3.1. Characterization of the Different Lignins
Table 2 shows the main characteristics of the lignins used in this study. As expected, the purity of the technical lignins SeolA, and OrgB was much higher (98.0 wt.% and 96.1 wt.%, respectively) than those of downstream biorefinery lignins, BioA and B (51.1 wt.% and 69.1 wt.%, respectively). It is important to note that BioA and B presented a relatively high sulfur content coming from the acidic thermal pretreatment with sulfuric acid [
50], which causes some sulfonation in the aromatic rings. Most of the impurities in biorefinery lignins, 38.8 wt.% in BioB and 23.2 wt.% in BioA, corresponded to glycans that were not fully removed upon enzymatic hydrolysis. Opposite to this, neither hemicelluloses nor celluloses or glycans were detected in SeolA and B, whereas only 0.4 wt.% of glycans were found in OrgB. In the case of SeolB, lignin content in the isolated solid was surprisingly low, 68.7 wt.%, with a high ash content, 30.1 wt.%. The reason for this high content in ashes could be a non-efficient removal of salts formed during lignin isolation.
According to the SEC analyses, downstream biorefinery lignins (see
Figure S2 in Supporting Information) presented much broader mass distributions than technical lignins. BioB presented its maximum at 1038 Da and BioA at 228 Da. However, fractions beyond 32000 Da were found in both cases. Although SEC for biorefinery lignins was carried out using 0.1% LiBr in
N,N-dimethylformamide, neither BioA nor B could be completely solubilized. This is probably the reason for the high dispersity index found for these solids. Presumably, low molecular weight fractions are much more soluble and provide the relatively low M
n in both cases, 535 Da and 414 Da, respectively. Size distributions in SeolA, B and OrgB were much narrower, as it is pointed by the dispersity index, which is, in all cases, in the range of 1.3 to 1.4. SEC chromatograms for OrgB and SeolB (see
Figure S2 in Supporting Information) presented similar profiles. M
n values were similar in both cases and no significant fractions could be detected at mass values beyond 4500 Da. SeolA also showed a much narrower distribution than BioA, presenting M
n values (2209 Da) which were much higher than those of OrgB and SeolB. Size distribution in SeolA, however, was broader and lignin fragments could be detected at mass values of ca. 8000 Da.
In a recent work [
6] we have shown that DOSY spectroscopy can be used to selectively determine the apparent mass of poly-(hydroxy)-aromatic fractions in BCD reaction mixtures. This estimation was carried out after proper calibration of log D
vs log MW of two different families of standards, polystyrene (PS) and poly-ethylene glycols and monomeric bioaromatics (PEG curve). These two families of standards accounted for different solute-solute and solute-solvent interactions. DOSY was particularly useful in the characterization of lignins. The Stejskal-Tanner equation (
Supporting Information SI.3) permitted estimating the diffusion coefficients (D) of the diffusion traces that appear in the aromatic, methoxy and aliphatic regions of the spectra and, through a proper calibration, correlating D to the apparent masses. This is particularly interesting in BioA and B lignins, whose SEC analyses were not reliable due to their low solubility in the eluent. The solubility of BioA and B was very low also in deuterated dimethylsulfoxide (DMSO-
d6) even after acetylation (see
Supporting Information for details), which complicated both the HSQC (see
Figure S13 in Supporting Information) and the corresponding DOSY analyses (see
Figure S3 in Supporting Information). Assuming that only lignin presents aromatic moieties in the analyzed samples, D for the aromatic region can be associated with the apparent masses of lignin. Apparent masses in the aromatic region for BioA were 1274 Da using PS calibration curves. In the case of acetylated BioB, apparent masses were graphically determined because experimental and theoretical area intensities determined by the Stejskal-Tanner equation did not present a good correlation (see
Table S2 in Supporting Information). Apparent masses in the aromatic region were 1630 Da, but cellulosic residues presented
1H signals in the range of 4.0–5.0 and 3.0–3.8 ppm, with higher intensity than those from lignin due to their higher solubility. Apparent masses for the saccharide region (4.5–3.0 ppm) ranged from 317 Da to 1796 Da in BioB, which is concordant with the SEC measurements (see
Table 2 and
Figure S2 in Supporting Information). Intense signals around 2.0 ppm appeared due to acetylation of lignin and the saccharide residues. Aliphatic traces at high fields (1.0 ppm–2.0 ppm) corresponded, almost exclusively to alkyl chains and solvent residues from acetylation (see
Figure S3 in Supporting Information).
Poplar lignins isolated via organosolv, OrgB, and pine and poplar lignins isolated by autohydrolysis followed by soda ethanosolv, SeolA and B respectively, were much more soluble in DMSO-
d6 than biorefinery lignins. Average diffusion coefficients were determined using the Stejskal-Tanner equation for the aromatic region of OrgB and SeolB that corresponded with apparent masses of 1445 Da and 1477 Da, respectively, which are in the range of M
n determined by SEC (1230 Da and 1295 Da respectively). Correlation in the Stejskal-Tanner equation was not optimal for SeolA and the diffusion coefficient was estimated graphically from the DOSY spectrum processed using TOPSPIN 3.6.2 corresponding to an apparent mass of 2122 Da. Most of the diffusion traces in DOSY spectra for technical lignins (see
Figure S4 in Supporting Information) turned up in the aromatic region and were thus assigned to the aromatic hydrogen atoms of lignins. Therefore, average apparent masses for lignins estimated by DOSY presented only slight differences with M
n determined by SEC. OrgB presented some traces in the 4.0 ppm–5.0 ppm range, which can be associated to hemicelluloses and/or celluloses, as it is shown in its compositional analysis (
Table 2), centered in in the range of 1000–1340 Da. Nevertheless, the aliphatic region presented hydrogen signals in the range of 540–914 Da according to PS calibration (see
Figure S4 in Supporting Information). These hydrogen signals may correspond to saccharides degradation produced upon treatment with isopropanol at 463 K. None of these traces were detected in either SeolA or B, probably due to the autohydrolysis step that removed the hemicellulose residue prior to soda ethanosolv extraction.
