, C.; Yang, L.; Lu, R.; Zhao, X.; Nie, G. A comparative genomics study of carbohydrate/glucose metabolic genes: From fish to mammals. BMC Genom. 2018, 19, 246. [Google Scholar] [CrossRef]
- Huang, S.M.; Xu, F.; Lam, S.H.; Gong, Z.; Ong, C.N. Metabolomics of developing zebrafish embryos using gas chromatography- and liquid chromatography-mass spectrometry. Mol. Biosyst. 2013, 9, 1372–1380. [Google Scholar] [CrossRef]
- Dhillon, S.S.; Torell, F.; Donten, M.; Lundstedt-Enkel, K.; Bennett, K.; Rannar, S.; Trygg, J.; Lundstedt, T. Metabolic profiling of zebrafish embryo development from blastula period to early larval stages. PLoS ONE 2019, 14, e0213661. [Google Scholar] [CrossRef]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 2018, 11, 294. [Google Scholar] [CrossRef]
- Eimon, P.M.; Rubinstein, A.L. The use of in vivo zebrafish assays in drug toxicity screening. Expert Opin. Drug Metab. Toxicol. 2009, 5, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Yoganantharjah, P.; Gibert, Y. The Use of the Zebrafish Model to Aid in Drug Discovery and Target Validation. Curr. Top Med. Chem. 2017, 17, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
- Basnet, R.M.; Zizioli, D.; Taweedet, S.; Finazzi, D.; Memo, M. Zebrafish Larvae as a Behavioral Model in Neuropharmacology. Biomedicines 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.A.; Aggad, D.; Martinez, J.; Tremblay, E.; Petrillo, J.; Armstrong, G.A.; La Fontaine, A.; Maios, C.; Liao, M.; Ciura, S.; et al. Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Rennekamp, A.J.; Peterson, R.T. 15 years of zebrafish chemical screening. Curr. Opin. Chem. Biol. 2015, 24, 58–70. [Google Scholar] [CrossRef]
- Costa, B.; Estrada, M.F.; Mendes, R.V.; Fior, R. Zebrafish Avatars towards Personalized Medicine-A Comparative Review between Avatar Models. Cells 2020, 9, 293. [Google Scholar] [CrossRef]
- Usai, A.; Di Franco, G.; Colucci, P.; Pollina, L.E.; Vasile, E.; Funel, N.; Palmeri, M.; Dente, L.; Falcone, A.; Morelli, L.; et al. A Model of a Zebrafish Avatar for Co-Clinical Trials. Cancers 2020, 12, 677. [Google Scholar] [CrossRef]
- Ozand, P.T.; Gascon, G.G. Organic acidurias: A review. Part 1. J. Child Neurol. 1991, 6, 196–219. [Google Scholar] [CrossRef]
- Vaidyanathan, K.; Narayanan, M.P.; Vasudevan, D.M. Organic acidurias: An updated review. Indian J. Clin. Biochem. 2011, 26, 319–325. [Google Scholar] [CrossRef]
- Yannicelli, S. Nutrition therapy of organic acidaemias with amino acid-based formulas: Emphasis on methylmalonic and propionic acidaemia. J. Inherit Metab. Dis. 2006, 29, 281–287. [Google Scholar] [CrossRef]
- Strauss, K.A.; Morton, D.H. Branched-chain Ketoacyl Dehydrogenase Deficiency: Maple Syrup Disease. Curr. Treat Options Neurol. 2003, 5, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Lambert, A.M.; Masino, M.A.; Downes, G.B. Mutation of zebrafish dihydrolipoamide branched-chain transacylase E2 results in motor dysfunction and models maple syrup urine disease. Dis. Models Mech. 2012, 5, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.L.; Outeiro, T.F.; Ferreira, J.J. Zebrafish as an Animal Model for Drug Discovery in Parkinson’s Disease and Other Movement Disorders: A Systematic Review. Front. Neurol. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Cornet, C.; Di Donato, V.; Terriente, J. Combining Zebrafish and CRISPR/Cas9: Toward a More Efficient Drug Discovery Pipeline. Front. Pharmacol. 2018, 9, 703. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, P. Zebrafish as a pharmacological tool: The how, why and when. Curr. Opin. Pharmacol. 2004, 4, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Wessler, L.B.; Farias, H.R.; Ronsani, J.F.; Candiotto, G.; Dos Santos, P.C.L.; de Oliveira, J.; Rico, E.P.; Streck, E.L. Acute exposure to leucine modifies behavioral parameters and cholinergic activity in zebrafish. Int. J. Dev. Neurosci. 2019, 78, 222–226. [Google Scholar] [CrossRef]
- Oberholzer, V.G.; Levin, B.; Burgess, E.A.; Young, W.F. Methylmalonic aciduria. An inborn error of metabolism leading to chronic metabolic acidosis. Arch. Dis. Child. 1967, 42, 492–504. [Google Scholar] [CrossRef]
- Chace, D.H.; DiPerna, J.C.; Kalas, T.A.; Johnson, R.W.; Naylor, E.W. Rapid diagnosis of methylmalonic and propionic acidemias: Quantitative tandem mass spectrometric analysis of propionylcarnitine in filter-paper blood specimens obtained from newborns. Clin. Chem. 2001, 47, 2040–2044. [Google Scholar] [CrossRef]
- Fenton, W.A.; Gravel, R.A.; Rosenblatt, D.S. The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2165–2193. [Google Scholar]
