Arsenic Methyltransferase and Methylation of Inorganic Arsenic


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
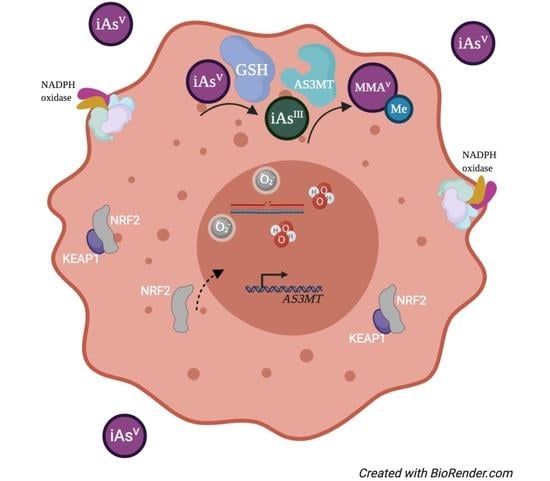
2. Arsenic Metabolism, Excretion, and Toxicity
3. Discovery of the AS3MT Gene and Protein
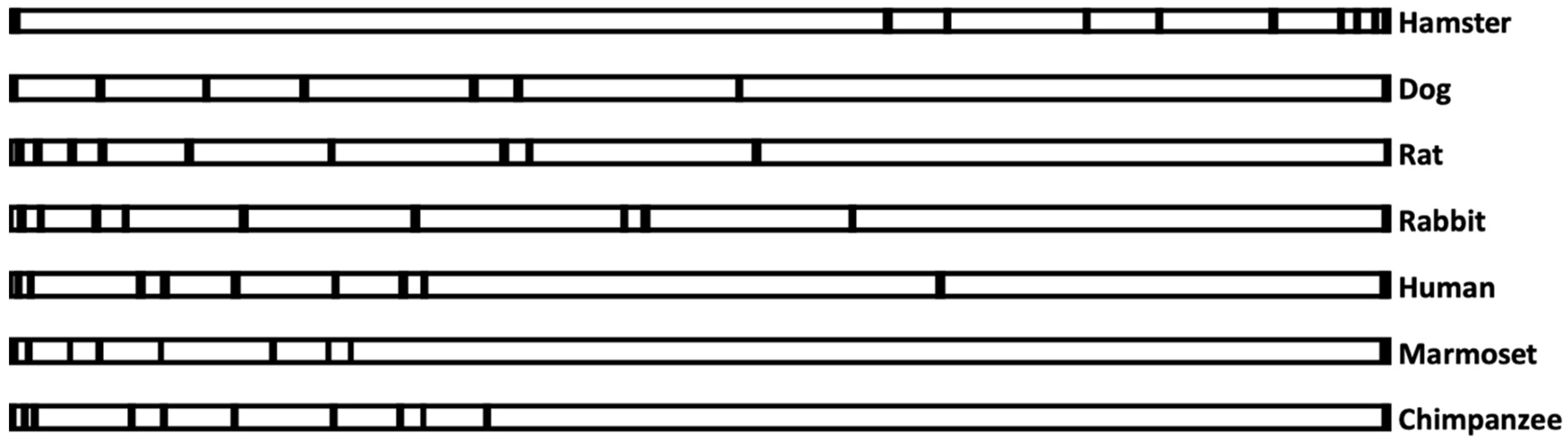
3.1. Analysis of AS3MT Genomic Sequences
3.2. The Relationship between AS3MT Promoter Sequences and the Ability to Methylate Arsenic
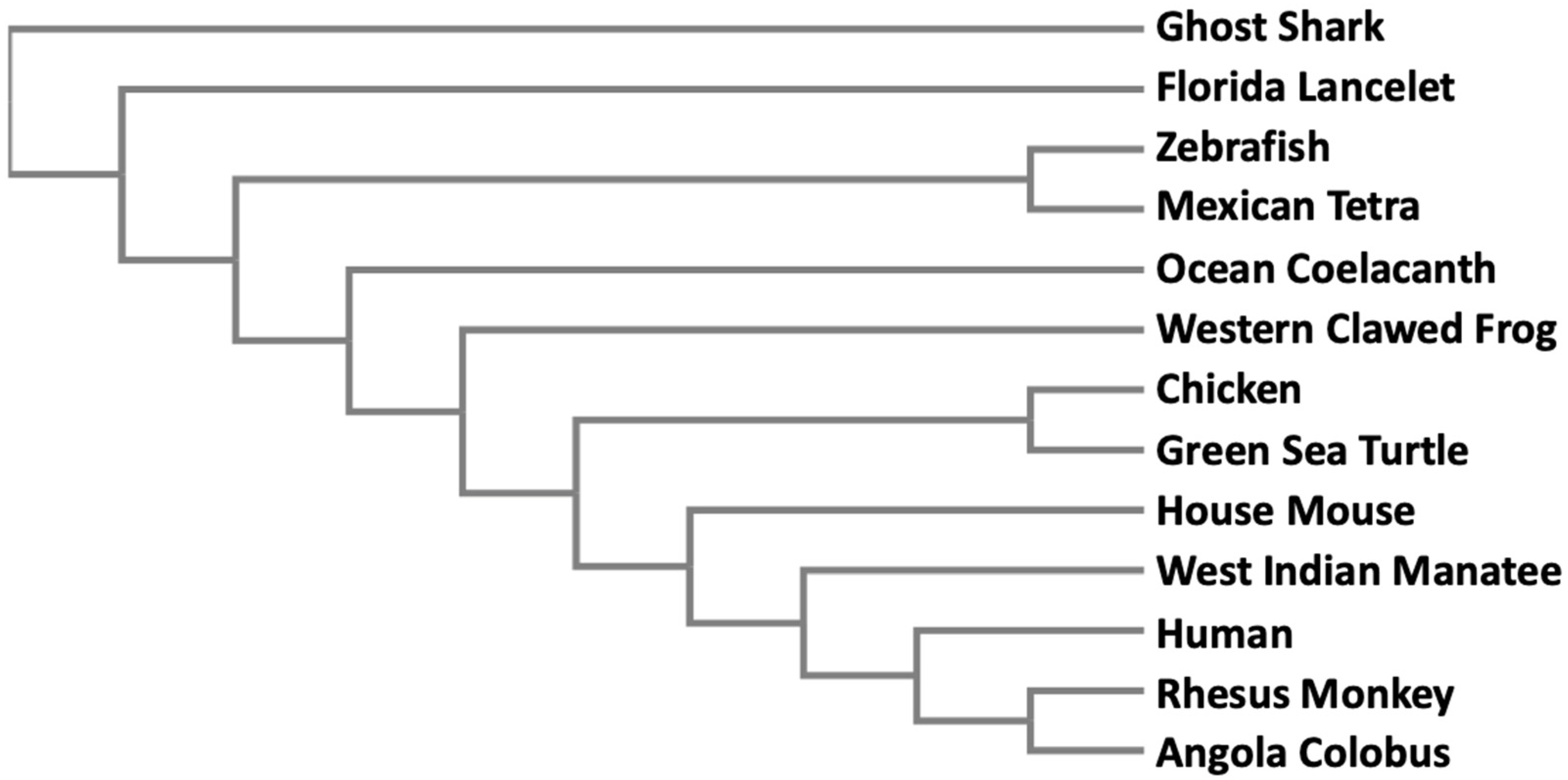
3.3. Evolution of the AS3MT Gene
4. DNA Methylation and Arsenic Exposure
5. Single Nucleotide Polymorphisms and AS3MT Activity
6. Conclusions
Funding
Conflicts of Interest
References
- Gurnari, C.; De Bellis, E.; Divona, M.; Ottone, T.; Lavorgna, S.; Voso, M.T. When Poisons Cure: The Case of Arsenic in Acute Promyelocytic Leukemia. Chemotherapy 2019, 64, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Sandell, E.B. Geochemistry of arsenic. Geochimica et Cosmochimica Acta 1955, 7, 1–33. [Google Scholar] [CrossRef]
- Chou, C.H.; Harper, C. Toxicological Profile for Arsenic; Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 2007.
