Computational Methods and Software Tools for Functional Analysis of miRNA Data



Abstract
1. Introduction
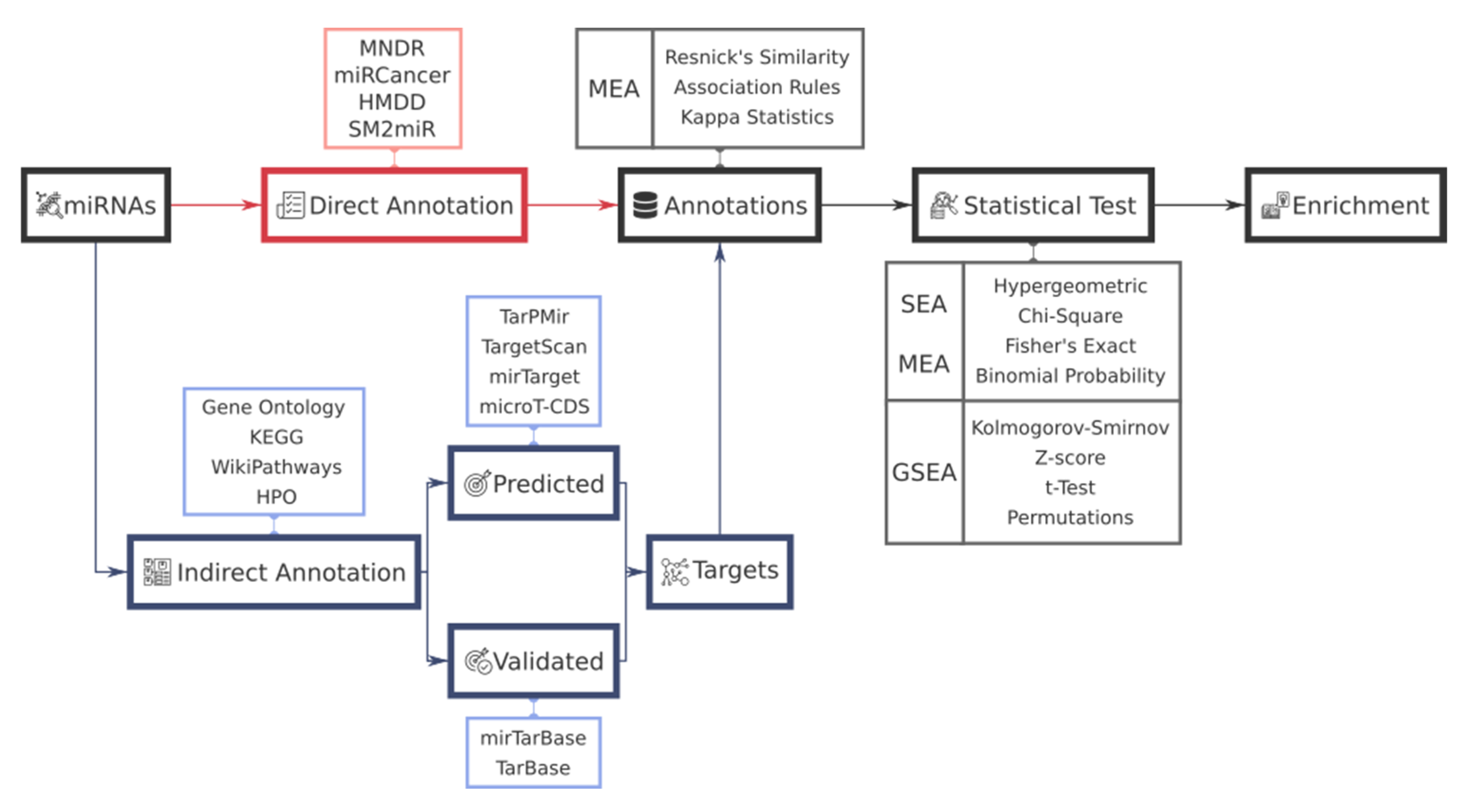
2. Functional Analysis Workflow in miRNAs
3. miRNA-Target Gene Annotation and Resources
3.1. miRNA-Target Gene Prediction Methods
3.2. Validated miRNA-Target Gene Resources
4. Functional Annotation Resources for miRNAs
4.1. miRNA-Based Annotation Resources
4.2. Gene-Based Annotation Resources
5. Tools for miRNA Functional Enrichment Analysis
5.1. MiRNet
5.2. GeneCodis
5.3. MiEAA
5.4. MIENTURNET
5.5. TAM
5.6. MiTALOS
5.7. MiRSystem
5.8. DIANA miRPath
6. Conclusions and Discussion
Supplementary Materials
Funding
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.-P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Backes, C.; Fehlmann, T.; Kern, F.; Kehl, T.; Lenhof, H.-P.; Meese, E.; Keller, A. miRCarta: A central repository for collecting miRNA candidates. Nucleic Acids Res. 2018, 46, D160–D167. [Google Scholar] [CrossRef]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The metazoan microRNA complement. Nucleic Acids Res. 2020, 48, D132–D141. [Google Scholar] [CrossRef] [PubMed]
- Fehlmann, T.; Backes, C.; Kahraman, M.; Haas, J.; Ludwig, N.; Posch, A.E.; Würstle, M.L.; Hübenthal, M.; Franke, A.; Meder, B.; et al. Web-based NGS data analysis using miRMaster: A large-scale meta-analysis of human miRNAs. Nucleic Acids Res. 2017, 45, 8731–8744. [Google Scholar] [CrossRef]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA-cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Zhang, L.; Huang, Y.; Yi, Y.; Tan, P.; Zhao, Y.; Hu, Y.; Xu, L.; Li, E.; Wang, D. MNDR v2.0: An updated resource of ncRNA-disease associations in mammals. Nucleic Acids Res. 2018, 46, D371–D374. [Google Scholar] [CrossRef]
- Guo, Z.; Kuang, Z.; Wang, Y.; Zhao, Y.; Tao, Y.; Cheng, C.; Yang, J.; Lu, X.; Hao, C.; Wang, T.; et al. PmiREN: A comprehensive encyclopedia of plant miRNAs. Nucleic Acids Res. 2020, 48, D1114–D1121. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- Huntley, R.P.; Kramarz, B.; Sawford, T.; Umrao, Z.; Kalea, A.; Acquaah, V.; Martin, M.J.; Mayr, M.; Lovering, R.C. Expanding the horizons of microRNA bioinformatics. RNA 2018, 24, 1005–1017. [Google Scholar] [CrossRef]
- Huntley, R.P.; Sitnikov, D.; Orlic-Milacic, M.; Balakrishnan, R.; D’Eustachio, P.; Gillespie, M.E.; Howe, D.; Kalea, A.Z.; Maegdefessel, L.; Osumi-Sutherland, D.; et al. Guidelines for the functional annotation of microRNAs using the Gene Ontology. RNA 2016, 22, 667–676. [Google Scholar] [CrossRef]
- Ziemann, M.; Kaspi, A.; El-Osta, A. Evaluation of microRNA alignment techniques. RNA 2016, 22, 1120–1138. [Google Scholar] [CrossRef]
- Bisgin, H.; Gong, B.; Wang, Y.; Tong, W. Evaluation of Bioinformatics Approaches for Next-Generation Sequencing Analysis of microRNAs with a Toxicogenomics Study Design. Front. Genet. 2018, 9, 22. [Google Scholar] [CrossRef]
- Bleazard, T.; Lamb, J.A.; Griffiths-Jones, S. Bias in microRNA functional enrichment analysis. Bioinformatics 2015, 31, 1592–1598. [Google Scholar] [CrossRef]
- Godard, P.; van Eyll, J. Pathway analysis from lists of microRNAs: Common pitfalls and alternative strategy. Nucleic Acids Res. 2015, 43, 3490–3497. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Riffo-Campos, Á.L.; Riquelme, I.; Brebi-Mieville, P. Tools for Sequence-Based miRNA Target Prediction: What to Choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of MicroRNA—Target Recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011, 18, 1139–1146. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef]