- Peters, H.; Nefedov, M.; Sarsero, J.; Pitt, J.; Fowler, K.J.; Gazeas, S.; Kahler, S.G.; Ioannou, P.A. A knock-out mouse model for methylmalonic aciduria resulting in neonatal lethality. J. Biol. Chem. 2003, 278, 52909–52913. [Google Scholar] [CrossRef]
- Luciani, A.; Schumann, A.; Berquez, M.; Chen, Z.; Nieri, D.; Failli, M.; Debaix, H.; Festa, B.P.; Tokonami, N.; Raimondi, A.; et al. Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl-CoA mutase deficiency. Nat. Commun. 2020, 11, 970. [Google Scholar] [CrossRef]
- Luciani, A.; Devuyst, O. Methylmalonyl acidemia: From mitochondrial metabolism to defective mitophagy and disease. Autophagy 2020, 16, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Berquez, M.; Luciani, A. Mitochondria, mitophagy, and metabolic disease: Towards assembling the puzzle. Cell Stress 2020, 4, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.L.; Achilly, N.P.; Arnold, M.L.; Catlett, J.L.; Blake, T.; Bishop, K.; Jones, M.; Harper, U.; English, M.A.; Anderson, S.; et al. The vitamin B12 processing enzyme, mmachc, is essential for zebrafish survival, growth and retinal morphology. Hum. Mol. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Selak, M.A.; Watson, C.T.; Coutts, C.; Scherer, P.C.; Panzer, J.A.; Gibbs, S.; Scott, M.O.; Willer, G.; Gregg, R.G.; et al. Mechanisms underlying metabolic and neural defects in zebrafish and human multiple acyl-CoA dehydrogenase deficiency (MADD). PLoS ONE 2009, 4, e8329. [Google Scholar] [CrossRef] [PubMed]
- Panzer, J.A.; Gibbs, S.M.; Dosch, R.; Wagner, D.; Mullins, M.C.; Granato, M.; Balice-Gordon, R.J. Neuromuscular synaptogenesis in wild-type and mutant zebrafish. Dev. Biol. 2005, 285, 340–357. [Google Scholar] [CrossRef] [PubMed]
- Khuchua, Z.; Yue, Z.; Batts, L.; Strauss, A.W. A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ. Res. 2006, 99, 201–208. [Google Scholar] [CrossRef]
- Jung, S.; Silvius, D.; Nolan, K.A.; Borchert, G.L.; Millet, Y.H.; Phang, J.M.; Gunn, T.M. Developmental cardiac hypertrophy in a mouse model of prolidase deficiency. Birth Defects Res. Clin. Mol. Teratol. 2011, 91, 204–217. [Google Scholar] [CrossRef]
- Shih, D.F.; Hsiao, C.D.; Min, M.Y.; Lai, W.S.; Yang, C.W.; Lee, W.T.; Lee, S.J. Aromatic L-amino acid decarboxylase (AADC) is crucial for brain development and motor functions. PLoS ONE 2013, 8, e71741. [Google Scholar] [CrossRef]
- Breuer, M.; Guglielmi, L.; Zielonka, M.; Hemberger, V.; Kolker, S.; Okun, J.G.; Hoffmann, G.F.; Carl, M.; Sauer, S.W.; Opladen, T. QDPR homologues in Danio rerio regulate melanin synthesis, early gliogenesis, and glutamine homeostasis. PLoS ONE 2019, 14, e0215162. [Google Scholar] [CrossRef]
- Hirata, H.; Carta, E.; Yamanaka, I.; Harvey, R.J.; Kuwada, J.Y. Defective glycinergic synaptic transmission in zebrafish motility mutants. Front. Mol. Neurosci. 2009, 2, 26. [Google Scholar] [CrossRef]
- Ogino, K.; Hirata, H. Defects of the Glycinergic Synapse in Zebrafish. Front. Mol. Neurosci. 2016, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Low, S.E.; Ito, D.; Hirata, H. Characterization of the Zebrafish Glycine Receptor Family Reveals Insights Into Glycine Receptor Structure Function and Stoichiometry. Front. Mol. Neurosci. 2018, 11, 286. [Google Scholar] [CrossRef] [PubMed]
- Samarut, E.; Chalopin, D.; Riche, R.; Allard, M.; Liao, M.; Drapeau, P. Individual knock out of glycine receptor alpha subunits identifies a specific requirement of glra1 for motor function in zebrafish. PLoS ONE 2019, 14, e0216159. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.N.; Gui, Y.H.; Wang, Y.X.; Qian, L.X.; Jiang, Q.; Liu, D.; Song, H.Y. Effect of dihydrofolate reductase gene knock-down on the expression of heart and neural crest derivatives expressed transcript 2 in zebrafish cardiac development. Chin. Med. J. 2007, 120, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.T.; Wang, K.C.; Chang, W.N.; Lin, C.Y.; Chen, B.H.; Wu, H.L.; Shi, G.Y.; Tsai, J.N.; Fu, T.F. Characterization and comparative studies of zebrafish and human recombinant dihydrofolate reductases--inhibition by folic acid and polyphenols. Drug Metab. Dispos. 2008, 36, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gui, Y.; Wang, Y.; Qian, L.; Liu, X.; Jiang, Q.; Song, H. Effects of methotrexate on the developments of heart and vessel in zebrafish. Acta Biochim. Biophys. Sin. 2009, 41, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.N.; Gui, Y.H.; Jiang, Q.; Song, H.Y. [Effects of folic acid on the development of heart of zebrafish]. Zhonghua Er Ke Za Zhi 2010, 48, 905–912. [Google Scholar]
