Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension


 and
and 
Abstract
1. Introduction
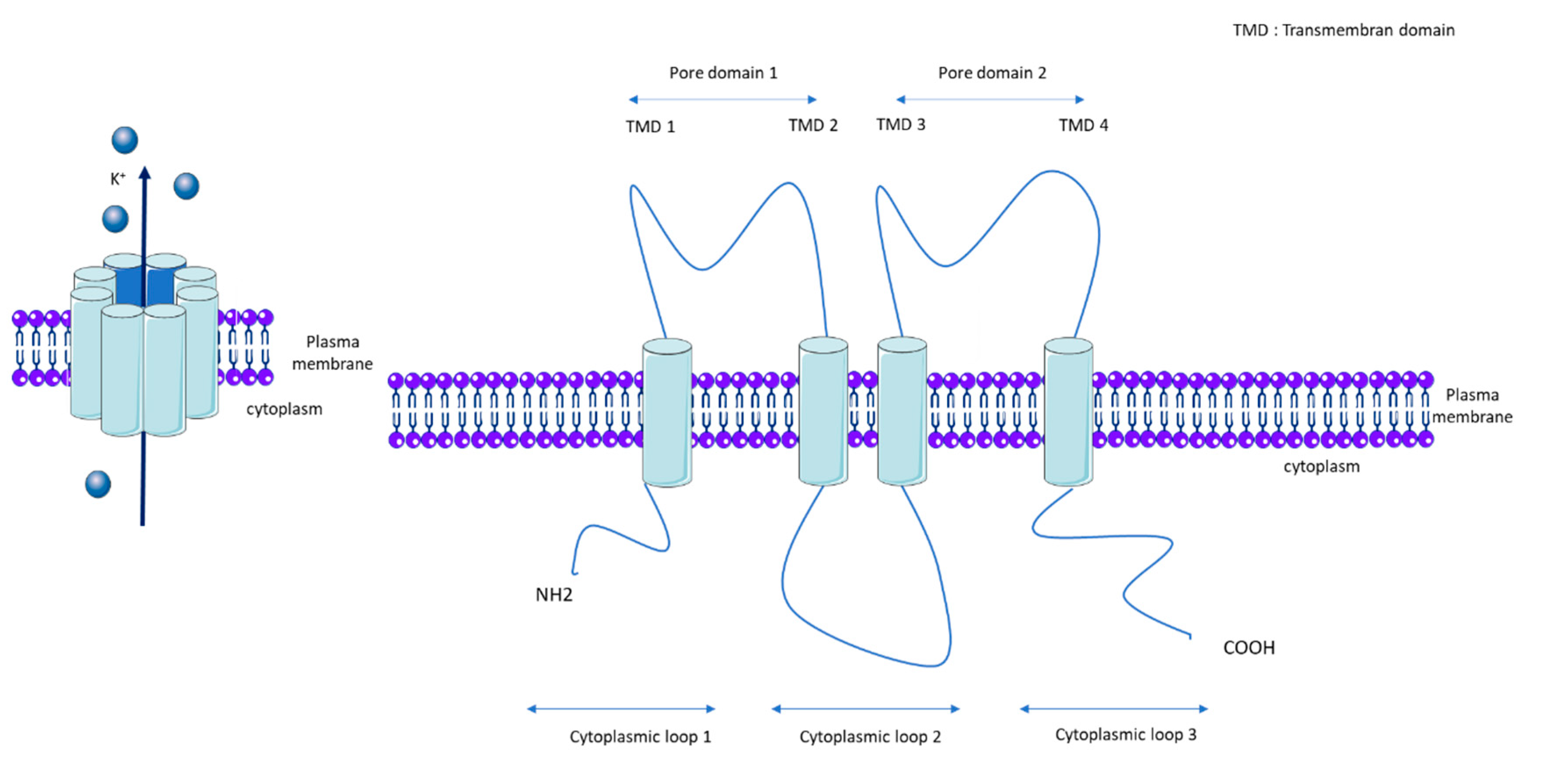
1.1. KCNK3
1.2. KCNK3 Properties and Regulatory Mechanisms
1.3. KCNK3 Function and Expression in Pulmonary Vasculature and RV
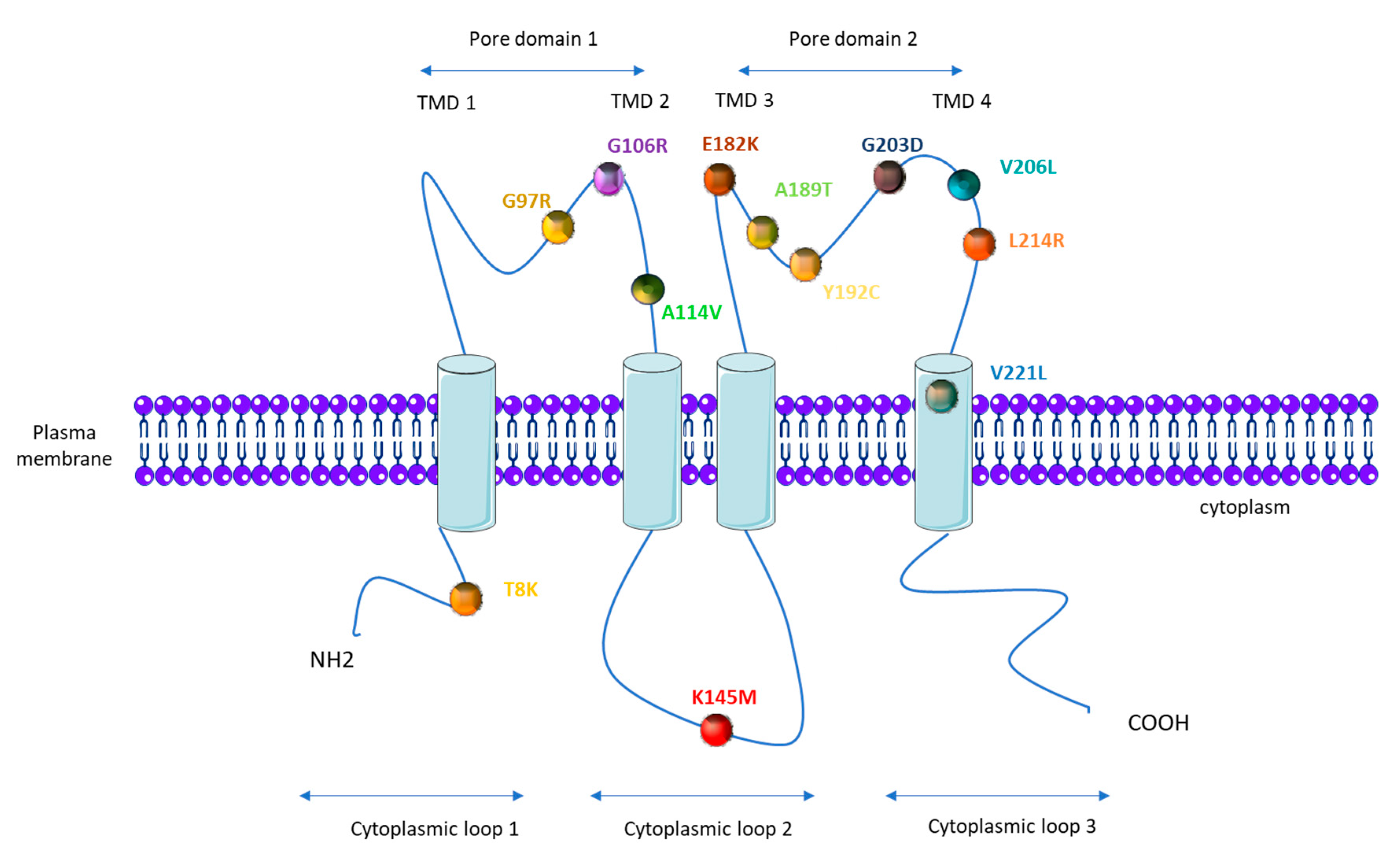
1.4. KCNK3 Mutations in PAH
1.5. Consequences of KCNK3 Dysfunction for the Physiopathology of PAH
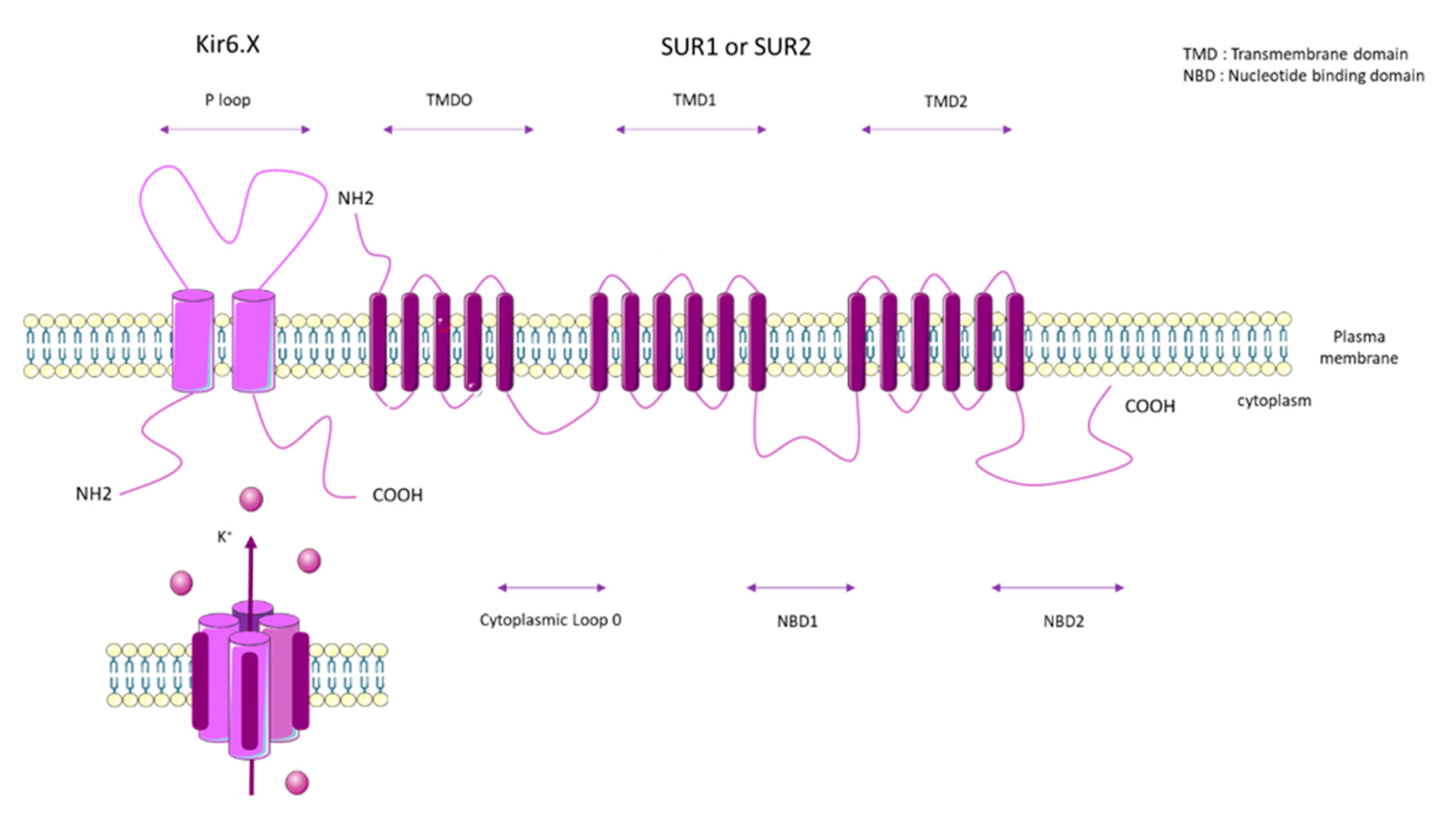
2. ATP Binding Cassette Subfamily C Member 8 (ABCC8)
2.1. SUR1 Properties and Mechanisms of Regulation
2.2. SUR1 Function and Expression in Pulmonary Vasculature and RV
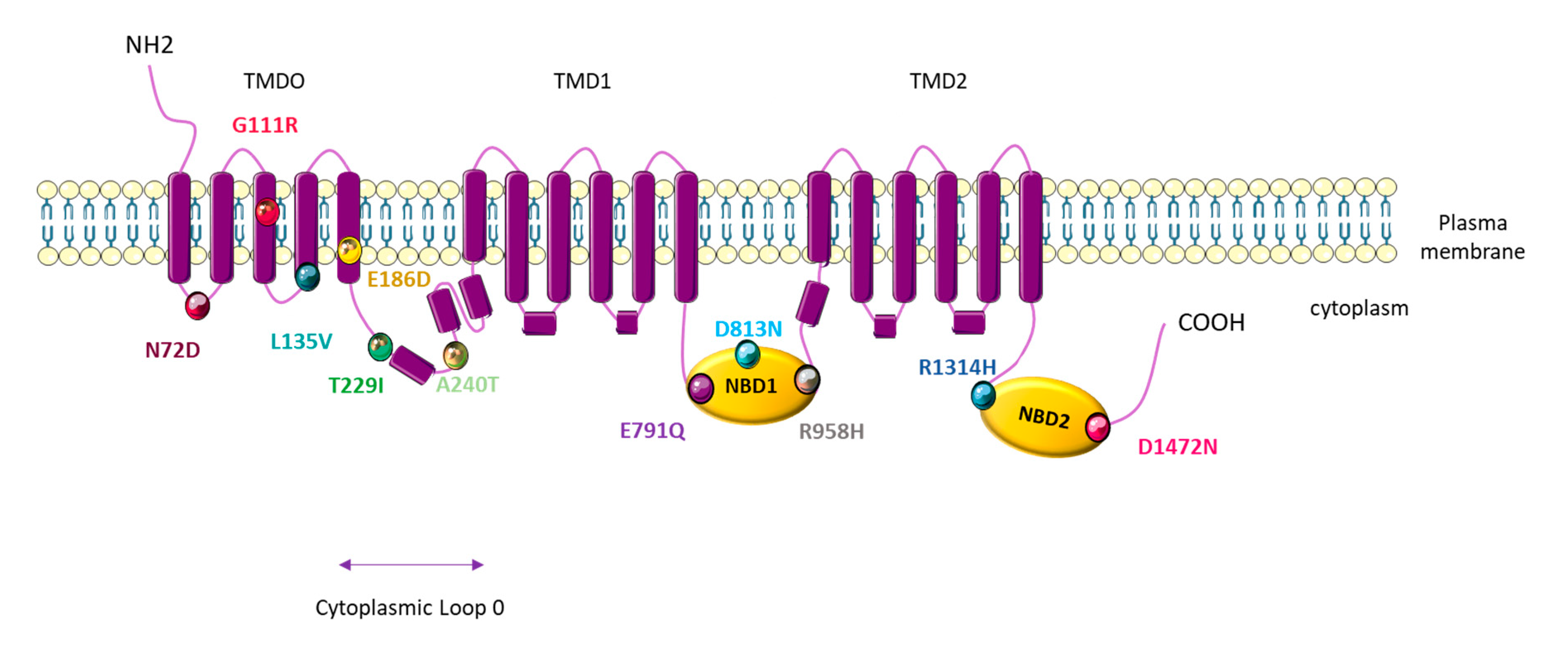
2.3. ABCC8 Mutations in PAH
2.4. Consequences of ABCC8 Dysfunction for the Physiopathology of PAH
3. Other Genetic Alterations in Genes Coding for K+ Channels
3.1. ATP Binding Cassette Subfamily C Member 9 (ABCC9)
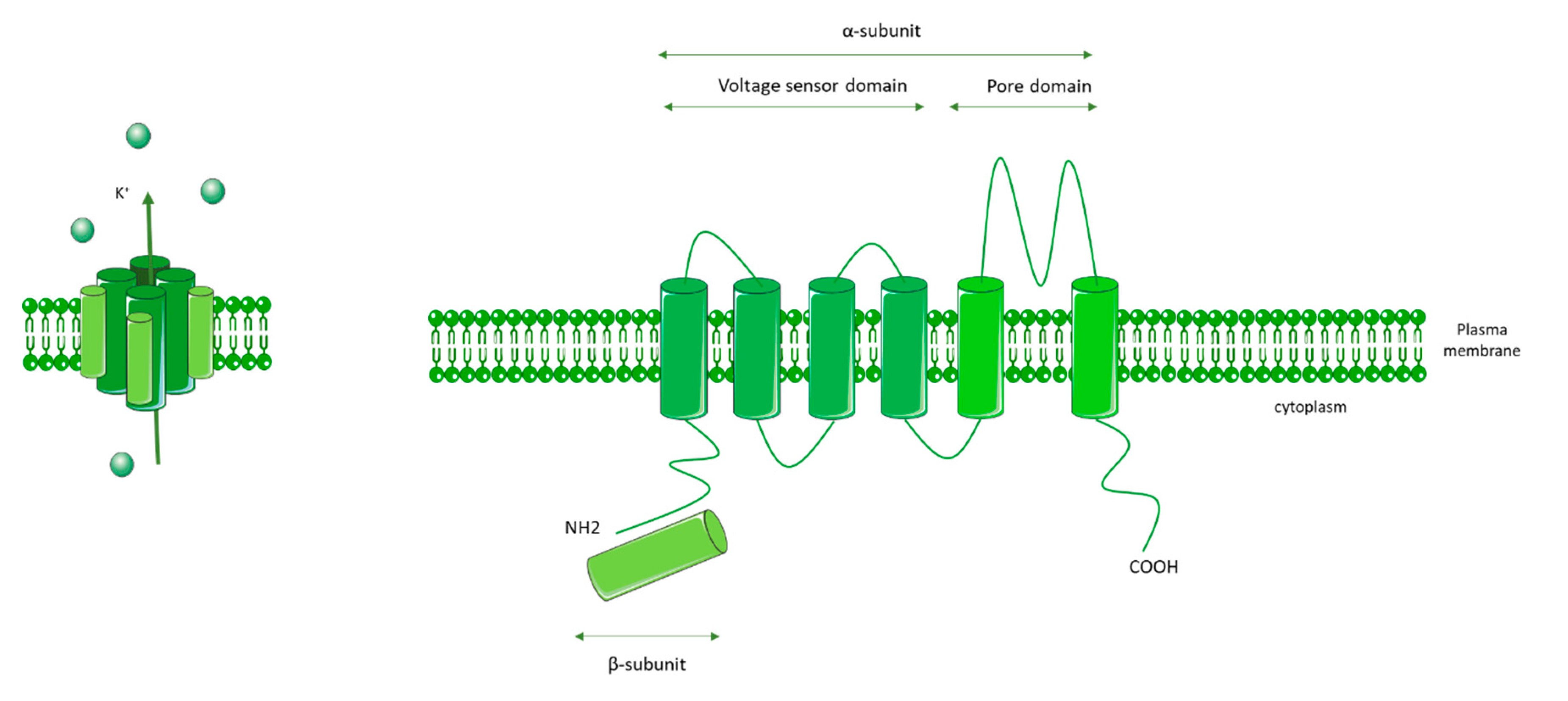
3.2. KCNA5 (Voltage-Gated K+ Channels 1.5: Kv1.5)
4. Potential Therapeutic Targets
4.1. KCNK3
4.2. KATP
4.3. Kv1.5
5. Conclusions
Funding
Conflicts of Interest
References
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiéry, J.-L.; Gibbs, S.; Lang, I.M.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2015, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Humbert, M. Pathobiology of pulmonary arterial hypertension: Understanding the roads less travelled. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Lau, E.M.; Montani, D.; Jaïs, X.; Sitbon, O.; Simonneau, G. Advances in Therapeutic Interventions for Patients with Pulmonary Arterial Hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Guignabert, C.; Barberà, J.A.; Bärtsch, P.; Bhattacharya, J.; Bhattacharya, S.; Bonsignore, M.R.; Dewachter, L.; Dinh-Xuan, A.T.; Dorfmüller, P.; et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur. Respir. J. 2018, 51, 1700745. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar] [CrossRef]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Eyries, M.; Montani, D.; Girerd, B.; Favrolt, N.; Riou, M.; Faivre, L.; Manaud, G.; Perros, F.; Grãf, S.; Morrell, N.W.; et al. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur. Respir. J. 2020, 55, 1902165. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Eccles, C.A.; Desai, A.A.; Gonzalez-Garay, M.L.; Yuan, J.X.-J.; Garcia, J.G.N.; Rafikova, O.; Rafikov, R. New cases of Glucose-6-Phosphate Dehydrogenase deficiency in Pulmonary Arterial Hypertension. PLoS ONE 2018, 13, e0203493. [Google Scholar] [CrossRef]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2019, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, D.H. Potassium channels—Multiplicity and challenges. Br. J. Pharmacol. 2006, 147, S63–S71. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirják, G.; Enyedi, P.; et al. TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 2017, 50, 1700754. [Google Scholar] [CrossRef] [PubMed]
- Mouratoglou, S.A.; Giannakoulas, G.; Deftereos, S.G.; Giannopoulos, G.; Angelidis, C.; Cleman, M.W.; Vassilikos, V.P. Intra-and Intercellular Calcium Handling in Pulmonary Arterial Hypertension. Med. Chem. 2016, 12, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. The Role of K+ Channels in Determining Pulmonary Vascular Tone, Oxygen Sensing, Cell Proliferation, and Apoptosis: Implications in Hypoxic Pulmonary Vasoconstriction and Pulmonary Arterial Hypertension. Microcirculation 2006, 13, 615–632. [Google Scholar] [CrossRef]
- Gardener, M.J.; Johnson, I.T.; Burnham, M.P.; Edwards, G.; Heagerty, A.M.; Weston, A.H. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br. J. Pharmacol. 2004, 142, 192–202. [Google Scholar] [CrossRef]
- Pandit, L.M.; Lloyd, E.E.; Reynolds, J.O.; Lawrence, W.S.; Reynolds, C.; Wehrens, X.H.T.; Bryan, R.M. TWIK-2 channel deficiency leads to pulmonary hypertension through a rho-kinase-mediated process. Hypertension 2014, 64, 1260–1265. [Google Scholar] [CrossRef]
- Lambert, M.; Capuano, V.; Olschewski, A.; Sabourin, J.; Nagaraj, C.; Girerd, B.; Weatherald, J.; Humbert, M.; Antigny, F. Ion Channels in Pulmonary Hypertension: A Therapeutic Interest? Int. J. Mol. Sci. 2018, 19, 3162. [Google Scholar] [CrossRef]
- Czirják, G.; Enyedi, P. Formation of Functional Heterodimers between the TASK-1 and TASK-3 Two-pore Domain Potassium Channel Subunits. J. Biol. Chem. 2002, 277, 5426–5432. [Google Scholar] [CrossRef]
- Lambert, M.; Capuano, V.; Boet, A.; Tesson, L.; Bertero, T.; Nakhleh, M.K.; Remy, S.; Anegon, I.; Péchoux, C.; Hautefort, A.; et al. Characterization of Kcnk3-Mutated Rat, a Novel Model of Pulmonary Hypertension. Circ. Res. 2019, 125, 678–695. [Google Scholar] [CrossRef]