Quantitative diffusion ordered spectroscopy NMR (q-DOSY) relies on the quantitative
1H NMR that requires of using an internal standard [
52,
53] and permits reliable quantification when using stimulated-echo DOSY sequences. Q-DOSY presents problems with complex mixtures that could have proton overlapping [
54] and thus different decays, hence making data fitting difficult [
55]. In this case, semi-quantitative DOSY combined with
31P NMR of the reaction mixtures has been used to make an estimation of the purity of starting lignins and the poly-(hydroxy)-aromatic ether fractions in the as-mentioned reaction mixtures and to evaluate the degree of the ethanol side-reactions (see below). This estimation has been carried by means of the calculation of the percent of aromatic carbon (
Table 2,
SI.3, Equation (S1) and Equation (S2) in Supporting Information) that reflects the proportion of aromatic carbon atoms (i.e., from lignin) in the reaction mixtures.
Most of the diffusion traces in downstream biorefinery lignins corresponded either to partially degraded cellulose after enzymatic hydrolysis, to degradation products upon pretreatment or to acetyl moieties derived from derivatization. The low solubility of the samples did not allow to obtain reliable results from DOSY integration. Semi-q-DOSY could be tested in the determination of the purity of technical lignins, SeolA, B and OrgB. The purity of lignins can be estimated using the positive integration values of the aromatic (7.1–6.0 ppm) and aliphatic regions (3.4–2.6 ppm and 2.4–0.5 ppm). The aliphatic region in the range 4.2–3.5 ppm was excluded for this estimation because hydrogen atoms either in methoxy and β-O-4 motifs appear in this range, which can lead to data misinterpretation. DOSY integration refers to the number of hydrogen atoms. However, the H/C ratio in aromatic and aliphatic moieties is different (2:1 in aliphatic, 1:3 in syringyl units and 1:2 in guaiacyl units, see
Supporting Information SI.3). Selected samples were also analyzed using
31P NMR after derivatization with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) to estimate the guaiacyl to syringyl ratio (G/S). This permitted a more accurate determination of H/C ratio for the aromatic compounds (See
Supporting Information SI.3, Seq.1 and 2). The integration values of aromatics are shown in
Table 2, resulting in 97% and 90% aromatic C for SeolA and OrgB respectively, that is consistent with the values showed in
Table 2. 98% of aromatic carbon was measured for SeolB while its purity was 69%. The difference accounts for the content in ashes (ca. 30%) that are not detected by NMR.
HSQC and HSQC-TOCSY analyses of the lignins were consistent with DOSY. Technical lignins presented intense H-C cross-peaks in the aromatic regions. As expected, SeolA (see
Figure S10) presented cross-peaks in the aromatic regions that corresponded to guaiacyl units together with signals corresponding to this guaiacyl units functionalized with double bonds. In the case of OrgB and SeolB (see
Figures S11 and S12 in Supporting Information), cross-peaks corresponding to both guaiacyl and syringyl units could be detected in the aromatic region. H-C cross-peaks corresponding to aromatic units could not be found in acetylated BioA nor in acetylated BioB (see
Figure S13 in the Supporting Information) due to their low solubility in DMSO-
d6. The most important cross-peak in all samples corresponded to the methoxy groups in 3.7–55.0 ppm region. This cross-peak was accompanied with cross-peaks corresponding to H
α-C
α, H
β-C
β and H
γ-C
γ in β-O-4 structures together with H
γ-C
γ in resinol and cinnamyl. In the case of BioA, some H-C cross peaks corresponding to the anomeric carbon of saccharides could be detected. This suggested that the detected signals in the range 3.0–5.0 ppm/ 60–100 ppm in the HSQC-TOCSY spectrum corresponded to H-C and long-distance correlations from partially degraded cellulose after incomplete enzymatic hydrolysis. Remarkably, no significant cross-peaks were found in the aliphatic region for SeolA or B, which suggests that degradation reactions did not occur during lignin isolation neither in the solvent nor in the saccharides, whereas in OrgB cross peaks corresponding to alkyl side chains were detected.
3.3. Aromatics Formed after Lignin Depolymerization
Depolymerization of BioA and B was first tried by catalytic hydrogenolysis using Ru/C (data not shown), in ethanol at 523 K. However, this treatment was not efficient in downstream biorefinery lignins because of their high recalcitrance. Recalcitrance in downstream biorefinery lignins is higher than in native lignins after acidic thermo-chemical pretreatment due to the formation of C-C bonds between aromatic units that hinders the release of low molecular weight units. In addition, the strong acidic thermo-chemical pretreatment reduces the number of β-O-4 motifs in the lignin [
42], preventing the nucleophilic attack of the ethanolic solvent to the α position of the β-O-4 motifs that triggers the hydrogenolysis reaction. Furthermore, both BioA and B were not soluble in ethanol, which caused diffusional limitations from the lignin into the catalyst.