- Sun, S.; Gui, Y.; Jiang, Q.; Song, H. Dihydrofolate reductase is required for the development of heart and outflow tract in zebrafish. Acta Biochim. Biophys. Sin. 2011, 43, 957–969. [Google Scholar] [CrossRef]
- Lee, M.S.; Bonner, J.R.; Bernard, D.J.; Sanchez, E.L.; Sause, E.T.; Prentice, R.R.; Burgess, S.M.; Brody, L.C. Disruption of the folate pathway in zebrafish causes developmental defects. BMC Dev. Biol. 2012, 12, 12. [Google Scholar] [CrossRef]
- Chang, W.N.; Chi, W.Y.; Kao, T.T.; Tsai, J.N.; Liu, W.; Liang, S.S.; Chiu, C.C.; Chen, B.H.; Fu, T.F. The Transgenic Zebrafish Display Fluorescence Reflecting the Expressional Dynamics of Dihydrofolate Reductase. Zebrafish 2017, 14, 223–235. [Google Scholar] [CrossRef]
- Thapliyal, C.; Jain, N.; Rashid, N.; Chaudhuri Chattopadhyay, P. Kinetics and thermodynamics of the thermal inactivation and chaperone assisted folding of zebrafish dihydrofolate reductase. Arch Biochem. Biophys. 2018, 637, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Matera, C.; Gomila, A.M.J.; Camarero, N.; Libergoli, M.; Soler, C.; Gorostiza, P. Photoswitchable Antimetabolite for Targeted Photoactivated Chemotherapy. J. Am. Chem. Soc. 2018, 140, 15764–15773. [Google Scholar] [CrossRef]
- Summar, M.L.; Mew, N.A. Inborn Errors of Metabolism with Hyperammonemia: Urea Cycle Defects and Related Disorders. Pediatr. Clin. North Am. 2018, 65, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.; Tuchman, M.; Caldovic, L. A zebrafish model of hyperammonemia. Mol. Genet. Metab. 2014, 113, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, M.; Breuer, M.; Okun, J.G.; Carl, M.; Hoffmann, G.F.; Kolker, S. Pharmacologic rescue of hyperammonemia-induced toxicity in zebrafish by inhibition of ornithine aminotransferase. PLoS ONE 2018, 13, e0203707. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, M.; Probst, J.; Carl, M.; Hoffmann, G.F.; Kolker, S.; Okun, J.G. Bioenergetic dysfunction in a zebrafish model of acute hyperammonemic decompensation. Exp. Neurol. 2019, 314, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Probst, J.; Kolker, S.; Okun, J.G.; Kumar, A.; Gursky, E.; Posset, R.; Hoffmann, G.F.; Peravali, R.; Zielonka, M. Chronic hyperammonemia causes a hypoglutamatergic and hyperGABAergic metabolic state associated with neurobehavioral abnormalities in zebrafish larvae. Exp. Neurol. 2020, 331, 113330. [Google Scholar] [CrossRef]
- Hicks, J.; Wartchow, E.; Mierau, G. Glycogen storage diseases: A brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct. Pathol. 2011, 35, 183–196. [Google Scholar] [CrossRef]
- Kanungo, S.; Wells, K.; Tribett, T.; El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 2018, 6, 474. [Google Scholar] [CrossRef]
- Wu, J.; Yang, Y.; Sun, C.; Sun, S.; Li, Q.; Yao, Y.; Fei, F.; Lu, L.; Chang, Z.; Zhang, W.; et al. Disruption of the gaa Gene in Zebrafish Fails to Generate the Phenotype of Classical Pompe Disease. DNA Cell Biol. 2017, 36, 10–17. [Google Scholar] [CrossRef]
- Bragato, C.; Carra, S.; Blasevich, F.; Salerno, F.; Brix, A.; Bassi, A.; Beltrame, M.; Cotelli, F.; Maggi, L.; Mantegazza, R.; et al. Glycogen storage in a zebrafish Pompe disease model is reduced by 3-BrPA treatment. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165662. [Google Scholar] [CrossRef]
- Castillo, J.; Crespo, D.; Capilla, E.; Diaz, M.; Chauvigne, F.; Cerda, J.; Planas, J.V. Evolutionary structural and functional conservation of an ortholog of the GLUT2 glucose transporter gene (SLC2A2) in zebrafish. Am. J. Physiol. Regul Integr. Comp. Physiol. 2009, 297, R1570–R1581. [Google Scholar] [CrossRef] [PubMed]
- Marin-Juez, R.; Rovira, M.; Crespo, D.; van der Vaart, M.; Spaink, H.P.; Planas, J.V. GLUT2-mediated glucose uptake and availability are required for embryonic brain development in zebrafish. J. Cereb. Blood Flow Metab. 2015, 35, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.; Vanegas, O.C.; Patel, M.; Rosti, V.; Li, H.; Waka, J.; Merghoub, T.; Pandolfi, P.P.; Notaro, R.; Manova, K.; et al. Maternally transmitted severe glucose 6-phosphate dehydrogenase deficiency is an embryonic lethal. EMBO J. 2002, 21, 4229–4239. [Google Scholar] [CrossRef] [PubMed]
- Patrinostro, X.; Carter, M.L.; Kramer, A.C.; Lund, T.C. A model of glucose-6-phosphate dehydrogenase deficiency in the zebrafish. Exp. Hematol. 2013, 41, 697–710. [Google Scholar] [CrossRef][Green Version]
- Hwang, S.; Mruk, K.; Rahighi, S.; Raub, A.G.; Chen, C.-H.; Dorn, L.E.; Horikoshi, N.; Wakatsuki, S.; Chen, J.K.; Mochly-Rosen, D. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. Nat. Commun. 2018, 9, 4045. [Google Scholar] [CrossRef]