- Putzke, C.; Wemhöner, K.; Sachse, F.B.; Rinné, S.; Schlichthörl, G.; Li, X.T.; Jaé, L.; Eckhardt, I.; Wischmeyer, E.; Wulf, H.; et al. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc. Res. 2007, 75, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Streit, A.K.; Netter, M.F.; Kempf, F.; Walecki, M.; Rinné, S.; Bollepalli, M.K.; Preisig-Müller, R.; Renigunta, V.; Daut, J.; Baukrowitz, T.; et al. A Specific Two-pore Domain Potassium Channel Blocker Defines the Structure of the TASK-1 Open Pore. J. Biol. Chem. 2011, 286, 13977–13984. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, D.P.; Simpson, D.S.; Miller, M.; Maki, B.E.; Zou, B.; Shi, J.; Wu, M.; McManus, O.B.; Aubé, J.; Li, M.; et al. Potent and selective inhibitors of the TASK-1 potassium channel through chemical optimization of a bis-amide scaffold. Bioorg. Med. Chem. Lett. 2014, 24, 3968–3973. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, M.S.; Roman-Campos, D.; Terrenoire, C.; Jnani, J.; Sampson, K.J.; Chung, W.K.; Kass, R.S. The Impact of Heterozygous KCNK3 Mutations Associated With Pulmonary Arterial Hypertension on Channel Function and Pharmacological Recovery. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kindler, C.H.; Yost, S.C.; Gray, A.T. Local Anesthetic Inhibition of Baseline Potassium Channels with Two Pore Domains in Tandem. Anesthesiology 1999, 90, 1092–1102. [Google Scholar] [CrossRef]
- Berg, A.P.; Talley, E.M.; Manger, J.P.; Bayliss, D.A. Motoneurons Express Heteromeric TWIK-Related Acid-Sensitive K+ (TASK) Channels Containing TASK-1 (KCNK3) and TASK-3 (KCNK9) Subunits. J. Neurosci. 2004, 24, 6693–6702. [Google Scholar] [CrossRef]
- Veale, E.L.; Buswell, R.; Clarke, C.E.; Mathie, A. Identification of a region in the TASK3 two pore domain potassium channel that is critical for its blockade by methanandamide. Br. J. Pharmacol. 2007, 152, 778–786. [Google Scholar] [CrossRef]
- Cotten, J.F. TASK-1 (KCNK3) and TASK-3 (KCNK9) Tandem Pore Potassium Channel Antagonists Stimulate Breathing in Isoflurane-Anesthetized Rats. Anesth. Analg. 2013, 116, 810–816. [Google Scholar] [CrossRef]
- Cotten, J.F.; Keshavaprasad, B.; Laster, M.J.; Eger, E.I.; Yost, C.S. The Ventilatory Stimulant Doxapram Inhibits TASK Tandem Pore (K2P) Potassium Channel Function but Does Not Affect Minimum Alveolar Anesthetic Concentration. Anesth. Analg. 2006, 102, 779–785. [Google Scholar] [CrossRef]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Péchoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Gurney, A.M.; Osipenko, O.N.; Macmillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E.J. Two-Pore Domain K Channel, TASK-1, in Pulmonary Artery Smooth Muscle Cells. Circ. Res. 2003, 93, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Czirják, G.; Enyedi, P. TASK-3 Dominates the Background Potassium Conductance in Rat Adrenal Glomerulosa Cells. Mol. Endocrinol. 2002, 16, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Li, Y.; Tang, B.; Hanze, J.; Eul, B.; Bohle, R.M.; Wilhelm, J.; Morty, R.E.; Brau, M.E.; Weir, E.K.; et al. Impact of TASK-1 in Human Pulmonary Artery Smooth Muscle Cells. Circ. Res. 2006, 98, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Talley, E.M.; Lei, Q.; Sirois, J.E.; Bayliss, D.A. TASK-1, a Two-Pore Domain K+ Channel, Is Modulated by Multiple Neurotransmitters in Motoneurons. Neuron 2000, 25, 399–410. [Google Scholar] [CrossRef]
- Tang, B.; Li, Y.; Nagaraj, C.; Morty, R.E.; Gabor, S.; Stacher, E.; Voswinckel, R.; Weissmann, N.; Leithner, K.; Olschewski, H.; et al. Endothelin-1 Inhibits Background Two-Pore Domain Channel TASK-1 in Primary Human Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2009, 41, 476–483. [Google Scholar] [CrossRef]
- Czirják, G.; Petheő, G.L.; Spät, A.; Enyedi, P. Inhibition of TASK-1 potassium channel by phospholipase C. Am. J. Physiol.-Cell Physiol. 2001, 281, C700–C708. [Google Scholar] [CrossRef]
- Lopes, C.M.B.; Rohacs, T.; Czirják, G.; Ballac, T.; Enyedi, P.; Logothetis, D.E. PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J. Physiol. 2005, 564, 117–129. [Google Scholar] [CrossRef]
- Wilke, B.U.; Lindner, M.; Greifenberg, L.; Albus, A.; Kronimus, Y.; Bünemann, M.; Leitner, M.G.; Oliver, D. Diacylglycerol mediates regulation of TASK potassium channels by Gq-coupled receptors. Nat. Commun. 2014, 5, 5540. [Google Scholar] [CrossRef]
- Seyler, C.; Duthil-Straub, E.; Zitron, E.; Gierten, J.; Scholz, E.P.; Fink, R.H.A.; Karle, C.A.; Becker, R.; Katus, H.A.; Thomas, D. TASK1 (K2P3.1) K+ channel inhibition by endothelin-1 is mediated through Rho kinase-dependent phosphorylation. Br. J. Pharmacol. 2012, 165, 1467–1475. [Google Scholar] [CrossRef]
- Toyoda, H.; Saito, M.; Okazawa, M.; Hirao, K.; Sato, H.; Abe, H.; Takada, K.; Funabiki, K.; Takada, M.; Kaneko, T.; et al. Protein Kinase G Dynamically Modulates TASK1-Mediated Leak K+ Currents in Cholinergic Neurons of the Basal Forebrain. J. Neurosci. 2010, 30, 5677–5689. [Google Scholar] [CrossRef]