- Yousef, M.; Jung, S.; Kossenkov, A.V.; Showe, L.C.; Showe, M.K. Naïve Bayes for microRNA target predictions—machine learning for microRNA targets. Bioinforma. Oxf. Engl. 2007, 23, 2987–2992. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Wang, X. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics 2014, 30, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, X.; Hu, H. TarPmiR: A new approach for microRNA target site prediction. Bioinformatics 2016, 32, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013, 41, W169–W173. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Bovolenta, L.A.; Nachtigall, P.G.; Herkenhoff, M.E.; Lemke, N.; Pinhal, D. Combining Results from Distinct MicroRNA Target Prediction Tools Enhances the Performance of Analyses. Front. Genet. 2017, 8, 59. [Google Scholar] [CrossRef]
- Tabas-Madrid, D.; Muniategui, A.; Sánchez-Caballero, I.; Martínez-Herrera, D.J.; Sorzano, C.O.S.; Rubio, A.; Pascual-Montano, A. Improving miRNA-mRNA interaction predictions. BMC Genom. 2014, 15 (Suppl. 10), S2. [Google Scholar] [CrossRef]
- Shirdel, E.A.; Xie, W.; Mak, T.W.; Jurisica, I. NAViGaTing the micronome—Using multiple microRNA prediction databases to identify signalling pathway-associated microRNAs. PLoS ONE 2011, 6, e17429. [Google Scholar] [CrossRef]
- Andrés-León, E.; González Peña, D.; Gómez-López, G.; Pisano, D.G. miRGate: A curated database of human, mouse and rat miRNA-mRNA targets. Database J. Biol. Databases Curation 2015, 2015, bav035. [Google Scholar] [CrossRef]
- Le, T.D.; Zhang, J.; Liu, L.; Li, J. Ensemble Methods for MiRNA Target Prediction from Expression Data. PLoS ONE 2015, 10, e0131627. [Google Scholar] [CrossRef] [PubMed]
- Quillet, A.; Saad, C.; Ferry, G.; Anouar, Y.; Vergne, N.; Lecroq, T.; Dubessy, C. Improving Bioinformatics Prediction of microRNA Targets by Ranks Aggregation. Front. Genet. 2020, 10, 1330. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA—Target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA—Gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef]
- Schriml, L.M.; Mitraka, E.; Munro, J.; Tauber, B.; Schor, M.; Nickle, L.; Felix, V.; Jeng, L.; Bearer, C.; Lichenstein, R.; et al. Human Disease Ontology 2018 update: Classification, content and workflow expansion. Nucleic Acids Res. 2019, 47, D955–D962. [Google Scholar] [CrossRef]
- Ruepp, A.; Kowarsch, A.; Schmidl, D.; Buggenthin, F.; Brauner, B.; Dunger, I.; Fobo, G.; Frishman, G.; Montrone, C.; Theis, F.J. PhenomiR: A knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010, 11, R6. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef]
- Liu, X.; Wang, S.; Meng, F.; Wang, J.; Zhang, Y.; Dai, E.; Yu, X.; Li, X.; Jiang, W. SM2miR: A database of the experimentally validated small molecules’ effects on microRNA expression. Bioinform. Oxf. Engl. 2013, 29, 409–411. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Vooren, S.V.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stöckel, D.; Meese, E.; Lenhof, H.-P.; Keller, A. miRPathDB 2.0: A novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef]