- WHO. Arsenic—World Health Organizaiton. Fact Sheets. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/arsenic (accessed on 1 June 2020).
- Ferguson, J.F.; Gavis, J. A review of the arsenic cycle in natural waters. Water Res. 1972, 6, 1259–1274. [Google Scholar] [CrossRef]
- Chung, J.-Y.; Yu, S.-D.; Hong, Y.-S. Environmental source of arsenic exposure. J. Prev. Med. Public Health 2014, 47, 253–257. [Google Scholar] [CrossRef]
- Ratnaike, R.N. Acute and chronic arsenic toxicity. Postgrad. Med. J. 2003, 79, 391. [Google Scholar] [CrossRef]
- Tseng, C.H.; Huang, Y.K.; Huang, Y.L.; Chung, C.J.; Yang, M.H.; Chen, C.J.; Hsueh, Y.M. Arsenic exposure, urinary arsenic speciation, and peripheral vascular disease in blackfoot disease-hyperendemic villages in Taiwan. Toxicol. Appl. Pharm. 2005, 206, 299–308. [Google Scholar] [CrossRef]
- Tseng, W.P. Blackfoot disease in Taiwan: A 30-year follow-up study. Angiology 1989, 40, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Straif, K.; Benbrahim-Tallaa, L.; Baan, R.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Cogliano, V. A review of human carcinogens—Part C: Metals, arsenic, dusts, and fibres. Lancet Oncol. 2009, 10, 453–454. [Google Scholar] [CrossRef]
- Zhu, Y.; Costa, M. Metals and Molecular Carcinogenesis. Carcinogenesis 2020. [Google Scholar] [CrossRef]
- Klein, C.B.; Leszczynska, J.; Hickey, C.; Rossman, T.G. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol. Appl. Pharm. 2007, 222, 289–297. [Google Scholar] [CrossRef]
- Hegedus, C.M.; Skibola, C.F.; Warner, M.; Skibola, D.R.; Alexander, D.; Lim, S.; Moore, L.E. Decreased urinary beta-defensin-1 expression as a biomarker of response to arsenic. Toxicol. Sci. 2008, 106, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, P.; Brammer, H.; Richards, K. Arsenic Pollution: A global Synthesis; John Wiley & Sons: Chichester, West Sussex, UK, 2009. [Google Scholar]
- Smith, A.H.; Lingas, E.O.; Rahman, M. Contamination of drinking-water by arsenic in Bangladesh: A public health emergency. Bull. World Health Organ. 2000, 78, 1093–1103. [Google Scholar] [PubMed]
- Bagchi, S. Arsenic threat reaching global dimensions. CMAJ 2007, 177, 1344–1345. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef]
- Uddin, R.; Huda, N.H. Arsenic poisoning in bangladesh. Oman Med. J. 2011, 26, 207. [Google Scholar] [CrossRef]
- Yu, W.H.; Harvey, C.M.; Harvey, C.F. Arsenic in groundwater in Bangladesh: A geostatistical and epidemiological framework for evaluating health effects and potential remedies. Water Resour. Res. 2003, 39. [Google Scholar] [CrossRef]
- Vahter, M.; Concha, G.; Nermell, B.; Nilsson, R.; Dulout, F.; Natarajan, A.T. A unique metabolism of inorganic arsenic in native Andean women. Eur. J. Pharm. 1995, 293, 455–462. [Google Scholar] [CrossRef]
- Moe, B.; Peng, H.; Lu, X.; Chen, B.; Chen, L.W.L.; Gabos, S.; Le, X.C. Comparative cytotoxicity of fourteen trivalent and pentavalent arsenic species determined using real-time cell sensing. J. Environ. Sci. (China) 2016, 49, 113–124. [Google Scholar] [CrossRef]
- Thomas, D.J.; Styblo, M.; Lin, S. The cellular metabolism and systemic toxicity of arsenic. Toxicol. Appl. Pharm. 2001, 176, 127–144. [Google Scholar] [CrossRef]