- Wu, Y.H.; Lee, Y.H.; Shih, H.Y.; Chen, S.H.; Cheng, Y.C.; Tsun-Yee Chiu, D. Glucose-6-phosphate dehydrogenase is indispensable in embryonic development by modulation of epithelial-mesenchymal transition via the NOX/Smad3/miR-200b axis. Cell Death Dis. 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Agusti, I.; Carecchio, M.; Bhatia, K.P.; Kojovic, M.; Parees, I.; Chandrashekar, H.S.; Footitt, E.J.; Burke, D.; Edwards, M.J.; Lachmann, R.H.; et al. Movement disorders in adult patients with classical galactosemia. Mov. Disord. 2013, 28, 804–810. [Google Scholar] [CrossRef]
- Vanoevelen, J.M.; van Erven, B.; Bierau, J.; Huang, X.; Berry, G.T.; Vos, R.; Coelho, A.I.; Rubio-Gozalbo, M.E. Impaired fertility and motor function in a zebrafish model for classic galactosemia. J. Inherit. Metab. Dis. 2018, 41, 117–127. [Google Scholar] [CrossRef]
- Leslie, N.D.; Yager, K.L.; McNamara, P.D.; Segal, S. A mouse model of galactose-1-phosphate uridyl transferase deficiency. Biochem. Mol. Med. 1996, 59, 7–12. [Google Scholar] [CrossRef]
- Tang, M.; Siddiqi, A.; Witt, B.; Yuzyuk, T.; Johnson, B.; Fraser, N.; Chen, W.; Rascon, R.; Yin, X.; Goli, H.; et al. Subfertility and growth restriction in a new galactose-1 phosphate uridylyltransferase (GALT)-deficient mouse model. Eur. J. Hum. Genet. 2014, 22, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Ryan, S.; Lambert, D.; Geoghegan, O.; Clark, A.; Rogers, Y.; Hendroff, U.; Monavari, A.; Twomey, E.; Treacy, E.P. Outcomes of siblings with classical galactosemia. J. Pediatr. 2009, 154, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Waisbren, S.E.; Potter, N.L.; Gordon, C.M.; Green, R.C.; Greenstein, P.; Gubbels, C.S.; Rubio-Gozalbo, E.; Schomer, D.; Welt, C.; Anastasoaie, V.; et al. The adult galactosemic phenotype. J. Inherit. Metab. Dis. 2012, 35, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Potter, N.L.; Nievergelt, Y.; Shriberg, L.D. Motor and speech disorders in classic galactosemia. JIMD Rep. 2013, 11, 31–41. [Google Scholar] [CrossRef]
- Haskovic, M.; Coelho, A.I.; Lindhout, M.; Zijlstra, F.; Veizaj, R.; Vos, R.; Vanoevelen, J.M.; Bierau, J.; Lefeber, D.J.; Rubio-Gozalbo, M.E. Nucleotide sugar profiles throughout development in wildtype and galt knockout zebrafish. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.J.; Gitlin, J.D.; Carayannopoulos, M.O. GLUT1 deficiency links nutrient availability and apoptosis during embryonic development. J. Biol. Chem. 2006, 281, 13382–13387. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Carten, J.D.; Farber, S.A. Using fluorescent lipids in live zebrafish larvae: From imaging whole animal physiology to subcellular lipid trafficking. Methods Cell Biol. 2016, 133, 165–178. [Google Scholar] [CrossRef]
- Fang, L.; Liu, C.; Miller, Y.I. Zebrafish models of dyslipidemia: Relevance to atherosclerosis and angiogenesis. Transl. Res. 2014, 163, 99–108. [Google Scholar] [CrossRef]
- Schlegel, A. Zebrafish Models for Dyslipidemia and Atherosclerosis Research. Front. Endocrinol. 2016, 7, 159. [Google Scholar] [CrossRef]
- Ishibashi, S.; Goldstein, J.L.; Brown, M.S.; Herz, J.; Burns, D.K. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Investig. 1994, 93, 1885–1893. [Google Scholar] [CrossRef]
- O’Hare, E.A.; Wang, X.; Montasser, M.E.; Chang, Y.P.; Mitchell, B.D.; Zaghloul, N.A. Disruption of ldlr causes increased LDL-c and vascular lipid accumulation in a zebrafish model of hypercholesterolemia. J. Lipid Res. 2014, 55, 2242–2253. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kim, Y.S.; Kim, J.; Pattison, J.; Kamaid, A.; Miller, Y.I. Modeling hypercholesterolemia and vascular lipid accumulation in LDL receptor mutant zebrafish. J. Lipid Res. 2018, 59, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, Q.Q.; Wu, N.; Shen, Y.G.; Liao, W.T.; Yang, Y.; Dong, E.; Zhang, G.M.; Liu, B.R.; Yue, X.Z.; et al. Chronological in vivo imaging reveals endothelial inflammation prior to neutrophils accumulation and lipid deposition in HCD-fed zebrafish. Atherosclerosis 2019, 290, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Kou, S.; He, K.; Han, Y.; Wang, Y.; Huang, T.; Zhou, X.; Xiao, Y.; Li, X.; Ye, X. Anti-hypercholesterolemic Effect of Berbamine Isolated from Rhizoma Coptidis in Hypercholesterolemic Zebrafish Induced by High-Cholesterol Diet. Iran J. Pharm. Res. 2018, 17, 292–306. [Google Scholar] [PubMed]