- Nagaraj, C.; Tang, B.; Bálint, Z.; Wygrecka, M.; Hrzenjak, A.; Kwapiszewska, G.; Stacher, E.; Lindenmann, J.; Weir, E.K.; Olschewski, H.; et al. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur. Respir. J. 2013, 41, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Weatherald, J.; Chaumais, M.-C.; Savale, L.; Jaïs, X.; Seferian, A.; Canuet, M.; Bouvaist, H.; Magro, P.; Bergeron, A.; Guignabert, C.; et al. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: A population-based study. Eur. Respir. J. 2017, 50, 1700217. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Mercier, O.; Humbert, M.; Sabourin, J. Excitation-contraction coupling and relaxation alteration in right ventricular remodelling caused by pulmonary arterial hypertension. Arch. Cardiovasc. Dis. 2020, 113, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Lamalle, C.; Oliveira, R.; Reid, J.; Gurney, A.M. Contractile and electrophysiological properties of pulmonary artery smooth muscle are not altered in TASK-1 knockout mice. J. Physiol. 2011, 589, 3231–3246. [Google Scholar] [CrossRef] [PubMed]
- Enyedi, P.; Czirják, G. Molecular Background of Leak K+ Currents: Two-Pore Domain Potassium Channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar] [CrossRef]
- Lambert, M.; Boet, A.; Rucker-Martin, C.; Mendes-Ferreira, P.; Capuano, V.; Hatem, S.; Adão, R.; Brás-Silva, C.; Hautefort, A.; Michel, J.-B.; et al. Loss of KCNK3 is a hallmark of RV hypertrophy/dysfunction associated with pulmonary hypertension. Cardiovasc. Res. 2018, 114, 880–893. [Google Scholar] [CrossRef]
- Schmidt, C.; Wiedmann, F.; Voigt, N.; Zhou, X.-B.; Heijman, J.; Lang, S.; Albert, V.; Kallenberger, S.; Ruhparwar, A.; Szabo, G.; et al. Upregulation of K2P3.1 K+ Current Causes Action Potential Shortening in Patients With Chronic Atrial Fibrillation. Circulation 2015, 132, 82–92. [Google Scholar] [CrossRef]
- Chai, S.; Wan, X.; Nassal, D.M.; Liu, H.; Moravec, C.S.; Ramirez-Navarro, A.; Deschênes, I. Contribution of two-pore K+ channels to cardiac ventricular action potential revealed using human iPSC-derived cardiomyocytes. Am. J. Physiol.-Heart Circ. Physiol. 2017, 312, H1144–H1153. [Google Scholar] [CrossRef]
- Rottlaender, D.; Motloch, L.J.; Schmidt, D.; Reda, S.; Larbig, R.; Wolny, M.; Dumitrescu, D.; Rosenkranz, S.; Erdmann, E.; Hoppe, U.C. Clinical Impact of Atrial Fibrillation in Patients with Pulmonary Hypertension. PLoS ONE 2012, 7, e33902. [Google Scholar] [CrossRef]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.-A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef]
- Navas Tejedor, P.; Tenorio Castaño, J.; Palomino Doza, J.; Arias Lajara, P.; Gordo Trujillo, G.; López Meseguer, M.; Román Broto, A.; Lapunzina, P.; Escribano Subías, P. An homozygous mutation in KCNK3 is associated with an aggressive form of hereditary pulmonary arterial hypertension. Clin. Genet. 2016, 91, 453–457. [Google Scholar] [CrossRef]
- Best, D.H.; Sumner, K.L.; Smith, B.P.; Damjanovich-Colmenares, K.; Nakayama, I.; Brown, L.M.; Ha, Y.; Paul, E.; Morris, A.; Jama, M.A.; et al. EIF2AK4 Mutations in Patients Diagnosed with Pulmonary Arterial Hypertension. Chest 2017, 151, 821–828. [Google Scholar] [CrossRef]
- Zhang, H.-S.; Liu, Q.; Piao, C.-M.; Zhu, Y.; Li, Q.-Q.; Du, J.; Gu, H. Genotypes and Phenotypes of Chinese Pediatric Patients With Idiopathic and Heritable Pulmonary Arterial Hypertension-A Single-Center Study. Can. J. Cardiol. 2019, 35, 1851–1856. [Google Scholar] [CrossRef] [PubMed]
- Haarman, M.G.; Kerstjens-Frederikse, W.S.; Vissia-Kazemier, T.R.; Breeman, K.T.N.; Timens, W.; Vos, Y.J.; Roofthooft, M.T.R.; Hillege, H.L.; Berger, R.M.F. The Genetic Epidemiology of Pediatric Pulmonary Arterial Hypertension. J. Pediatr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Higasa, K.; Ogawa, A.; Terao, C.; Shimizu, M.; Kosugi, S.; Yamada, R.; Date, H.; Matsubara, H.; Matsuda, F. A burden of rare variants in BMPR2 and KCNK3 contributes to a risk of familial pulmonary arterial hypertension. BMC Pulm. Med. 2017, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galié, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Cunningham, K.P.; Holden, R.G.; Escribano-Subias, P.M.; Cogolludo, Á.; Veale, E.L.; Mathie, A. Characterization and regulation of wild-type and mutant TASK-1 two pore domain potassium channels indicated in pulmonary arterial hypertension. J. Physiol. 2019, 597, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- US Patent for Method of Treating a Condition Associated with Phosphorylation of TASK-1 Patent (Patent # 8,097,650 Issued 17 January 2012)—Justia Patents Search. Available online: https://patents.justia.com/patent/8097650 (accessed on 22 May 2020).