- Challenger, F. Biological methylation. Sci. Prog. 1947, 35, 396–416. [Google Scholar]
- Li, J.; Duan, X.; Dong, D.; Zhang, Y.; Zhao, L.; Li, W.; Li, B. Tissue-specific distributions of inorganic arsenic and its methylated metabolites, especially in cerebral cortex, cerebellum and hippocampus of mice after a single oral administration of arsenite. J. Trace Elem. Med. Biol. 2017, 43, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Vahter, M. Methylation of inorganic arsenic in different mammalian species and population groups. Sci. Prog. 1999, 82, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Styblo, M.; Del Razo, L.M.; Vega, L.; Germolec, D.R.; LeCluyse, E.L.; Hamilton, G.A.; Thomas, D.J. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 2000, 74, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Buchet, J.P.; Lauwerys, R.; Roels, H. Comparison of the urinary excretion of arsenic metabolites after a single oral dose of sodium arsenite, monomethylarsonate, or dimethylarsinate in man. Int. Arch. Occup. Environ. Health 1981, 48, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Buchet, J.P.; Lauwerys, R.; Roels, H. Urinary excretion of inorganic arsenic and its metabolites after repeated ingestion of sodium metaarsenite by volunteers. Int. Arch. Occup. Environ. Health 1981, 48, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Li, X.-F.; Cullen, W.R.; Weinfeld, M.; Le, X.C. Arsenic binding to proteins. Chem. Rev. 2013, 113, 7769–7792. [Google Scholar] [CrossRef]
- Yokohira, M.; Arnold, L.L.; Pennington, K.L.; Suzuki, S.; Kakiuchi-Kiyota, S.; Herbin-Davis, K.; Cohen, S.M. Severe systemic toxicity and urinary bladder cytotoxicity and regenerative hyperplasia induced by arsenite in arsenic (+3 oxidation state) methyltransferase knockout mice. A preliminary report. Toxicol. Appl. Pharm. 2010, 246, 1–7. [Google Scholar] [CrossRef]
- Naranmandura, H.; Chen, X.; Tanaka, M.; Wang, W.W.; Rehman, K.; Xu, S.; Suzuki, N. Release of apoptotic cytochrome C from mitochondria by dimethylarsinous acid occurs through interaction with voltage-dependent anion channel in vitro. Toxicol. Sci. 2012, 128, 137–146. [Google Scholar] [CrossRef]
- Rehman, K.; Fu, Y.J.; Zhang, Y.F.; Wang, Q.Q.; Wu, B.; Wu, Y.; Naranmandura, H. Trivalent methylated arsenic metabolites induce apoptosis in human myeloid leukemic HL-60 cells through generation of reactive oxygen species. Metallomics 2014, 6, 1502–1512. [Google Scholar] [CrossRef]
- Mass, M.J.; Tennant, A.; Roop, B.C.; Cullen, W.R.; Styblo, M.; Thomas, D.J.; Kligerman, A.D. Methylated trivalent arsenic species are genotoxic. Chem. Res. Toxicol. 2001, 14, 355–361. [Google Scholar] [CrossRef]