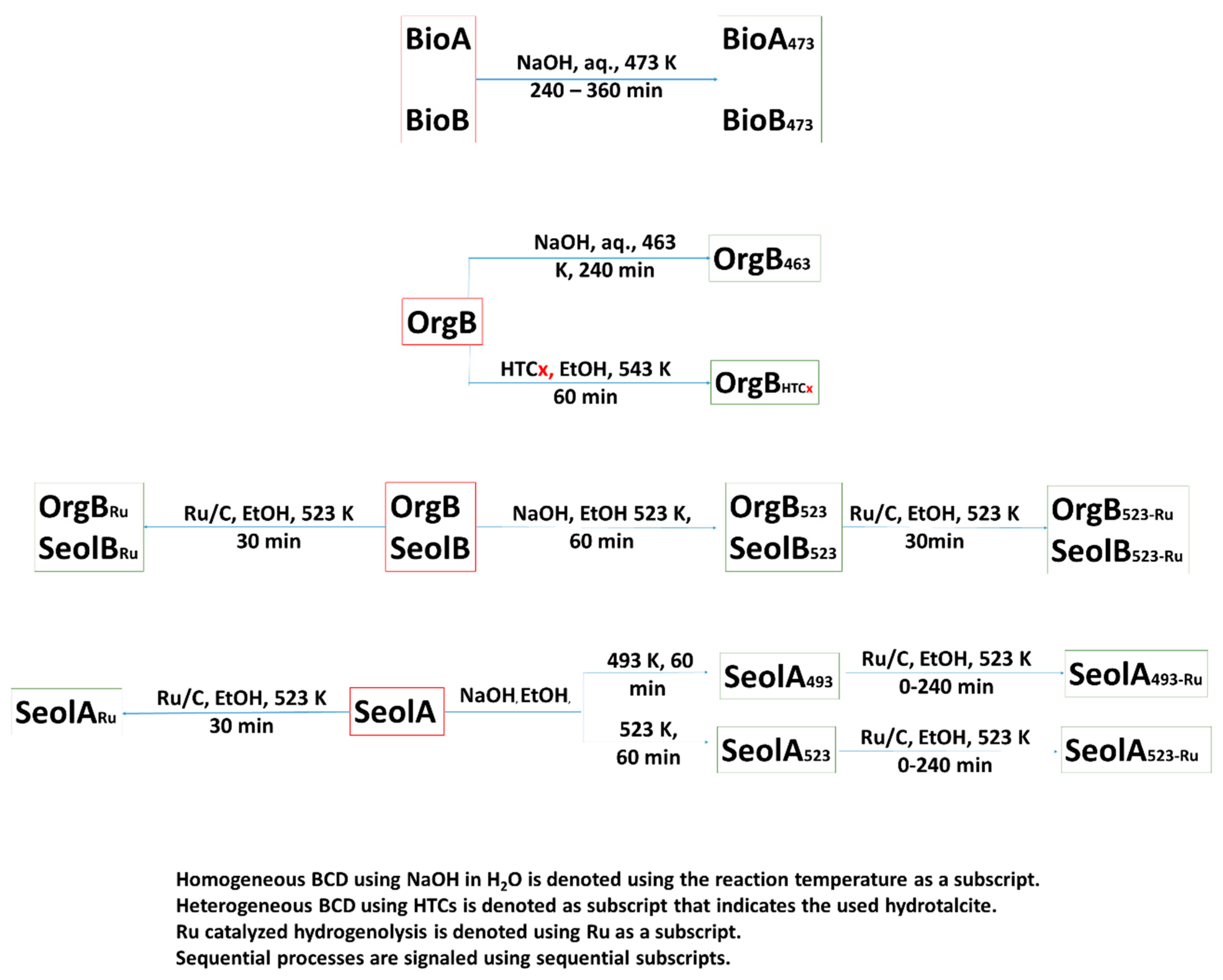
Thus, the BCD reaction conditions for BioB were carefully optimized instead in aqueous medium [
6]. Basic aqueous medium enhanced the solubility of lignin upon formation of phenolates that also initiates the depolymerization process. Amongst the different bases, aqueous NaOH was found to be effective at relatively low temperatures (473 K) and reaction times in the 240 min–360 min range (see
Table 4), whereas ethanolic NaOH proved to be ineffective. Monomer weight yields (wt.%) under these conditions ranged between 9.1 wt.% and 9.7 wt.%, whereas longer reaction times provoked a decrease in monomer yields due to repolymerization. Guaiacol, 10, was the major monomer (30%), followed by syringol, 20, and phenol, 1 (25% and 18% respectively). BCD of BioA under the same conditions only produced 4.1 wt.% of monomeric aromatics. As expected, guaiacol, 10, was the major monomer (66%), followed by other guaiacol derivatives such as methylguaiacol, 11, ethylguaiacol, 12 and acetovanillone, 18 (ca. 6% each).
Given the excellent yields obtained with downstream biorefinery lignins, the optimized reaction conditions were tested in the BCD of OrgB at 463 K in H
2O. Low monomer yields were obtained, 5.2 wt.%, which was due to the low solubility of OrgB in the reaction medium. Hence, the same reaction conditions were tested using ethanol as solvent, yielding 2.1 wt.% of aromatic monomers. Monomer distribution was different, though, being 1 (47%) the most abundant monomer when BCD was carried in ethanol and 20 (45%) when the reaction was carried in H
2O. Noteworthy, in the case of aqueous BCD of OrgB, syringol derived monomers were predominant (ca. 69%) whereas in the case of ethanolic BCD, phenolic derivatives (60%) were predominant suggesting that, under these operating conditions, severe demethoxylation took place. Homogeneous BCD was finally carried in ethanol at 543 K to test the effect of reaction temperature, OrgB
543-EtOH, but only 1.8 wt.% of aromatics was obtained, despite the high solubility of OrgB, accounting for severe repolymerization (see
Table 4) due to harsh reaction conditions.
Thus, HTC-M catalysts were deemed a reasonable option to perform BCD on OrgB in ethanol. Besides the intrinsic advantages of using heterogeneous catalysts, HTC-Ni catalysts have already proved their efficiency in lignin BCD. Heterogeneous BCD of OrgB was tested in supercritical ethanol (543 K, 110 bar, see
Table 4). Non-catalyzed depolymerization of OrgB under these conditions provided slightly higher monomer yields than NaOH ethanolic depolymerization at 543 K, 3.2 wt.%, that is presumably due to ethanolic fractionation of OrgB. In addition, catalytic depolymerization in scEtOH (543 K) of OrgB using the calcined HTC support (without impregnation) provided slightly lower aromatic monomers yields than in the blank reaction. It is worth mentioning that homosyringaldehyde, 26, (48%) was the most abundant monomer. Hydroxy-3-methoxyphenylacetone, 15, (26%) and phenol 1, (14%) yields were also relatively high. Monomer yield increased beyond 5 wt.% with HTC-0, HTC-2.5 and HTC-4, reaching its maximum with HTC-1, 6.8 wt.%, and decreased with the highest nickel content in the solid, HTC-5, 2.4 wt.%. Monomer distribution was somewhat stable at different Ni/Cu loadings though, being in all cases homosyringaldehyde 26 the most abundant aromatic monomer (44–56%) followed by 15 (15–20%). This suggest that, besides the basic activity of HTC-M, which is enhanced by nitrate anions as described by Kruger et. al. [
44], using scEtOH as solvent promotes the oxidation of the alkyl side chains providing 15 and 26 as mayor aromatic monomers.
HTC/Ni-Cu catalyzed depolymerization of BioB in aqueous media provided negligible monomer yields due to mass transfer limitations as mentioned before for Ru/C. BCD of SeolA lignins using Ni and/or Cu catalysts supported on HTC proved to be unsuccessful. Monomer yields were in all cases under 3.0 wt.%. Almost 85% of the monomers under these conditions were phenol (85%) and syringyl monomers (15%).