- Kitagawa, M.G.; Reynolds, J.O.; Wehrens, X.H.T.; Bryan, R.M.J.; Pandit, L.M. Hemodynamic and Pathologic Characterization of the TASK-1−/− Mouse Does Not Demonstrate Pulmonary Hypertension. Front. Med. 2017, 4, 177. [Google Scholar] [CrossRef]
- Anandharajan, R.; Eric, A.; Harikrishna, T.; Ling, Y.; Gladson, M.; Christy, M.; Sheila, S.; Tom, B.; Santhi, G.; Taylor, S.; et al. Abstract 16143: Inflammation Triggers the Onset of Hereditary Pulmonary Arterial Hypertension in Kcnk3−/− Animals. Circulation 2019, 140, A16143. [Google Scholar] [CrossRef]
- Hautefort, A.; Mendes-Ferreira, P.; Sabourin, J.; Manaud, G.; Bertero, T.; Rücker-Martin, C.; Riou, M.; Adão, R.; Manoury, B.; Lambert, M.; et al. Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension. Circulation 2019, 139, 932–948. [Google Scholar] [CrossRef]
- Han, L.; Song, N.; Hu, X.; Zhu, A.; Wei, X.; Liu, J.; Yuan, S.; Mao, W.; Chen, X. Inhibition of RELM-β prevents hypoxia-induced overproliferation of human pulmonary artery smooth muscle cells by reversing PLC-mediated KCNK3 decline. Life Sci. 2020, 246, 117419. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Teng, X.; Hassoun, P.M.; Yang, S.C.; Champion, H.C.; Tuder, R.M.; Johns, R.A. Resistin-Like Molecule-β in Scleroderma-Associated Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2009, 41, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Aittoniemi, J.; Fotinou, C.; Craig, T.J.; De Wet, H.; Proks, P.; Ashcroft, F.M. SUR1: A unique ATP-binding cassette protein that functions as an ion channel regulator. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.K.; Chen, J.; MacKinnon, R. Molecular structure of human KATP in complex with ATP and ADP. eLife 2017, 6, e32481. [Google Scholar] [CrossRef]
- Gribble, F.M.; Ashfield, R.; Ammala, C.; Ashcroft, F.M. Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J. Physiol. 1997, 498, 87–98. [Google Scholar] [CrossRef]
- McClenaghan, C.; Woo, K.V.; Nichols, C.G. Pulmonary Hypertension and ATP-Sensitive Potassium Channels. Hypertension 2019, 74, 14–22. [Google Scholar] [CrossRef]
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef]
- Foster, M.N.; Coetzee, W.A. KATP Channels in the Cardiovascular System. Physiol. Rev. 2016, 96, 177–252. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Sulphonylurea action revisited: The post-cloning era. Diabetologia 2003, 46, 875–891. [Google Scholar] [CrossRef]
- Rorsman, P.; Eliasson, L.; Kanno, T.; Zhang, Q.; Göpel, S.O. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog. Biophys. Mol. Biol. 2011, 107, 224–235. [Google Scholar] [CrossRef]
- Prost, A.-L.; Bloc, A.; Hussy, N.; Dérand, R.; Vivaudou, M.B. Zinc is both an intracellular and extracellular regulator of KATP channel function. J. Physiol. 2004, 559, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Fedinec, A.L.; Liu, J.; Weiss, M.A.; Pourcyrous, M.; Harsono, M.; Parfenova, H.; Leffler, C.W. H2S mediates the vasodilator effect of endothelin-1 in the cerebral circulation. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H1759–H1764. [Google Scholar] [CrossRef] [PubMed]
- Thorneloe, K.S.; Maruyama, Y.; Malcolm, A.T.; Light, P.E.; Walsh, M.P.; Cole, W.C. Protein kinase C modulation of recombinant ATP-sensitive K+ channels composed of Kir6.1 and/or Kir6.2 expressed with SUR2B. J. Physiol. 2002, 541, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Hayabuchi, Y.; Davies, N.W.; Standen, N.B. Angiotensin II inhibits rat arterial KATP channels by inhibiting steady-state protein kinase A activity and activating protein kinase Cε. J. Physiol. 2001, 530, 193–205. [Google Scholar] [CrossRef]
- Tykocki, N.R.; Boerman, E.M.; Jackson, W.F. Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr. Physiol. 2017, 7, 485–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, C.; Zhang, T.; Ge, Y.; Han, X.; Sun, S.; Ding, J.; Lu, M.; Hu, G. Deletion of Kir6.2/SUR1 potassium channels rescues diminishing of DA neurons via decreasing iron accumulation in PD. Mol. Cell. Neurosci. 2018, 92, 164–176. [Google Scholar] [CrossRef]
- Lefer, D.J.; Nichols, C.G.; Coetzee, W.A. Sulfonylurea Receptor 1 Subunits of ATP-Sensitive Potassium Channels and Myocardial Ischemia/Reperfusion Injury. Trends Cardiovasc. Med. 2009, 19, 61–67. [Google Scholar] [CrossRef]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom. Precis. Med. 2018, 11. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; Boët, A.; Lambert, M.; Chung, W.K.; Montani, D.; Humbert, M.; Antigny, F. Sur1/kir6.2 Potassium Channel a New Actor Involved in Pulmonary Arterial Hypertension. Circulation 2019, 140, A10804. [Google Scholar] [CrossRef]