- Tokar, E.J.; Kojima, C.; Waalkes, M.P. Methylarsonous acid causes oxidative DNA damage in cells independent of the ability to biomethylate inorganic arsenic. Arch. Toxicol. 2014, 88, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Konkel, L. Organoarsenic Drugs over Time: The Pharmacokinetics of Roxarsone in Chicken Meat. Environ. Health Perspect. 2016, 124, A150. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Shi, Q.; Nix, F.B.; Styblo, M.; Beck, M.A.; Herbin-Davis, K.M.; Thomas, D.J. A novel S-adenosyl-L-methionine:arsenic(III) methyltransferase from rat liver cytosol. J. Biol. Chem. 2002, 277, 10795–10803. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Waters, S.B.; Drobna, Z.; Devesa, V.; Styblo, M.; Thomas, D.J. Arsenic (+3 oxidation state) methyltransferase and the inorganic arsenic methylation phenotype. Toxicol. Appl. Pharm. 2005, 204, 164–169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zimin, A.V.; Cornish, A.S.; Maudhoo, M.D.; Gibbs, R.M.; Zhang, X.; Pandey, S.; Norgren, R.B., Jr. A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol. Direct 2014, 9, 20. [Google Scholar] [CrossRef]
- Thomas, D.J.; Waters, S.B.; Styblo, M. Elucidating the pathway for arsenic methylation. Toxicol. Appl. Pharm. 2004, 198, 319–326. [Google Scholar] [CrossRef]
- Zakharyan, R.; Wu, Y.; Bogdan, G.M.; Aposhian, H.V. Enzymatic methylation of arsenic compounds: Assay, partial purification, and properties of arsenite methyltransferase and monomethylarsonic acid methyltransferase of rabbit liver. Chem. Res. Toxicol. 1995, 8, 1029–1038. [Google Scholar] [CrossRef]
- Aposhian, H.V. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Annu. Rev. Pharm. Toxicol. 1997, 37, 397–419. [Google Scholar] [CrossRef]
- Hughes, M.F.; Beck, B.D.; Chen, Y.; Lewis, A.S.; Thomas, D.J. Arsenic exposure and toxicology: A historical perspective. Toxicol. Sci. 2011, 123, 305–332. [Google Scholar] [CrossRef]
- Zakharyan, R.A.; Ayala-Fierro, F.; Cullen, W.R.; Carter, D.M.; Aposhian, H.V. Enzymatic methylation of arsenic compounds. VII. Monomethylarsonous acid (MMAIII) is the substrate for MMA methyltransferase of rabbit liver and human hepatocytes. Toxicol. Appl. Pharm. 1999, 158, 9–15. [Google Scholar] [CrossRef]
- Rosen, B.P.; Packianathan, C.; Li, J. Mechanism of as(III) S-adenosylmethionine methyltransferases and the consequences of human polymorphisms in hAS3MT. In Environmental Arsenic in a Changing World, 1st ed.; Zhu, Y., Guo, H., Bhattacharya, P., Ahmad, A., Bundschuh, J., Naidu, R., Eds.; CRC Press: London, UK, 2019; pp. 9–11. [Google Scholar]
- Qin, J.; Lehr, C.R.; Yuan, C.; Le, X.C.; McDermott, T.R.; Rosen, B.P. Biotransformation of arsenic by a Yellowstone thermoacidophilic eukaryotic alga. Proc. Natl. Acad. Sci. USA 2009, 106, 5213–5217. [Google Scholar] [CrossRef] [PubMed]
- Marapakala, K.; Qin, J.; Rosen, B.P. Identification of catalytic residues in the as(III) S-adenosylmethionine methyltransferase. Biochemistry 2012, 51, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Ajees, A.A.; Marapakala, K.; Packianathan, C.; Sankaran, B.; Rosen, B.P. Structure of an As(III) S-adenosylmethionine methyltransferase: Insights into the mechanism of arsenic biotransformation. Biochemistry 2012, 51, 5476–5485. [Google Scholar] [CrossRef] [PubMed]