- Ebrahimkhani, S.; Vafaee, F.; Hallal, S.; Wei, H.; Lee, M.Y.T.; Young, P.E.; Satgunaseelan, L.; Beadnall, H.; Barnett, M.H.; Shivalingam, B.; et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis. Oncol. 2018, 2, 28. [Google Scholar] [CrossRef]
- Li, J.; Han, X.; Wan, Y.; Zhang, S.; Zhao, Y.; Fan, R.; Cui, Q.; Zhou, Y. TAM 2.0: Tool for MicroRNA set analysis. Nucleic Acids Res. 2018, 46, W180–W185. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E.; The Mouse Genome Database Group; Anagnostopoulos, A.; Asabor, R.; Baldarelli, R.M.; Beal, J.S.; et al. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef] [PubMed]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A.; et al. LINCS Data Portal 2.0: Next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020, 48, D431–D439. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Russo, F.; Di Bella, S.; Vannini, F.; Berti, G.; Scoyni, F.; Cook, H.V.; Santos, A.; Nigita, G.; Bonnici, V.; Laganà, A.; et al. miRandola 2017: A curated knowledge base of non-invasive biomarkers. Nucleic Acids Res. 2018, 46, D354–D359. [Google Scholar] [CrossRef]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2019, 48, D160–D165. [Google Scholar] [CrossRef]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C.; et al. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 2017, 45, D135–D138. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Tong, Z.; Cui, Q.; Wang, J.; Zhou, Y. TransmiR v2.0: An updated transcription factor-microRNA regulation database. Nucleic Acids Res. 2019, 47, D253–D258. [Google Scholar] [CrossRef]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef] [PubMed]
- Tabas-Madrid, D.; Nogales-Cadenas, R.; Pascual-Montano, A. GeneCodis3: A non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012, 40, W478–W483. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Saez, P.; Chagoyen, M.; Tirado, F.; Carazo, J.M.; Pascual-Montano, A. GENECODIS: A web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 2007, 8, R3. [Google Scholar] [CrossRef]
- Kern, F.; Fehlmann, T.; Solomon, J.; Schwed, L.; Backes, C.; Meese, E.; Keller, A. miEAA 2.0: Integrating multi-species microRNA enrichment analysis and workflow management systems. Nucleic Acids Res. 2020, 48, W521–W528. [Google Scholar] [CrossRef] [PubMed]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef]
- Preusse, M.; Theis, F.J.; Mueller, N.S. miTALOS v2: Analyzing Tissue Specific microRNA Function. PLoS ONE 2016, 11, e0151771. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein—RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef]
- Lu, T.-P.; Lee, C.-Y.; Tsai, M.-H.; Chiu, Y.-C.; Hsiao, C.K.; Lai, L.-C.; Chuang, E.Y. miRSystem: An Integrated System for Characterizing Enriched Functions and Pathways of MicroRNA Targets. PLoS ONE 2012, 7, e42390. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Hosack, D.A.; Dennis, G.; Sherman, B.T.; Lane, H.C.; Lempicki, R.A. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003, 4, R70. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Tool | Learn Attributes Remark | Organisms | URL | Last Up-Date |
|---|---|---|---|---|
| TarPmiR | Novel features from CLASH data | Homo sapiens | http://hulab.ucf.edu/research/projects/miRNA/TarPmiR/ | 2016 |