- Kurland, D.B.; Gerzanich, V.; Karimy, J.K.; Woo, S.K.; Vennekens, R.; Freichel, M.; Nilius, B.; Bryan, J.; Simard, J.M. The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J. Neuroinflamm. 2016, 13, 130. [Google Scholar] [CrossRef]
- Yang, X.-R.; Lin, M.-J.; McIntosh, L.S.; Sham, J.S.K. Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L1267–L1276. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Waldron, B.J.; Brayden, J.E. Critical Role for Transient Receptor Potential Channel TRPM4 in Myogenic Constriction of Cerebral Arteries. Circ. Res. 2004, 95, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Blödow, A.; Begandt, D.; Bader, A.; Becker, A.; Burghard, A.; Kühne, D.; Kral, A.; Ngezahayo, A. ATP-sensitive K+ channels (Kir6.1/SUR1) regulate gap junctional coupling in cochlear-supporting cells. Pflügers Arch.-Eur. J. Physiol. 2016, 468, 1215–1222. [Google Scholar] [CrossRef]
- Soundarapandian, M.M.; Wu, D.; Zhong, X.; Petralia, R.S.; Peng, L.; Tu, W.; Lu, Y. Expression of functional Kir6.1 channels regulates glutamate release at CA3 synapses in generation of epileptic form of seizures. J. Neurochem. 2007, 103, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Dart, C.; Standen, N.B. Activation of ATP-dependent K+ channels by hypoxia in smooth muscle cells isolated from the pig coronary artery. J. Physiol. 1995, 483, 29–39. [Google Scholar] [CrossRef]
- Quayle, J.M.; Turner, M.R.; Burrell, H.E.; Kamishima, T. Effects of hypoxia, anoxia, and metabolic inhibitors on KATP channels in rat femoral artery myocytes. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H71–H80. [Google Scholar] [CrossRef][Green Version]
- Nichols, C.G. ATP-sensitive Potassium Currents in Heart Disease and Cardioprotection. Card. Electrophysiol. Clin. 2016, 8, 323–335. [Google Scholar] [CrossRef]
- Fedorov, V.V.; Glukhov, A.V.; Ambrosi, C.M.; Kostecki, G.; Chang, R.; Janks, D.; Schuessler, R.B.; Moazami, N.; Nichols, C.G.; Efimov, I.R. Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts. J. Mol. Cell. Cardiol. 2011, 51, 215–225. [Google Scholar] [CrossRef]
- Movahed, M.-R.; Hashemzadeh, M.; Jamal, M.M. The prevalence of pulmonary embolism and pulmonary hypertension in patients with type II diabetes mellitus. Chest 2005, 128, 3568–3571. [Google Scholar] [CrossRef]
- Lang, I.M.; Palazzini, M. The burden of comorbidities in pulmonary arterial hypertension. Eur. Heart J. Suppl. 2019, 21, K21–K28. [Google Scholar] [CrossRef]
- Abernethy, A.D.; Stackhouse, K.; Hart, S.; Devendra, G.; Bashore, T.M.; Dweik, R.; Krasuski, R.A. Impact of diabetes in patients with pulmonary hypertension. Pulm. Circ. 2015, 5, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Puljung, M.C.; Vedovato, N. Neonatal Diabetes and the KATP Channel: From Mutation to Therapy. Trends Endocrinol. Metab. 2017, 28, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Galcheva, S.; Demirbilek, H.; Al-Khawaga, S.; Hussain, K. The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism. Front. Endocrinol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Seino, S.; Iwanaga, T.; Nagashima, K.; Miki, T. Diverse roles of K(ATP) channels learned from Kir6.2 genetically engineered mice. Diabetes 2000, 49, 311–318. [Google Scholar] [CrossRef]
- Zingman, L.V.; Hodgson, D.M.; Bast, P.H.; Kane, G.C.; Perez-Terzic, C.; Gumina, R.J.; Pucar, D.; Bienengraeber, M.; Dzeja, P.P.; Miki, T.; et al. Kir6.2 is required for adaptation to stress. Proc. Natl. Acad. Sci. USA 2002, 99, 13278–13283. [Google Scholar] [CrossRef]
- Yamada, S.; Kane, G.C.; Behfar, A.; Liu, X.-K.; Dyer, R.B.; Faustino, R.S.; Miki, T.; Seino, S.; Terzic, A. Protection conferred by myocardial ATP-sensitive K+ channels in pressure overload-induced congestive heart failure revealed in KCNJ11 Kir6.2-null mutant. J. Physiol. 2006, 577, 1053–1065. [Google Scholar] [CrossRef]
- Kane, G.C.; Behfar, A.; Dyer, R.B.; O’Cochlain, D.F.; Liu, X.-K.; Hodgson, D.M.; Reyes, S.; Miki, T.; Seino, S.; Terzic, A. KCNJ11 gene knockout of the Kir6.2 K ATP channel causes maladaptive remodeling and heart failure in hypertension. Hum. Mol. Genet. 2006, 15, 2285–2297. [Google Scholar] [CrossRef]
- Seghers, V.; Nakazaki, M.; DeMayo, F.J.; Aguilar-Bryan, L.; Bryan, J. Sur1Knockout Mice. J. Biol. Chem. 2000, 275, 9270–9277. [Google Scholar] [CrossRef]
- Shiota, C.; Larsson, O.; Shelton, K.D.; Shiota, M.; Efanov, A.M.; Høy, M.; Lindner, J.; Kooptiwut, S.; Juntti-Berggren, L.; Gromada, J.; et al. Sulfonylurea Receptor Type 1 Knock-out Mice Have Intact Feeding-stimulated Insulin Secretion despite Marked Impairment in Their Response to Glucose. J. Biol. Chem. 2002, 277, 37176–37183. [Google Scholar] [CrossRef]
- Melamed-Frank, M.; Terzic, A.; Carrasco, A.J.; Nevo, E.; Avivi, A.; Levy, A.P. Reciprocal regulation of expression of pore-forming KATP channel genes by hypoxia. Mol. Cell. Biochem. 2001, 225, 145–150. [Google Scholar] [CrossRef]