- Li, F.; Wu, Z.; Sui, X. Biotransformation of ginsenoside Rb1 with wild Cordyceps sinensis and Ascomycota sp. and its antihyperlipidemic effects on the diet-induced cholesterol of zebrafish. J. Food Biochem. 2020, 44, e13192. [Google Scholar] [CrossRef]
- Hoppstadter, J.; Valbuena Perez, J.V.; Linnenberger, R.; Dahlem, C.; Legroux, T.M.; Hecksteden, A.; Tse, W.K.F.; Flamini, S.; Andreas, A.; Herrmann, J.; et al. The glucocorticoid-induced leucine zipper mediates statin-induced muscle damage. FASEB J. 2020, 34, 4684–4701. [Google Scholar] [CrossRef]
- Liu, C.; Gates, K.P.; Fang, L.; Amar, M.J.; Schneider, D.A.; Geng, H.; Huang, W.; Kim, J.; Pattison, J.; Zhang, J.; et al. Apoc2 loss-of-function zebrafish mutant as a genetic model of hyperlipidemia. Dis. Model. Mech. 2015, 8, 989–998. [Google Scholar] [CrossRef]
- Matthijs, G.; Schollen, E.; Pardon, E.; Veiga-Da-Cunha, M.; Jaeken, J.; Cassiman, J.J.; Van Schaftingen, E. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat. Genet. 1997, 16, 88–92. [Google Scholar] [CrossRef]
- Iyer, S.; Sam, F.S.; DiPrimio, N.; Preston, G.; Verheijen, J.; Murthy, K.; Parton, Z.; Tsang, H.; Lao, J.; Morava, E.; et al. Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef]
- Himmelreich, N.; Kaufmann, L.T.; Steinbeisser, H.; Korner, C.; Thiel, C. Lack of phosphomannomutase 2 affects Xenopus laevis morphogenesis and the non-canonical Wnt5a/Ror2 signalling. J. Inherit Metab. Dis. 2015, 38, 1137–1146. [Google Scholar] [CrossRef]
- Lao, J.P.; DiPrimio, N.; Prangley, M.; Sam, F.S.; Mast, J.D.; Perlstein, E.O. Yeast Models of Phosphomannomutase 2 Deficiency, a Congenital Disorder of Glycosylation. G3 Genes Genomes Genet. 2019, 9, 413–423. [Google Scholar] [CrossRef]
- Cline, A.; Gao, N.; Flanagan-Steet, H.; Sharma, V.; Rosa, S.; Sonon, R.; Azadi, P.; Sadler, K.C.; Freeze, H.H.; Lehrman, M.A.; et al. A zebrafish model of PMM2-CDG reveals altered neurogenesis and a substrate-accumulation mechanism for N-linked glycosylation deficiency. Mol. Biol. Cell 2012, 23, 4175–4187. [Google Scholar] [CrossRef] [PubMed]
- Mukaigasa, K.; Tsujita, T.; Nguyen, V.T.; Li, L.; Yagi, H.; Fuse, Y.; Nakajima-Takagi, Y.; Kato, K.; Yamamoto, M.; Kobayashi, M. Nrf2 activation attenuates genetic endoplasmic reticulum stress induced by a mutation in the phosphomannomutase 2 gene in zebrafish. Proc. Natl. Acad. Sci. USA 2018, 115, 2758–2763. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Mir, A.; Gao, N.; Rosa, S.; Monson, C.; Sharma, V.; Steet, R.; Freeze, H.H.; Lehrman, M.A.; Sadler, K.C. A zebrafish model of congenital disorders of glycosylation with phosphomannose isomerase deficiency reveals an early opportunity for corrective mannose supplementation. Dis. Models Mech. 2013, 6, 95–105. [Google Scholar] [CrossRef] [PubMed]
- DeRossi, C.; Bode, L.; Eklund, E.A.; Zhang, F.; Davis, J.A.; Westphal, V.; Wang, L.; Borowsky, A.D.; Freeze, H.H. Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J. Biol. Chem. 2006, 281, 5916–5927. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, L.R.; Spencer, Z.T.; Nayak, A.; Cselenyi, C.S.; Benchabane, H.; Youngblood, C.Q.; Zouaoui, A.; Ng, V.; Stephens, L.; Hann, T.; et al. Developmental regulation of Wnt signaling by Nagk and the UDP-GlcNAc salvage pathway. Mech. Dev. 2019, 156, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Cuellar, G.; Gauthier, J.; Desilets, V.; Lachance, C.; Lemire-Girard, M.; Rypens, F.; Le Deist, F.; Decaluwe, H.; Duval, M.; Bouron-Dal Soglio, D.; et al. A Novel PGM3 Mutation Is Associated With a Severe Phenotype of Bone Marrow Failure, Severe Combined Immunodeficiency, Skeletal Dysplasia, and Congenital Malformations. J. Bone Min. Res. 2017, 32, 1853–1859. [Google Scholar] [CrossRef]
- Stray-Pedersen, A.; Backe, P.H.; Sorte, H.S.; Morkrid, L.; Chokshi, N.Y.; Erichsen, H.C.; Gambin, T.; Elgstoen, K.B.; Bjoras, M.; Wlodarski, M.W.; et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am. J. Hum. Genet. 2014, 95, 96–107. [Google Scholar] [CrossRef]
- Bammens, R.; Mehta, N.; Race, V.; Foulquier, F.; Jaeken, J.; Tiemeyer, M.; Steet, R.; Matthijs, G.; Flanagan-Steet, H. Abnormal cartilage development and altered N-glycosylation in Tmem165-deficient zebrafish mirrors the phenotypes associated with TMEM165-CDG. Glycobiology 2015, 25, 669–682. [Google Scholar] [CrossRef]
- Zeevaert, R.; de Zegher, F.; Sturiale, L.; Garozzo, D.; Smet, M.; Moens, M.; Matthijs, G.; Jaeken, J. Bone Dysplasia as a Key Feature in Three Patients with a Novel Congenital Disorder of Glycosylation (CDG) Type II Due to a Deep Intronic Splice Mutation in TMEM165. JIMD Rep. 2013, 8, 145–152. [Google Scholar] [CrossRef]