As mentioned before, most of the studies on catalytic depolymerization of technical lignins rely on heterogeneous catalysis of hydrogenolysis reactions. Direct hydrogenolysis of technical lignins using commercially available catalysts was tried in ethanol because of its ability to prevent repolymerization [
39,
40]. SeolA was reacted in ethanol in the presence of Pd/C catalyst at 473 K, though monomer yields were under 3.0 wt.% at the selected operating conditions. Hydrogenolysis at 523 K under hydrogen in ethanolic medium in the presence of Ru/C, SeolA
Ru, performed only slightly better (3.5 wt.%), being guaiacyl monomers (86%) the major monomers followed by phenols (3%). Among them, 4-ethylguaicol, 12, was the most abundant monomer followed by 4-propylguaicol, 13. It is worth noting that gigantol, 19, was present with ca. 6%. Hydrogenolysis of SeolB and OrgB under the same conditions provided slightly higher bioaromatics yields, 3.9 wt.% and 6.3 wt.% respectively. However, as could be expected, the monomers distribution changed, being the most relevant monomers homosyringaldehyde (20% and 37%), 26, and 4-hydroxy-3-methoxyphenylacetone (32% and 31%), 15 (see
Table 5).
The alleged synergistic effect of Ru/C and basic catalysis is still a matter of controversy [
17,
41]. Therefore, homogeneous BCD and Ru/C hydrogenolysis were carried out in separate processes (see
Scheme 2). Homogeneous BCD using NaOH seemed to be a plausible option. However, in view of the results obtained with OrgB, reaction conditions were slightly modified. The low solubility of SeolA in H
2O was circumvented using NaOH in EtOH/H
2O mixtures at lower temperatures, 493 K and 523 K, for 60 min, to obtain mixtures SeolA
493 and A
523, respectively, in almost quantitative yields. Low monomer yields were obtained under these conditions, 0.6 wt.% and 1.1 wt.%, respectively. The monomer distribution showed that, as expected, the major monomers were guaiacyl monomers (81% and 88%, respectively) followed by syringyl monomers (10% and 5%, respectively). As a thumbnail rule, syringyl monomers are not present in pine lignins; however, it has been reported that small amounts can be found in pine barks [
57].
31P NMR spectrum of derivatized SeolA confirmed the presence of syringyl units by the presence of a shoulder at 143.1 ppm. These syringyl units are more easily detached from lignin than guaiacyl units since the formation of C-C bonds occurs more easily in the latter. Nevertheless, both DOSY and SEC experiments (see below) evidenced that, although the monomer yield was relatively low, M
n and M
w in SeolA
493 and A
523 were much lower than in SeolA, which indicates that some depolymerization had occurred.
SeolA493 and A523 were subjected to hydrogenolysis in the presence of Ru/C in ethanol at 523 K, SeolA493-Ru and A523-Ru, respectively. Monomer yields in SeolA493-Ru steadily increased with reaction time from 2.4 wt.% (0 min) to 4.2 wt.% (after 120 min) and remained constant thereafter. Guaiacyl monomers were the most abundant (ca. 82%) again and, amongst them, 12 was the most abundant. However, the content in 12 decreased from 53% to 42% over reaction time (0 to 240 min), with a concomitant increase in guaiacol, 10, whose content was raised from 12% to 20%, whereas the proportions of 11 and 4-acetovanillone, 18, remained almost constant with reaction time (10% and 7% respectively). This suggests that increasing reaction times produced an increase in 10 due to alkyl side-chain breakage in 12. A noticeable increase in the aromatic monomer yields was observed when SeolA523 was subjected to hydrogenolysis under the same conditions, SeolA523-Ru. Consequently, the monomer yield increased from 5.0 wt.% at 0 min to 7.3 wt.% after 30 min and remained constant (6.7 wt.%) at longer reaction times. The proportion of guaiacyl monomers rose beyond 90% in all cases, being 12 the most abundant amongst them (ca. 45%) followed by 10 (ca. 30%). Noteworthy, any change in the monomer distribution in the reaction mixture could be found regardless of the reaction time. It can be observed that the relative amounts of 11 and 12 are very similar in SeolA493-Ru and A523-Ru, but there is a strong increase in 10 (from 20% to 3%) together with a decrease in 18 (10% to 2%, at 30 min of reaction time), and gigantol, 19, (6% to 2%, at 30 min of reaction time).
Next, SeolB was reacted following the optimized procedure for SeolA. Monomer yields for SeolB523 (9.7 wt.%) were, however, much higher than for SeolA523-Ru, because of lower cross-linking in the starting lignin. The most abundant monomers were 4-hydroxy-3-methoxyphenylacetone (36%), 15, followed by syringol, 20 (17%). Further hydrogenolysis in the presence of Ru/C produced a slight increase in the aromatics yield up to 11.2 wt.% with 15 being the most abundant bioaromatic (45%) followed by 12 (16%) and 20 (15%). Finally, treatment of OrgB with NaOH at 523 K in EtOH/H2O, OrgB523, provided 7.5 wt.% yield to phenolic monomers, being 20 (34%) the most abundant followed by phenols, while 15 was not detected and 26 accounted for just 11% of the bioaromatics. Hydrogenation of OrgB523 also produced a slight increase in aromatic monomers, 8.7 wt.%, with 15 (36%) and 20 (19%) being the most abundant monomers. This suggested that most of the depolymerization in SeolB and OrgB occurred under BCD in EtOH/H2O at 523 K, while Ru/C catalyzed hydrogenolysis had little effect in monomer yields.