- Yokoshiki, H.; Sunagawa, M.; Seki, T.; Sperelakis, N. ATP-sensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells. Am. J. Physiol.-Cell Content 1998, 274, C25–C37. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Tran, S.; Tinker, A.; Clapp, L.H. The Molecular Composition of KATP Channels in Human Pulmonary Artery Smooth Muscle Cells and Their Modulation by Growth. Am. J. Respir. Cell Mol. Biol. 2002, 26, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.C.; Malcolm, T.; Walsh, M.P.; Light, P.E. Inhibition by Protein Kinase C of the KNDP Subtype of Vascular Smooth Muscle ATP-Sensitive Potassium Channel. Circ. Res. 2000, 87, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Mehrke, G.; Pohl, U.; Daut, J. Effects of vasoactive agonists on the membrane potential of cultured bovine aortic and guinea-pig coronary endothelium. J. Physiol. 1991, 439, 277–299. [Google Scholar] [CrossRef]
- Yoshida, H.; Feig, J.E.; Morrissey, A.; Ghiu, I.A.; Artman, M.; Coetzee, W.A. KATP channels of primary human coronary artery endothelial cells consist of a heteromultimeric complex of Kir6.1, Kir6.2, and SUR2B subunits. J. Mol. Cell. Cardiol. 2004, 37, 857–869. [Google Scholar] [CrossRef]
- Stoller, D.A.; Fahrenbach, J.P.; Chalupský, K.; Tan, B.-H.; Aggarwal, N.; Metcalfe, J.; Hadhazy, M.; Shi, N.-Q.; Makielski, J.C.; McNally, E.M. Cardiomyocyte sulfonylurea receptor 2-KATP channel mediates cardioprotection and ST segment elevation. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H1100–H1108. [Google Scholar] [CrossRef]
- Harakalova, M.; Van Harssel, J.J.T.; Terhal, P.A.; Van Lieshout, S.; Duran, K.; Renkens, I.; Amor, D.J.; Wilson, L.C.; Kirk, E.P.; Turner, C.L.S.; et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat. Genet. 2012, 44, 793–796. [Google Scholar] [CrossRef]
- Ma, A.; Gurnasinghani, S.; Kirk, E.P.; McClenaghan, C.; Singh, G.K.; Grange, D.K.; Pandit, C.; Zhu, Y.; Roscioli, T.; Elakis, G.; et al. Glibenclamide treatment in a Cantú syndrome patient with a pathogenic ABCC9 gain-of-function variant: Initial experience. Am. J. Med. Genet. Part A 2019, 179, 1585–1590. [Google Scholar] [CrossRef]
- Park, J.Y.; Koo, S.H.; Jung, Y.J.; Lim, Y.-J.; Chung, M.L. A patient with Cantú syndrome associated with fatal bronchopulmonary dysplasia and pulmonary hypertension. Am. J. Med. Genet. Part A 2014, 164, 2118–2120. [Google Scholar] [CrossRef]
- Kobayashi, D.; Cook, A.L.; Williams, D.A. Pulmonary hypertension secondary to partial pulmonary venous obstruction in a child with Cantu syndrome. Pediatr. Pulmonol. 2010, 45, 727–729. [Google Scholar] [CrossRef]
- McClenaghan, C.; Hanson, A.; Sala-Rabanal, M.; Roessler, H.I.; Josifova, D.; Grange, D.K.; Van Haaften, G.; Nichols, C.G. Cantu syndrome–associated SUR2 (ABCC9) mutations in distinct structural domains result in KATP channel gain-of-function by differential mechanisms. J. Biol. Chem. 2018, 293, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; McClenaghan, C.; Harter, T.M.; Hinman, K.; Halabi, C.M.; Matkovich, S.J.; Zhang, H.; Brown, G.S.; Mecham, R.P.; England, S.K.; et al. Cardiovascular consequences of KATP overactivity in Cantu syndrome. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.; Pardo, L.A.; Dominguez, P.; Sierra, L.M.; De La Peña, P. New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question. Int. J. Mol. Sci. 2019, 20, 248. [Google Scholar] [CrossRef] [PubMed]
- Wipff, J.; Dieude, P.; Guedj, M.; Ruiz, B.; Riemekasten, G.; Cracowski, J.L.; Matucci-Cerinic, M.; Melchers, I.; Humbert, M.; Hachulla, E.; et al. Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthritis Rheum. 2010, 62, 3093–3100. [Google Scholar] [CrossRef] [PubMed]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations ofKCNA5in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol.-Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef]
- Jiao, Y.-R.; Wang, W.; Lei, P.-C.; Jia, H.-P.; Dong, J.; Gou, Y.-Q.; Chen, C.-L.; Cao, J.; Wang, Y.-F.; Zhu, Y. 5-HTT, BMPR2, EDN1, ENG, KCNA5 gene polymorphisms and susceptibility to pulmonary arterial hypertension: A meta-analysis. Gene 2019, 680, 34–42. [Google Scholar] [CrossRef]
- Yuan, X.-J. Voltage-Gated K+ Currents Regulate Resting Membrane Potential and [Ca2+]i in Pulmonary Arterial Myocytes. Circ. Res. 1995, 77, 370–378. [Google Scholar] [CrossRef]
- Platoshyn, O.; Golovina, V.A.; Bailey, C.L.; Limsuwan, A.; Krick, S.; Juhaszova, M.; Seiden, J.E.; Rubin, L.J.; Yuan, J.X.-J. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am. J. Physiol. Physiol. 2000, 279, C1540–C1549. [Google Scholar] [CrossRef]