- Dheeman, D.S.; Packianathan, C.; Pillai, J.K.; Rosen, B.P. Pathway of human AS3MT arsenic methylation. Chem. Res. Toxicol. 2014, 27, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.; Engström, K.; Hallström, B.M.; Wahlberg, K.; Søndergaard, D.A.; Säll, T.; Broberg, K. AS3MT-mediated tolerance to arsenic evolved by multiple independent horizontal gene transfers from bacteria to eukaryotes. PLoS ONE 2017, 12, e0175422. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, R.J.; Drobna, Z.; Walton, F.S.; Xun, P.; Thomas, D.J.; Styblo, M. Methylation of arsenic by recombinant human wild-type arsenic (+3 oxidation state) methyltransferase and its methionine 287 threonine (M287T) polymorph: Role of glutathione. Toxicol. Appl. Pharm. 2012, 264, 121–130. [Google Scholar] [CrossRef]
- Hernandez, A.; Xamena, N.; Sekaran, C.; Tokunaga, H.; Sampayo-Reyes, A.; Quinteros, D.; Marcos, R. High arsenic metabolic efficiency in AS3MT287Thr allele carriers. Pharm. Genom. 2008, 18, 349–355. [Google Scholar] [CrossRef]
- Li, J.; Packianathan, C.; Rossman, T.G.; Rosen, B.P. Nonsynonymous Polymorphisms in the Human AS3MT Arsenic Methylation Gene: Implications for Arsenic Toxicity. Chem. Res. Toxicol. 2017, 30, 1481–1491. [Google Scholar] [CrossRef]
- Mishra, D.; Flora, S.J. Differential oxidative stress and DNA damage in rat brain regions and blood following chronic arsenic exposure. Toxicol. Ind. Health 2008, 24, 247–256. [Google Scholar] [CrossRef]
- Mathers, J.; Fraser, J.A.; McMahon, M.; Saunders, R.D.; Hayes, J.D.; McLellan, L.I. Antioxidant and cytoprotective responses to redox stress. Biochem. Soc. Symp. 2004, 157–176. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Ki, S.H. Nrf2: A Key Regulator of Redox Signaling in Liver Diseases. In Liver Pathophysiology; Muriel, P., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 355–374. [Google Scholar]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Tian, M.; Spirohn, K.; Boutros, M.; Bohmann, D. Keap1-Independent Regulation of Nrf2 Activity by Protein Acetylation and a BET Bromodomain Protein. PLoS Genet. 2016, 12, e1006072. [Google Scholar] [CrossRef]
- He, X.; Chen, M.G.; Lin, G.X.; Ma, Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 x Keap1 x Cul3 complex and recruiting Nrf2 x Maf to the antioxidant response element enhancer. J. Biol. Chem. 2006, 281, 23620–23631. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Chen, W.; Li, Y.; Villeneuve, N.F.; Zhang, D.D. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: Enhanced Keap1-Cul3 interaction. Toxicol. Appl. Pharm. 2008, 230, 383–389. [Google Scholar] [CrossRef]
- Li, W.; Yu, S.W.; Kong, A.N. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J. Biol. Chem. 2006, 281, 27251–27263. [Google Scholar] [CrossRef]
- Stamatelos, S.K.; Androulakis, I.P.; Kong, A.-N.T.; Georgopoulos, P.G. A semi-mechanistic integrated toxicokinetic-toxicodynamic (TK/TD) model for arsenic(III) in hepatocytes. J. Theor. Biol. 2013, 317, 244–256. [Google Scholar] [CrossRef]
- McNeill, E.; Crabtree, M.J.; Sahgal, N.; Patel, J.; Chuaiphichai, S.; Iqbal, A.J.; Channon, K.M. Regulation of iNOS function and cellular redox state by macrophage Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2 activation. Free Radic. Biol. Med. 2015, 79, 206–216. [Google Scholar] [CrossRef]
- Clustal Omega. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 1 June 2020).
- Simple Phylogeny. Available online: https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny/ (accessed on 1 June 2020).