3.4. Poly-(Hydroxy)-Aromatic Fractions
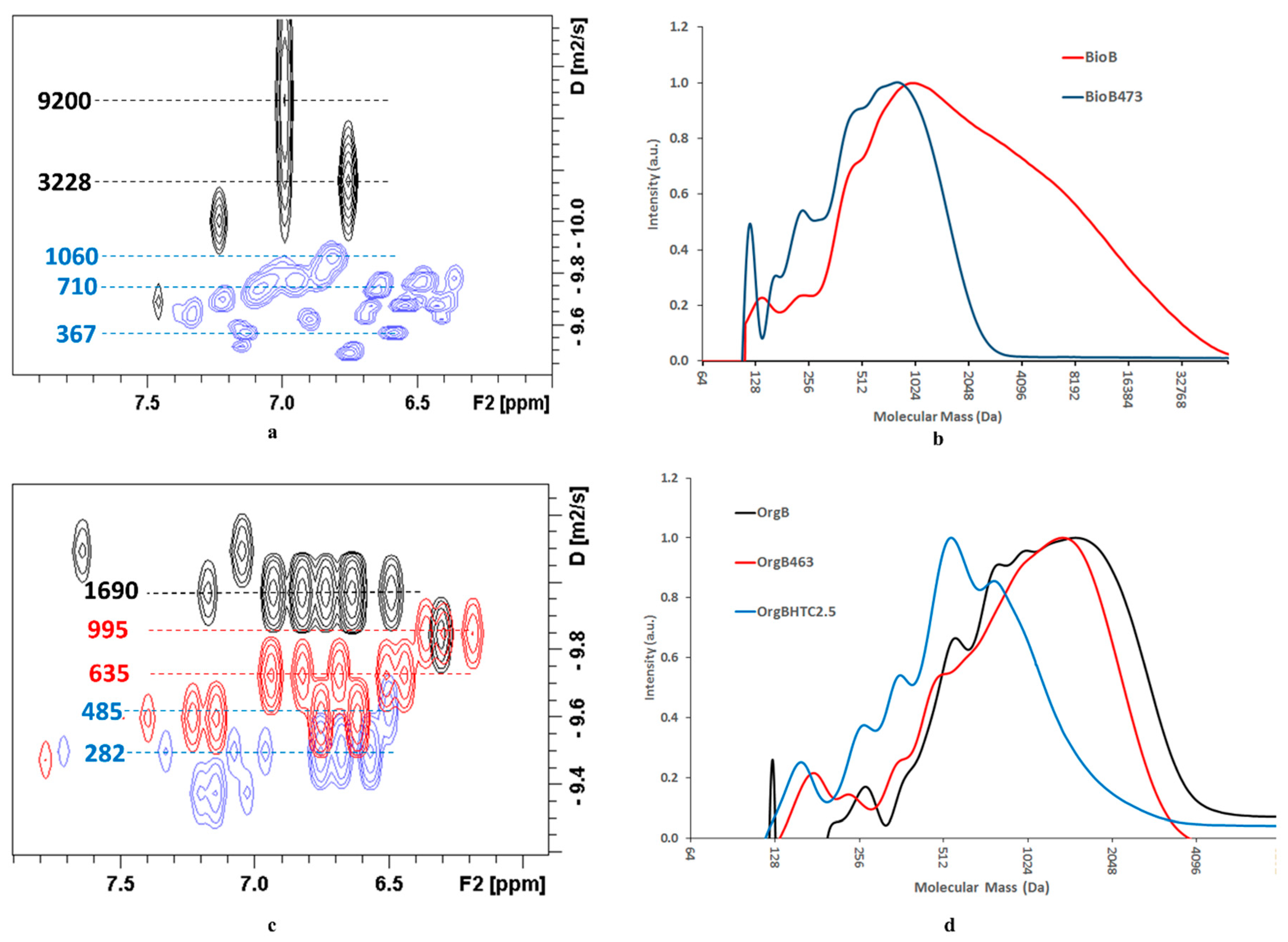
BioB was subjected to BCD in the presence of NaOH at 473 K, BioB
473. DOSY spectra (see
Figure 1a) clearly showed the difference in apparent mass between the aromatic hydrogen atoms in the starting lignin and those corresponding to the BCD reaction mixture, with fractions in the range of 360 Da–1060 Da. Moreover, some diffusion traces were detected in the aliphatic region (see
Figure S5 in Supporting Information), which correspond to degradation products from the peeling reactions of saccharides in the basic medium [
58]. A similar observation can be made when OrgB is reacted under similar reaction conditions at 463 K (see
Figure 1c), OrgB
463. The apparent mass decreased from 1454 Da to 709 Da (see
Table 6) according to DOSY measurements in the aromatic region, although the SEC analyses did not show any noticeable change but a slight shift to lower masses (see
Figure 1d). However, when the same reaction was carried out in ethanol under the same conditions, the mass distribution changed drastically and the size distribution narrowed with a prominent peak at ca. 530 Da, although the monomer yield decreased to 2.1 wt.% (see
Table 4,
Table 6 and
Figure S9). The maximum monomer yield in the depolymerization of OrgB was obtained using HTC-1 in scEtOH (543 K), with an increase in the monomers yield up to 6.8 wt.%. M
n determined by SEC was 694 Da while the apparent mass determined by DOSY was 428 Da, which represents a noticeable decrease in apparent mass from the starting OrgB and B
463 in H
2O. Size distribution was clearly shifted to lower masses when BCD was catalyzed by HTC-M, OrgB
HTCx, whatever the Ni and/or Cu loading. In addition, DOSY measurements (see
Figure 1) showed traces with noticeable lower apparent masses than OrgB
463, which evidenced that depolymerization is much more extensive when the reaction is carried in ethanol than when using H
2O as solvent, which can be due to the highest solubility of OrgB in ethanol than in H
2O.