- Boucherat, O.; Chabot, S.; Antigny, F.; Perros, F.; Provencher, S.; Bonnet, S. Potassium channels in pulmonary arterial hypertension. Eur. Respir. J. 2015, 46, 1167–1177. [Google Scholar] [CrossRef]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade -Navarro, M.A.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An Abnormal Mitochondrial–Hypoxia Inducible Factor-1α–Kv Channel Pathway Disrupts Oxygen Sensing and Triggers Pulmonary Arterial Hypertension in Fawn Hooded Rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef]
- Rao, J.N.; Platoshyn, O.; Li, L.; Guo, X.; Golovina, V.A.; Yuan, J.X.-J.; Wang, J.-Y. Activation of K+ channels and increased migration of differentiated intestinal epithelial cells after wounding. Am. J. Physiol.-Cell Physiol. 2002, 282, C885–C898. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Wu, X.-C.; Thébaud, B.; Nsair, A.; Bonnet, S.; Tyrrell, B.; McMurtry, M.S.; Hashimoto, K.; Harry, G.; Michelakis, E.D. Preferential Expression and Function of Voltage-Gated, O2-Sensitive K+ Channels in Resistance Pulmonary Arteries Explains Regional Heterogeneity in Hypoxic Pulmonary Vasoconstriction: Ionic diversity in smooth muscle cells. Circ. Res. 2004, 95, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; London, B.; Hampl, V.; Wu, X.; Nsair, A.; Puttagunta, L.; Hashimoto, K.; Waite, R.E.; Michelakis, E.D. Impairment of hypoxic pulmonary vasoconstriction in mice lacking the voltage-gated potassium channel Kv1. FASEB J. 2001, 15, 1801–1803. [Google Scholar] [CrossRef]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J. Clin. Investig. 1998, 101, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Powell, F.; Haddad, G.H.; Yuan, J.X.-J. Hypoxia Selectively Inhibits KCNA5 Channels in Pulmonary Artery Smooth Muscle Cells. Ann. N. Y. Acad. Sci. 2009, 1177, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-J.; Wang, J.; Juhaszova, M.; Gaine, S.P.; Rubin, L.J. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 1998, 351, 726–727. [Google Scholar] [CrossRef]
- Young, K.A.; Ivester, C.; West, J.; Carr, M.; Rodman, D.M. BMP signaling controls PASMC KV channel expression in vitro and in vivo. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L841–L848. [Google Scholar] [CrossRef]
- Brevnova, E.E.; Platoshyn, O.; Zhang, S.; Yuan, J.X.-J. Overexpression of human KCNA5 increases IK(V) and enhances apoptosis. Am. J. Physiol.-Cell Physiol. 2004, 287, C715–C722. [Google Scholar] [CrossRef]
- Nesti, E.; Everill, B.; Morielli, A.D. Endocytosis as a Mechanism for Tyrosine Kinase-dependent Suppression of a Voltage-gated Potassium Channel. Mol. Biol. Cell 2004, 15, 4073–4088. [Google Scholar] [CrossRef]
- Hogg, D.S.; McMurray, G.; Kozlowski, R.Z. Endothelial Cells Freshly Isolated from Small Pulmonary Arteries of the Rat Possess Multiple Distinct K+ Current Profiles. Lung 2002, 180, 203–214. [Google Scholar] [CrossRef]
- Fedida, D.; Eldstrom, J.; Hesketh, J.C.; Lamorgese, M.; Castel, L.; Steele, D.F.; Van Wagoner, D.R. Kv1.5 Is an Important Component of Repolarizing K+ Current in Canine Atrial Myocytes. Circ. Res. 2003, 93, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Jeevaratnam, K.; Chadda, K.R.; Huang, C.L.-H.; Camm, A.J. Cardiac Potassium Channels: Physiological Insights for Targeted Therapy. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.V.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- London, B.; Guo, W.; Pan, X.-H.; Lee, J.S.; Shusterman, V.; Rocco, C.J.; Logothetis, D.A.; Nerbonne, J.M.; Hill, J.A. Targeted Replacement of Kv1.5 in the Mouse Leads to Loss of the 4-Aminopyridine–Sensitive Component of IK,slow and Resistance to Drug-Induced QT Prolongation. Circ. Res. 2001, 88, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Demirbilek, H.; Hussain, K. Congenital Hyperinsulinism: Diagnosis and Treatment Update. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Galcheva, S.V.; Al-Khawaga, S.; Hussain, K. Diagnosis and management of hyperinsulinaemic hypoglycaemia. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 551–573. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.S.; McLay, J.; Kenmure, A.C. Reversibility of primary pulmonary hypertension during six years of treatment with oral diazoxide. Heart 1987, 57, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Klinke, W.P.; Gilbert, J.A.L. Diazoxide in Primary Pulmonary Hypertension. N. Engl. J. Med. 1980, 302, 91–92. [Google Scholar] [CrossRef]
- Chen, S.C.; Dastamani, A.; Pintus, D.; Yau, D.; Aftab, S.; Bath, L.; Swinburne, C.; Hunter, L.; Giardini, A.; Christov, G.; et al. Diazoxide-induced pulmonary hypertension in hyperinsulinaemic hypoglycaemia: Recommendations from a multicentre study in the United Kingdom. Clin. Endocrinol. 2019, 91, 770–775. [Google Scholar] [CrossRef]