- Sadler, K.C.; Amsterdam, A.; Soroka, C.; Boyer, J.; Hopkins, N. A genetic screen in zebrafish identifies the mutants vps18, nf2 and foie gras as models of liver disease. Development 2005, 132, 3561–3572. [Google Scholar] [CrossRef] [PubMed]
- Cinaroglu, A.; Gao, C.; Imrie, D.; Sadler, K.C. Activating transcription factor 6 plays protective and pathological roles in steatosis due to endoplasmic reticulum stress in zebrafish. Hepatology 2011, 54, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.C.; Zhu, W.; Mitsuhashi, S.; Noguchi, S.; Sacher, M.; Ogawa, M.; Shih, H.H.; Jong, Y.J.; Nishino, I. Congenital muscular dystrophy with fatty liver and infantile-onset cataract caused by TRAPPC11 mutations: Broadening of the phenotype. Skelet Muscle 2015, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- DeRossi, C.; Vacaru, A.; Rafiq, R.; Cinaroglu, A.; Imrie, D.; Nayar, S.; Baryshnikova, A.; Milev, M.P.; Stanga, D.; Kadakia, D.; et al. trappc11 is required for protein glycosylation in zebrafish and humans. Mol. Biol. Cell 2016, 27, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Matalonga, L.; Bravo, M.; Serra-Peinado, C.; Garcia-Pelegri, E.; Ugarteburu, O.; Vidal, S.; Llambrich, M.; Quintana, E.; Fuster-Jorge, P.; Gonzalez-Bravo, M.N.; et al. Mutations in TRAPPC11 are associated with a congenital disorder of glycosylation. Hum. Mutat. 2017, 38, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Peanne, R. What is new in CDG? J. Inherit Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef]
- Taylor, M.R.; Hurley, J.B.; Van Epps, H.A.; Brockerhoff, S.E. A zebrafish model for pyruvate dehydrogenase deficiency: Rescue of neurological dysfunction and embryonic lethality using a ketogenic diet. Proc. Natl. Acad. Sci. USA 2004, 101, 4584–4589. [Google Scholar] [CrossRef]
- Brockerhoff, S.E.; Hurley, J.B.; Janssen-Bienhold, U.; Neuhauss, S.C.; Driever, W.; Dowling, J.E. A behavioral screen for isolating zebrafish mutants with visual system defects. Proc. Natl. Acad. Sci. USA 1995, 92, 10545–10549. [Google Scholar] [CrossRef] [PubMed]
- Wijburg, F.A.; Barth, P.G.; Bindoff, L.A.; Birch-Machin, M.A.; van der Blij, J.F.; Ruitenbeek, W.; Turnbull, D.M.; Schutgens, R.B. Leigh syndrome associated with a deficiency of the pyruvate dehydrogenase complex: Results of treatment with a ketogenic diet. Neuropediatrics 1992, 23, 147–152. [Google Scholar] [CrossRef]
- Maurer, C.M.; Schonthaler, H.B.; Mueller, K.P.; Neuhauss, S.C. Distinct retinal deficits in a zebrafish pyruvate dehydrogenase-deficient mutant. J. Neurosci. 2010, 30, 11962–11972. [Google Scholar] [CrossRef]
- Ferriero, R.; Manco, G.; Lamantea, E.; Nusco, E.; Ferrante, M.I.; Sordino, P.; Stacpoole, P.W.; Lee, B.; Zeviani, M.; Brunetti-Pierri, N. Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci. Transl. Med. 2013, 5, 175ra131. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Luciani, A.; Mateos, J.M.; Barmettler, G.; Giles, R.H.; Neuhauss, S.C.F.; Devuyst, O. Transgenic zebrafish modeling low-molecular-weight proteinuria and lysosomal storage diseases. Kidney Int. 2020, 97, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Peterson, R.T. Modeling Lysosomal Storage Diseases in the Zebrafish. Front. Mol. Biosci. 2020, 7, 82. [Google Scholar] [CrossRef]
- Grabowski, G.A. Gaucher disease and other storage disorders. Hematol. Am. Soc. Hematol. Educ. Program. 2012, 2012, 13–18. [Google Scholar] [CrossRef]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. 2014, 12, 72–81. [Google Scholar]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood. Cells Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef]
- Toni, M.; Cioni, C. Fish Synucleins: An Update. Mar. Drugs 2015, 13, 6665–6686. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.; Keatinge, M.; Gegg, M.; Bai, Q.; Sandulescu, M.C.; Vardi, A.; Futerman, A.H.; Schapira, A.H.V.; Burton, E.A.; Bandmann, O. Ablation of the pro-inflammatory master regulator miR-155 does not mitigate neuroinflammation or neurodegeneration in a vertebrate model of Gaucher’s disease. Neurobiol. Dis. 2019, 127, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Zancan, I.; Bellesso, S.; Costa, R.; Salvalaio, M.; Stroppiano, M.; Hammond, C.; Argenton, F.; Filocamo, M.; Moro, E. Glucocerebrosidase deficiency in zebrafish affects primary bone ossification through increased oxidative stress and reduced Wnt/beta-catenin signaling. Hum. Mol. Genet. 2015, 24, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Artola, M.; Kuo, C.L.; Lelieveld, L.T.; Rowland, R.J.; van der Marel, G.A.; Codee, J.D.C.; Boot, R.G.; Davies, G.J.; Aerts, J.; Overkleeft, H.S. Functionalized Cyclophellitols Are Selective Glucocerebrosidase Inhibitors and Induce a Bona Fide Neuropathic Gaucher Model in Zebrafish. J. Am. Chem. Soc. 2019, 141, 4214–4218. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, L.T.; Mirzaian, M.; Kuo, C.L.; Artola, M.; Ferraz, M.J.; Peter, R.E.A.; Akiyama, H.; Greimel, P.; van den Berg, R.; Overkleeft, H.S.; et al. Role of beta-glucosidase 2 in aberrant glycosphingolipid metabolism: Model of glucocerebrosidase deficiency in zebrafish. J. Lipid Res. 2019, 60, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T.; Millat, G. Structure and function of the NPC2 protein. Biochim. Biophys. Acta 2004, 1685, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.A.; Portmann, B.; Mowat, A.P.; Sherlock, S.; Lake, B.D. Niemann-Pick disease type C: Diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 1993, 123, 242–247. [Google Scholar] [CrossRef]
- Patterson, M.C.; Mengel, E.; Wijburg, F.A.; Muller, A.; Schwierin, B.; Drevon, H.; Vanier, M.T.; Pineda, M. Disease and patient characteristics in NP-C patients: Findings from an international disease registry. Orphanet J. Rare Dis 2013, 8, 12. [Google Scholar] [CrossRef]
- Mengel, E.; Pineda, M.; Hendriksz, C.J.; Walterfang, M.; Torres, J.V.; Kolb, S.A. Differences in Niemann-Pick disease Type C symptomatology observed in patients of different ages. Mol. Genet. Metab. 2017, 120, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.C.; Loeb, H.E.; Pei, W.; Tsai-Morris, C.H.; Xu, L.; Cluzeau, C.V.; Wassif, C.A.; Feldman, B.; Burgess, S.M.; Pavan, W.J.; et al. Modeling Niemann-Pick disease type C1 in zebrafish: A robust platform for in vivo screening of candidate therapeutic compounds. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cai, X.; Wang, G.; Ouyang, G.; Cao, H. Model construction of Niemann-Pick type C disease in zebrafish. Biol. Chem. 2018, 399, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Schwend, T.; Loucks, E.J.; Snyder, D.; Ahlgren, S.C. Requirement of Npc1 and availability of cholesterol for early embryonic cell movements in zebrafish. J. Lipid Res. 2011, 52, 1328–1344. [Google Scholar] [CrossRef] [PubMed]
- Louwette, S.; Regal, L.; Wittevrongel, C.; Thys, C.; Vandeweeghde, G.; Decuyper, E.; Leemans, P.; De Vos, R.; Van Geet, C.; Jaeken, J.; et al. NPC1 defect results in abnormal platelet formation and function: Studies in Niemann-Pick disease type C1 patients and zebrafish. Hum. Mol. Genet. 2013, 22, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U.; Suzuki, K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta 2004, 1685, 48–62. [Google Scholar] [CrossRef]
- Liu, B.; Turley, S.D.; Burns, D.K.; Miller, A.M.; Repa, J.J.; Dietschy, J.M. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. USA 2009, 106, 2377–2382. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1-2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef]
- Ehlert, K.; Frosch, M.; Fehse, N.; Zander, A.; Roth, J.; Vormoor, J. Farber disease: Clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 2007, 5, 15. [Google Scholar] [CrossRef]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef]
- Zhou, J.; Tawk, M.; Tiziano, F.D.; Veillet, J.; Bayes, M.; Nolent, F.; Garcia, V.; Servidei, S.; Bertini, E.; Castro-Giner, F.; et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am. J. Hum. Genet. 2012, 91, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Trauger, S.A.; Vidoudez, C.; Doane, K.P.; Pluimer, B.R.; Peterson, R.T. Parallel Reaction Monitoring reveals structure-specific ceramide alterations in the zebrafish. Sci. Rep. 2019, 9, 19939. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F.; Muenzer, J. The mucopolysaccharidosis. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Giugliani, R.; Federhen, A.; Rojas, M.V.; Vieira, T.; Artigalas, O.; Pinto, L.L.; Azevedo, A.C.; Acosta, A.; Bonfim, C.; Lourenco, C.M.; et al. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genet. Mol. Biol. 2010, 33, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: From pathophysiology to therapy. Ital. J. Pediatr. 2018, 44, 124. [Google Scholar] [CrossRef] [PubMed]
- Moro, E.; Tomanin, R.; Friso, A.; Modena, N.; Tiso, N.; Scarpa, M.; Argenton, F. A novel functional role of iduronate-2-sulfatase in zebrafish early development. Matrix Biol. 2010, 29, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Urbani, A.; Salvalaio, M.; Bellesso, S.; Cieri, D.; Zancan, I.; Filocamo, M.; Bonaldo, P.; Szabo, I.; Tomanin, R.; et al. Perturbations in cell signaling elicit early cardiac defects in mucopolysaccharidosis type II. Hum. Mol. Genet. 2017, 26, 1643–1655. [Google Scholar] [CrossRef]