SeolA and B depolymerization reactions were conducted using a different strategy that consisted of a two-step approach. Firstly, NaOH treatment in EtOH/H
2O was carried out at 493 K or 523 K for 60 min runs, SeolA
493 and A
523. The monomer yields in both cases were low, 0.6 wt.% and 1.0 wt.%, respectively (see
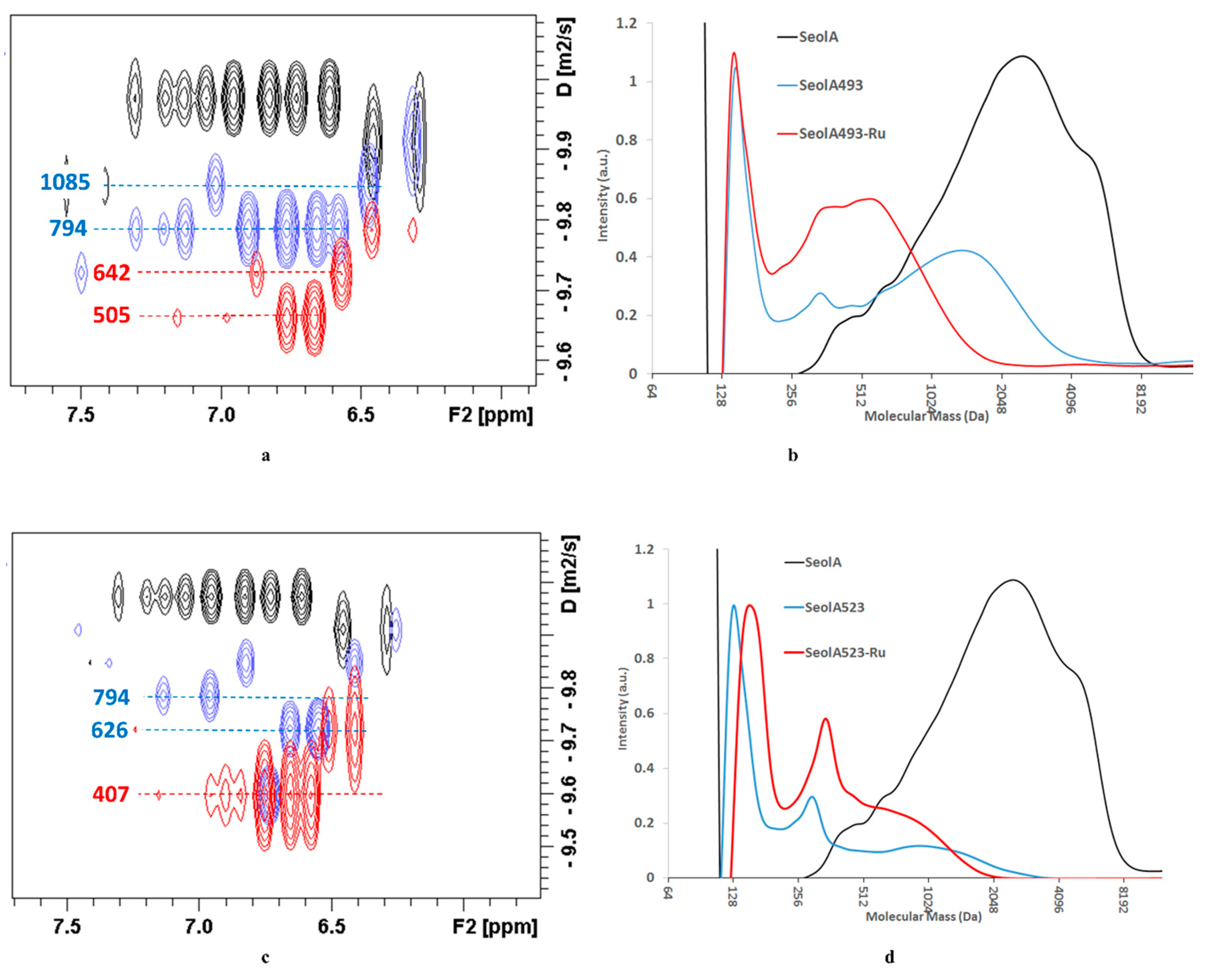
Table 5). However, DOSY for SeolA
493 and A
523 showed in both cases that depolymerization of SeolA did occur, being more extensive in SeolA
523, 660 Da, than in SeolA
493 1039 Da (see
Table 6). The most representative diffusion trace for SeolA
493 was centred at 800 Da but representative traces were present in the 800–1600 Da range, whereas in the case of SeolA
523, the most representative diffusion traces appeared centred at 630 Da with some small diffusion traces around 1024 Da (see
Figure 2).
Hydrogenolysis of SeolA
493 and A
523 was done in scEtOH at 523 K in the presence of Ru/C (20 bar). As expected, the monomer yields were higher than in starting SeolA
493 and A
523, reaching 4.2 wt.% in SeolA
493-Ru and 7.3 wt.% in SeolA
523-Ru. DOSY spectra showed in this case a more pronounced effect of the treatment in SeolA
493-Ru, since its apparent mass decreased from 1039 Da to 691 Da (see
Table 6). In the case of SeolA
523-Ru the apparent mass was similar to that of SeolA
523 (ca. 660 Da) and, although DOSY spectra showed some slight differences between SeolA
523 and A
523-Ru, it can be appreciated that an important fraction of the diffusion in SeolA
523 overlaps with those of SeolA
523-Ru (see
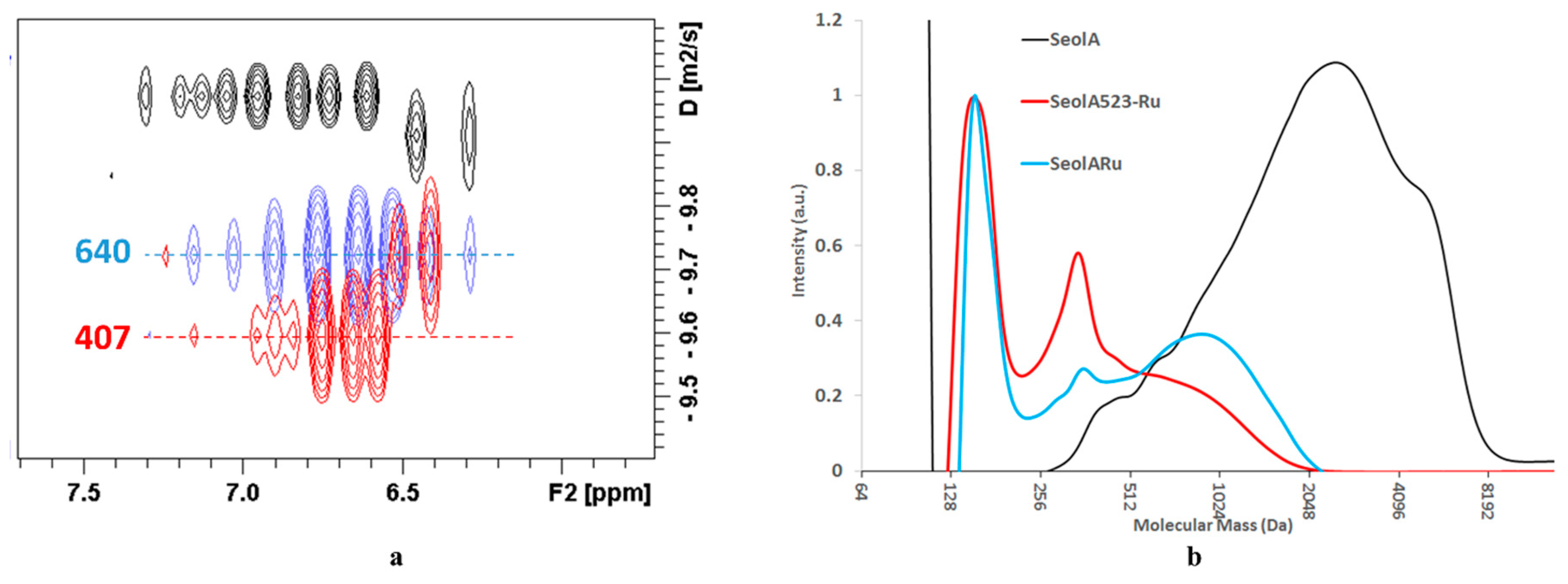
Figure 2). Noteworthy, the estimated apparent masses, 746 Da, were only slightly higher when SeolA was directly treated with Ru/C at 523 K in ethanol and diffusion traces were essentially similar to those of SeolA
523-Ru (see
Table 6 and
Figure 3) although its chemical monomer yield was much lower.