- Kylat, R. Pulmonary hypertension occurring with diazoxide use in a preterm infant with hypoglycemia. Drug Healthc. Patient Saf. 2019, 11, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Nebesio, T.D.; Hoover, W.C.; Caldwell, R.L.; Nitu, M.E.; Eugster, E.A. Development of Pulmonary Hypertension in an Infant Treated with Diazoxide. J. Pediatr. Endocrinol. Metab. 2007, 20, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Timlin, M.R.; Black, A.B.; Delaney, H.M.; Matos, R.I.; Percival, C.S. Development of Pulmonary Hypertension During Treatment with Diazoxide: A Case Series and Literature Review. Pediatr. Cardiol. 2017, 38, 1247–1250. [Google Scholar] [CrossRef]
- FDA. FDA Drug Safety Communication: FDA Warns about a Serious Lung Condition in Infants and Newborns Treated with Proglycem (Diazoxide); FDA: White Oak, MD, USA, 2019. [Google Scholar]
- Zhu, R.; Bi, L.-Q.; Wu, S.-L.; Li, L.; Kong, H.; Xie, W.-P.; Wang, H.; Meng, Z.-L. Iptakalim attenuates hypoxia-induced pulmonary arterial hypertension in rats by endothelial function protection. Mol. Med. Rep. 2015, 12, 2945–2952. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, Y.; Zhang, S.; Xie, W.; Li, Q.; Zhou, Y.; Wang, H. Iptakalim inhibited endothelin-1-induced proliferation of human pulmonary arterial smooth muscle cells through the activation of KATP channel. Vasc. Pharmacol. 2008, 48, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Y.; Cui, W.-Y.; Wang, H. The new antihypertensive drug iptakalim activates ATP-sensitive potassium channels in the endothelium of resistance blood vessels. Acta Pharmacol. Sin. 2015, 36, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thébaud, B.; Wu, X.-C.; Dyck, J.R.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In Vivo Gene Transfer of the O2-Sensitive Potassium Channel Kv1.5 Reduces Pulmonary Hypertension and Restores Hypoxic Pulmonary Vasoconstriction in Chronically Hypoxic Rats. Circualtion 2003, 107, 2037–2044. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KCNK3 Mutation (AA) | KCNK3 Mutation (Nucleic Acid) | Number of PAH Patients Carrying the Mutation | Number of Healthy Carrier | Zygosity | Function (Patch Clamp) | Function Restored by ONO-RS-082 | References |
|---|---|---|---|---|---|---|---|
| T8K | 1 | Heterozygous | loss | yes | [50] | ||
| G97R | 289 G > A | 2 | 1 | Heterozygous | loss | / | [50] |
| G106R | 316 G > C | 2 | 1 | Heterozygous and homozygous | loss | no | [51] |
| A114V | 341C > T | 1 | Heterozygous | / | / | [53] | |
| K145M | 434 A > T | 1 | Heterozygous | / | / | [52] | |
| E182K | 1 | Heterozygous | loss | yes | [50] | ||
| A189T | 565 G > A | 1 | Heterozygous | / | / | [52] | |
| Y192C | 1 | Heterozygous | loss | / | [50] | ||
| G203D | 608 G > A | 6 | 1 | Heterozygous | loss | no | [50,55] |
| V206L | 616 G > T | 1 | Heterozygous | / | / | [54] | |
| L214R | 641 T > G | 1 | Heterozygous | loss | no | [51] | |
| V221L | 661 G > C | 1 | Heterozygous | loss | / | [50] |
| ABCC8 Mutation (AA) | ABCC8 Mutation (Nucleic Acid) | Number of PAH Patients Carrying the Mutation | Zygosity | Function | Function Restored by Diazoxide | |
|---|---|---|---|---|---|---|
| (Patch Clamp) | (Rubidium (86Rb+) Efflux Assays) | |||||
| N72D | 214 A > G | 1 | Heterozygous | / | / | / |
| G111R | 331 G > A | 1 | Heterozygous | / | / | / |
| L135V | 403 C > G | 1 | Heterozygous | loss | decrease | yes |
| E186D | 558 G > T | 1 | Heterozygous | loss | not decrease | yes |
| T229I | 686 C > T | 1 | Heterozygous | / | / | / |
| A240T | 718 G > A | 1 | Heterozygous | loss | decrease | yes |
| E791Q | 2371 G > C | 1 | Heterozygous | loss | small decrease | yes |
| D813N | 2437 G > A | 1 | Heterozygous | loss | decrease | yes |
| R958H | 2873 G > A | 1 | Heterozygous | not loss | decrease | yes |
| R1314H | 3941 G > A | 1 | Heterozygous | loss | small decrease | yes |
| D1472N | 4414 G > A | 1 | Heterozygous | loss | decrease | yes |
| / | 2694 T > 2G | 1 | Heterozygous | / | / | / |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Ribeuz, H.; Capuano, V.; Girerd, B.; Humbert, M.; Montani, D.; Antigny, F. Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules 2020, 10, 1261. https://doi.org/10.3390/biom10091261
Le Ribeuz H, Capuano V, Girerd B, Humbert M, Montani D, Antigny F. Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules. 2020; 10(9):1261. https://doi.org/10.3390/biom10091261
Chicago/Turabian StyleLe Ribeuz, Hélène, Véronique Capuano, Barbara Girerd, Marc Humbert, David Montani, and Fabrice Antigny. 2020. "Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension" Biomolecules 10, no. 9: 1261. https://doi.org/10.3390/biom10091261
APA StyleLe Ribeuz, H., Capuano, V., Girerd, B., Humbert, M., Montani, D., & Antigny, F. (2020). Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules, 10(9), 1261. https://doi.org/10.3390/biom10091261