- Bellesso, S.; Salvalaio, M.; Lualdi, S.; Tognon, E.; Costa, R.; Braghetta, P.; Giraudo, C.; Stramare, R.; Rigon, L.; Filocamo, M.; et al. FGF signaling deregulation is associated with early developmental skeletal defects in animal models for mucopolysaccharidosis type II (MPSII). Hum. Mol. Genet. 2018, 27, 2407. [Google Scholar] [CrossRef]
- Konno, M.; Asai, A.; Kitagawa, T.; Yabumoto, M.; Ofusa, K.; Arai, T.; Hirotsu, T.; Doki, Y.; Eguchi, H.; Ishii, H. State-of-the-Art Technology of Model Organisms for Current Human Medicine. Diagnostics 2020, 10, 392. [Google Scholar] [CrossRef]
- Patterson, V.L.; Burdine, R.D. Swimming toward solutions: Using fish and frogs as models for understanding RASopathies. Birth Defects Res. 2020, 112, 749–765. [Google Scholar] [CrossRef]
- Xiao, J.; Glasgow, E.; Agarwal, S. Zebrafish Xenografts for Drug Discovery and Personalized Medicine. Trends Cancer 2020, 6, 569–579. [Google Scholar] [CrossRef]
- Shrestha, R.; Lieberth, J.; Tillman, S.; Natalizio, J.; Bloomekatz, J. Using Zebrafish to Analyze the Genetic and Environmental Etiologies of Congenital Heart Defects. Adv. Exp. Med. Biol. 2020, 1236, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.A.; Armstrong, G.A.; Lissouba, A.; Kabashi, E.; Parker, J.A.; Drapeau, P. Fishing for causes and cures of motor neuron disorders. Dis. Model. Mech. 2014, 7, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Unal, I.; Emekli-Alturfan, E. Fishing for Parkinson’s Disease: A review of the literature. J. Clin. Neurosci. 2019, 62, 1–6. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Metabolism | Disorders | Zebrafish Genes | Approach | Reference |
|---|---|---|---|---|
| Amino acid and peptide metabolism | Maple syrup urine disorder | dbt | CRISPR/Cas9; Metabolite exposure | [64,68] |
| Methylmalonic acidemia | mmut, mmachc | CRISPR/Cas9 | [73,74,75,76] | |
| Glutaric aciduria type II | etfdh | CRISPR/Cas9 | [77,78] | |
| Barth Syndrome | taz | Morpholino | [79] | |
| Prolidase deficiency | pepd | Morpholino | [80] | |
| Neurotransmission | AADC deficiency | ddc | Inhibitor, Morpholino | [81] |
| DHPR deficiency | qdpra/qdprb1/qdprb2 | Morpholino | [82] | |
| Hyperekplexia | glra1, glrbb | ENU | [83,84,85,86] | |
| Metabolism of Vitamins and Co-factors | DHFR deficiency | dhfr | Inhibitor, Morpholino | [87,88,89,90,91,92] |
| Urea Cycle Disorders | Hyperammonemia | / | Metabolite exposure | [97,98,99,100] |
| Carbohydrate Metabolism | GSD Type II/Pompe disease | gaa | Morpholino | [103,104] |
| GSD type XI/Fanconi-Bickel disease | slc2a2 | Morpholino | [106] | |
| G6PD deficiency | g6pd | CRISPR/Cas9; Morpholino | [108,109,110] | |
| Galactosemia | galt | TALEN | [112,118] | |
| GLUT1 deficiency | glut1 | Morpholino | [119] | |
| Lipoprotein Metabolism | Hypercholesterolemia | ldlr | CRISPR/Cas9, Morpholino, Diet | [124,125,126,127,128] |
| Hypertriglyceridemia | apoc2 | CRISPR/Cas9 | [130] | |
| Congenital disorders of glycosylation (CDG) | PMM2-CDG | pmm2 | ENU, Morpholino | [135,136] |
| MPI-CDG | mpi | Morpholino | [137] | |
| TMEM165-CDG | tmem165 | Morpholino | [142] | |
| TRAPPC11-CDG | trappc11c | gene-trap cassette | [144,145,146,147] | |
| Energy and Pyruvate Metabolism | Pyruvate dehydrogenase complex deficiency | dlat, pdh1b | ENU | [150,151,153,154] |
| Lysosomal Storage Disorders | Gaucher’s disease | gba1 | TALEN; CRISPR/Cas9; Morpholino | [163,165,166,167,168] |
| Niemann–Pick disease C | npc1 | CRISPR/Cas9; Morpholino | [175,176,177,178] | |
| Farber disease | asah1a, asah1b | CRISPR/Cas9; Morpholino | [184,185] | |
| MPS II/Hunter Syndrome | ids | CRISPR/Cas9; Morpholino | [189,190,191] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breuer, M.; Patten, S.A. A Great Catch for Investigating Inborn Errors of Metabolism—Insights Obtained from Zebrafish. Biomolecules 2020, 10, 1352. https://doi.org/10.3390/biom10091352
Breuer M, Patten SA. A Great Catch for Investigating Inborn Errors of Metabolism—Insights Obtained from Zebrafish. Biomolecules. 2020; 10(9):1352. https://doi.org/10.3390/biom10091352
Chicago/Turabian StyleBreuer, Maximilian, and Shunmoogum A. Patten. 2020. "A Great Catch for Investigating Inborn Errors of Metabolism—Insights Obtained from Zebrafish" Biomolecules 10, no. 9: 1352. https://doi.org/10.3390/biom10091352
APA StyleBreuer, M., & Patten, S. A. (2020). A Great Catch for Investigating Inborn Errors of Metabolism—Insights Obtained from Zebrafish. Biomolecules, 10(9), 1352. https://doi.org/10.3390/biom10091352