Data provided by DOSY spectroscopy shed light about the somewhat strange SEC chromatograms obtained after depolymerization of SeolA (see
Figure 2 and
Figure 3). Apparent masses determined by DOSY do not seem to match with the most prominent peaks determined by SEC. Thus, in all analysed samples, a peak at ca. 130 Da appeared. This peak is the most intense in the samples treated with Ru/C in EtOH, but it is also important in OrgB samples depolymerized using HTC in EtOH. This is consistent with the detection of diffusion traces in the aliphatic region of the DOSY spectra that might correspond to compounds arising from ethanol via hydrogen transfer mechanism (see
Figure 4 and
Figure S7).
Besides this peak, the rest of SEC chromatograms presented a broad distribution which is centred at similar mass values than those estimated by DOSY for the aromatic region (i.e., poly-(hydroxy)-aromatic fraction). In the case of SeolA
493 and A
493-Ru, broad mass fractions were present, which were more important in SeolA
493, centred at 1360 Da, and at 570 Da in SeolA
493-Ru. In SeolA
523, A
523-Ru and A
Ru mass distributions were much narrower and presented a prominent peak at ca. 360 Da (see
Figure 2). OrgB
HTC presented also a broad distribution with two major peaks centred at 526 Da and 758 Da that may correspond to three and four-unit aromatic compounds.
It is important to note that in all depolymerization reactions of OrgB, carried out in EtOH in the presence of either NaOH or HTCs with different Ni-Cu loadings, no significant differences in M
n (ca. 700 Da) could be found among the different Ni-Cu loadings or in the blank reaction. However, when BCD was carried out in H
2O in the presence of NaOH, M
n was much higher, ca. 1200 Da, which suggests that fractionation of lignin is caused by the ethanolic solvent at high temperatures (viz. thermos-solvolysis). Similarly, direct hydrogenolysis in the presence of Ru/C in ethanol and BCD in ethanol/H2O at 523 K of SeolA (see
Figure 3), B and OrgB, M
n was in the range of 600–700 Da, that also suggest the essential role of thermos-solvolysis in lignin depolymerization.
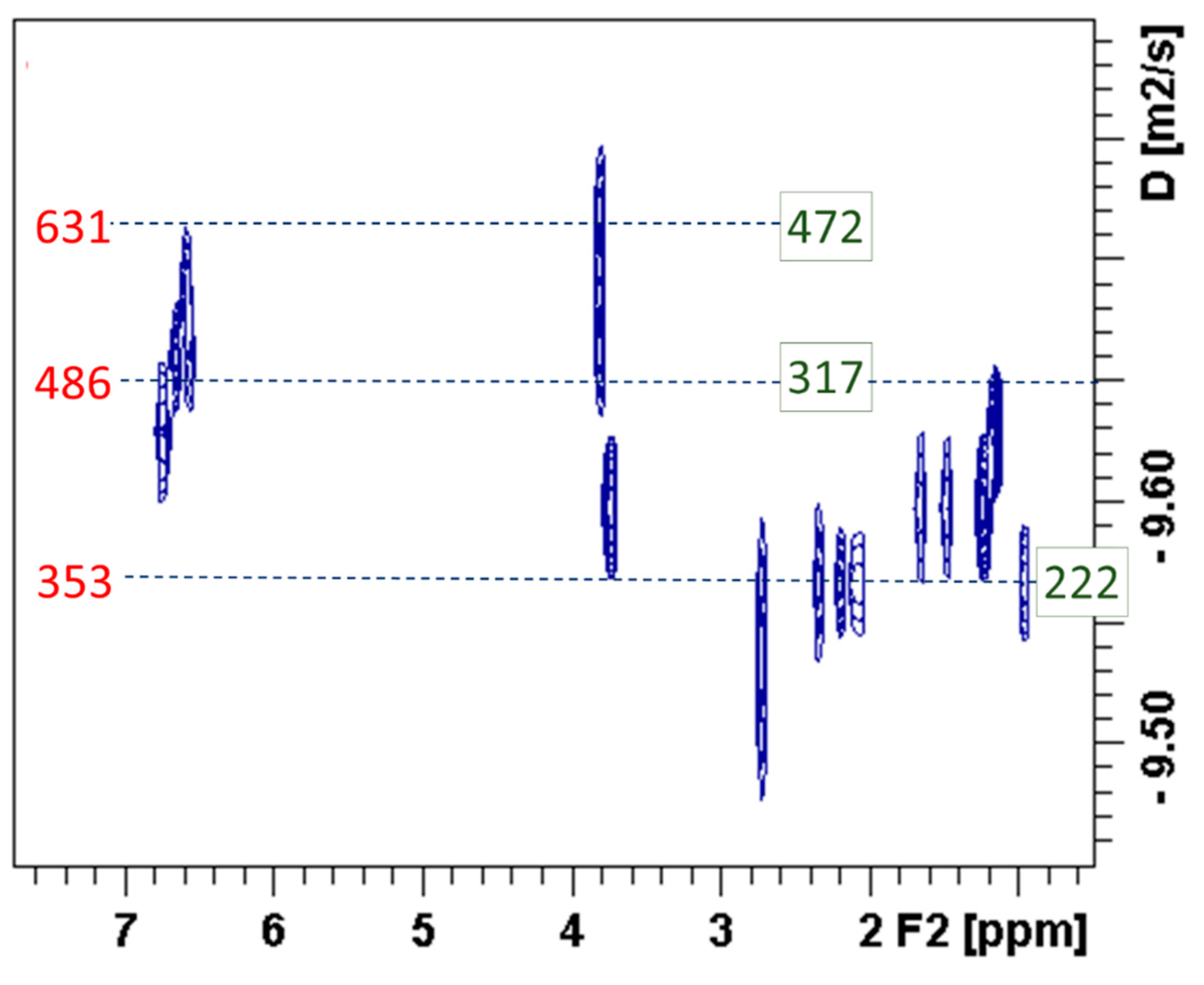
Diffusion traces in the range of 300–330 Da using the PS calibration curve or in the range of 185–300 Da using PEG curves, could be detected in all the DOSY spectra. This agrees with the peak that can be observed in most SEC chromatograms in the range of 275–350 Da. These diffusion traces in the aromatic region can be attributed to the presence of dimers (e.g., guaiacylglycerol-β-guaiacyl ether, gigantol, etc.). It must be highlighted that in all the studied samples derived from technical lignins, SeolA, B and OrgB, diffusion traces in this apparent mass range could be detected in the aromatic region, which suggests that most of this peak may come from the presence of two-unit aromatic compounds. However (see
Figure 4 and below), diffusion traces corresponding to these relatively low apparent masses were much more intense in the aliphatic regions.