- Thomas, D.J.; Li, J.; Waters, S.B.; Xing, W.; Adair, B.M.; Drobna, Z.; Styblo, M. Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp. Biol. Med. (Maywood) 2007, 232, 3–13. [Google Scholar]
- Miyashita, S.; Fujiwara, S.; Tsuzuki, M.; Kaise, T. Rapid biotransformation of arsenate into oxo-arsenosugars by a freshwater unicellular green alga, Chlamydomonas reinhardtii. Biosci. Biotechnol. Biochem. 2011, 75, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; McHale, C.M.; Skibola, C.F.; Smith, A.H.; Smith, M.T.; Zhang, L. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect. 2011, 119, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Hung, W.C.; Chen, W.T.; Jiang, W.H.; Yu, H.S.; Chai, C.Y. Effects of MEK and DNMT inhibitors on arsenic-treated human uroepithelial cells in relation to Cyclin-D1 and p16. Toxicol. Lett. 2011, 200, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef]
- Hossain, M.B.; Vahter, M.; Concha, G.; Broberg, K. Environmental arsenic exposure and DNA methylation of the tumor suppressor gene p16 and the DNA repair gene MLH1: Effect of arsenic metabolism and genotype. Metallomics 2012, 4, 1167–1175. [Google Scholar] [CrossRef]
- Engstrom, K.; Vahter, M.; Mlakar, S.J.; Concha, G.; Nermell, B.; Raqib, R.; Broberg, K. Polymorphisms in arsenic(+III oxidation state) methyltransferase (AS3MT) predict gene expression of AS3MT as well as arsenic metabolism. Environ. Health Perspect. 2011, 119, 182–188. [Google Scholar] [CrossRef]
- Schlawicke Engstrom, K.; Broberg, K.; Concha, G.; Nermell, B.; Warholm, M.; Vahter, M. Genetic polymorphisms influencing arsenic metabolism: Evidence from Argentina. Environ. Health Perspect. 2007, 115, 599–605. [Google Scholar] [CrossRef]
- Engstrom, K.S.; Hossain, M.B.; Lauss, M.; Ahmed, S.; Raqib, R.; Vahter, M.; Broberg, K. Efficient arsenic metabolism—The AS3MT haplotype is associated with DNA methylation and expression of multiple genes around AS3MT. PLoS ONE 2013, 8, e53732. [Google Scholar] [CrossRef]
- Wood, T.C.; Salavagionne, O.E.; Mukherjee, B.; Wang, L.; Klumpp, A.F.; Thomae, B.A.; Weinshilboum, R.M. Human arsenic methyltransferase (AS3MT) pharmacogenetics: Gene resequencing and functional genomics studies. J. Biol. Chem. 2006, 281, 7364–7373. [Google Scholar] [CrossRef]
- Apata, M.; Pfeifer, S.P. Recent population genomic insights into the genetic basis of arsenic tolerance in humans: The difficulties of identifying positively selected loci in strongly bottlenecked populations. Heredity (Edinb.) 2020, 124, 253–262. [Google Scholar] [CrossRef]
- Apata, M.; Arriaza, B.; Llop, E.; Moraga, M. Human adaptation to arsenic in Andean populations of the Atacama Desert. Am. J. Phys. Anthr. 2017, 163, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Arriaza, B.; Amarasiriwardena, D.; Standen, V.; Yanez, J.; Van Hoesen, J.; Figueroa, L. Living in poisoning environments: Invisible risks and human adaptation. Evol. Anthr. 2018, 27, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Ariazza, B.T. Arseniasis as an environmental hypothetical explanation for the origin of the oldest artificial mummification practice in the world. Chungara Revista de Antropoĺogia Chilena 2005, 37, 255–260. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, N.K.; Murphy, A.; Costa, M. Arsenic Methyltransferase and Methylation of Inorganic Arsenic. Biomolecules 2020, 10, 1351. https://doi.org/10.3390/biom10091351
Roy NK, Murphy A, Costa M. Arsenic Methyltransferase and Methylation of Inorganic Arsenic. Biomolecules. 2020; 10(9):1351. https://doi.org/10.3390/biom10091351
Chicago/Turabian StyleRoy, Nirmal K., Anthony Murphy, and Max Costa. 2020. "Arsenic Methyltransferase and Methylation of Inorganic Arsenic" Biomolecules 10, no. 9: 1351. https://doi.org/10.3390/biom10091351
APA StyleRoy, N. K., Murphy, A., & Costa, M. (2020). Arsenic Methyltransferase and Methylation of Inorganic Arsenic. Biomolecules, 10(9), 1351. https://doi.org/10.3390/biom10091351