DOSY spectra presented in most cases a remarkable difference between the diffusion coefficients associated to the aromatic and the aliphatic regions, being the latter significantly higher. Diffusion traces in the DOSY spectra (see
Figure 4) associated to the aromatic region are related to apparent masses above 400 Da, and centered in the range of 486–630 Da, whereas the aliphatic regions are below 500 Da, according to PS calibration, and below 300 Da according to PEG calibration (for the comparison of average apparent masses in both regions see
Table 6). In a previous work [
6], these traces in the aliphatic region were associated to the peeling reaction of saccharide impurities in the starting lignins, as is the case for BioA and B. However, diffusion traces at low apparent masses were also observed in the depolymerization of technical lignins, as it can be clearly seen for SeolA
523-Ru (see
Figure 4). GC-MS analysis from the depolymerization reactions carried in SeolA, B and OrgB presented products whose origin can be associated to side reactions from ethanol (viz. hexanol, 2-ethyl-butan-1-ol, 4-methyltetrahydro-2
H-pyran-2-one, etc.). Both Ru/C and Cu-containing HTC catalysts are able to promote hydrogen transfer reactions from ethanol that prompt the Guerbet reaction [
47,
59] that produces long chain alcohols. Given the reaction conditions, it can be expected that higher mass ethanol derivatives, that were not detected by GC-MS were produced.
Semi-q-DOSY was used in
Section 3.1 to estimate the purity of isolated lignin. A similar approach was used to estimate the percent of C atoms that correspond to aromatic rings in the reaction mixtures (see
Table 6). The percentage of aromatic C is relatively low in the depolymerization of BioA and B (54% and 58%) because of the peeling reaction of partially degraded saccharides in the starting lignins. OrgB
463 (73%) presents higher aromatic C content than BioA
473 and B
473 because no degradation of saccharides occurred upon BCD.
SeolARu, BRu and OrgBRu also presented relatively low contents in aromatic C (65%, 71% and 68% respectively) according to DOSY integration. However, in this case the decrease in the aromatic percentage of C atoms accounts for the formation of aliphatic compounds upon reaction of ethanol at high temperatures, following the hydrogen transfer mechanism in a similar fashion than in the Guerbet reaction. The estimated aromatic C is higher in SeolA493 and A523 (83% and 66%) than in SeolA493-Ru and A523-Ru (61% and 56%) respectively that is due to more extensive ethanol side reaction upon reaction at 523 K. A similar trend was observed in SeolA523 and A523-Ru (66% and 56%) and OrgB523 and B523-Ru (61% and 44%).
This effect was more pronounced when OrgB reacted with NaOH. 73% and 62% of aromatic C were estimated when the reaction was run at 463 K in H
2O and ethanol respectively, whereas only 16% was estimated when the reaction was run in ethanol at 543 K. Even more interesting, when the BCD was carried out using HTCs at 543 K, the estimated aromatic carbon increased with increasing nickel contents. Thus, 38% of aromatic C was estimated for treatments with HTC-0 and HTC-1, 44% for HTC-2.5, 49% for HTC-4 and 57% for HTC-5. This percentage of aromatic carbons was low, suggesting that the Guerbet reaction took place to a great extent, which is consistent with previous reports that described that the Guerbet reaction is catalyzed by HTC-Cu catalysts [
47].
31P NMR analyses after derivatization of some reaction mixtures provided the number of aromatic hydroxy groups per gram of sample (see
Table 6). This can be also a good parameter to evaluate the purity of the mixture in terms of poly-(hydroxy)-aromatic content. Indeed, this parameter followed the same trend that the aromatic C content in comparable reaction mixtures. In the case of the depolymerization of OrgB it can be observed that OrgB
463 carried out in H
2O presented 5.09 mmol aromatic OH/g while the same reaction in ethanolic medium provided a reaction mixture with 2.20 mmol aromatic OH/g. Similarly, OrgB
HTC2.5 contained 2.61 mmol aromatic OH/g. In the reaction mixtures of the SeolA series it can be also observed that the content of aromatic OH/g decreased from SeolA
493 to A
493-Ru and from SeolA
523 to A
523-Ru, which can be attributed to the increase in aliphatic carbon derived from side reactions produced by the presence of ethanol.
Some of the diffusion traces detected in the aliphatic region may also come from C-and O-alkylation under reaction conditions with scEtOH, as it has been previously described [
39,
47,
60]. HSQC of SeolA
523 and A
523-Ru in the aliphatic region (see
Figure S14 in Supporting Information) presented cross-peaks that are compatible with alkylated aromatic compounds. A similar observation can be made for OrgB
HTC2.5 (see
Figure S15 in Supporting Information), but not in those reactions carried in H
2O. However, the intensity of these signals was relatively low and was only detected after selective peak-picking treatment of the raw spectrum, and their diffusion coefficient was slightly higher than that of the corresponding aromatic region. Finally, O-alkylated and C-alkylated (others than gathered in
Table 4 and
Table 5) products were not detected by GC-MS.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}