| TargetScan | Score for mammal predictions | H. sapiens, Mus musculus, Rattus norvegicus, Pan troglodytes, Macaca mulatta, Canis familiaris, M. domestica, Bos taurus, C. elegans, Drosophila melanogaster, Danio rerio, Gallus gallus, Xenopus tropicalis | http://www.targetscan.org | 2015 |
| MiRTarget | Functional targets from RNA-seq | H. sapiens, M. musculus, R. norvegicus, C. familiaris, G. gallus | http://mirdb.org/ | 2019 |
| DIANA microT-CDS | PAR-CLIP data, targets in CDS and 3′ UTR | H. sapiens, M. musculus, C. elegans, D. melanogaster | http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS | 2013 |
| Tool | Curation | Target-miRNA | Organisms | URL | Last Update |
|---|---|---|---|---|---|
| miRTarBase | 11,021 articles, 331 CLIP-seq datasets | 479,340 | 32 | http://mirtarbase.cuhk.edu.cn | 2020 |
| DIANA-TarBase | 1208 articles, 353 datasets, 34 methods | 665,843 | 18 | https://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8 | 2017 |
| Tool | Annotation/Bias Handling | Method | Targets | Sources of Annotations | Organism |
|---|---|---|---|---|---|
| miRNet | Indirect, Direct/Empirical sampling | SEA | Validated, predicted | GO, KEGG, Reactome, TAM [58] | H. sapiens, M. musculus, R. norvegicus, B. taurus, Sus scrofa, G. gallus, D. melanogaster, C. elegans, D. rerio, Schistosoma mansoni |
| GeneCodis | Indirect/Empirical sampling, co-annotation | MEA, SEA | Validated | DoRothEA [59], miRTarBase, GO, KEGG Pathways, MGI [60], Panther, Reactome, WikiPathways, CTD [61], HPO, LINCS [62], OMIM, PharmGKB [63] | D. melanogaster, D. rerio, H. sapiens, M. musculus, R. norvegicus. |
| miEAA | Indirect, Direct/None | SEA, GSEA | Validated, redicted | GO, HMDD, KEGG, miRandola [64], miRBase, miRPathDB, miRTarBase, miRWalk [65], MNDR, NPInter [66], RNALocate [67], SM2miR, TAM, TissueAtlas [68], TransmiR [69], Literature | H. sapiens, M. musculus. R. norvegicus, Arabidopsis thaliana, B. taurus, C. elegans, D. melanogaster, D. rerio, G. gallus, S. scrofa |
| MIENTURNET | Indirect/None | SEA | Validated, predicted | KEGG, Reactome, WikiPathways, Disease Ontology | H sapiens, M. musculus, R. norvegicus, C. elegans, D. melanogaster, D. rerio |
| TAM | Direct/Mask cancer and unspecific terms | SEA | - | Literature | H. sapiens |
| miTALOS | Indirect/Background specificity | SEA | Validated, predicted | KEGG, WikiPathways, Reactome | H. sapiens, M. musculus |
| miRSystem | Indirect/Empirical sampling | SEA | Validated, predicted | KEGG, GO, BioCarta [70], Pathway Interaction Database [71], Reactome. | H. sapiens, M. musculus |
| DIANA miRPath | Indirect/Empirical sampling | SEA | Validated, predicted | GO, KEGG | H. sapiens, M. musculus, R. norvegicus, D. melanogaster, C. elegans, G. gallus, D. rerio. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Moreno, A.; Carmona-Saez, P. Computational Methods and Software Tools for Functional Analysis of miRNA Data. Biomolecules 2020, 10, 1252. https://doi.org/10.3390/biom10091252
Garcia-Moreno A, Carmona-Saez P. Computational Methods and Software Tools for Functional Analysis of miRNA Data. Biomolecules. 2020; 10(9):1252. https://doi.org/10.3390/biom10091252
Chicago/Turabian StyleGarcia-Moreno, Adrian, and Pedro Carmona-Saez. 2020. "Computational Methods and Software Tools for Functional Analysis of miRNA Data" Biomolecules 10, no. 9: 1252. https://doi.org/10.3390/biom10091252
APA StyleGarcia-Moreno, A., & Carmona-Saez, P. (2020). Computational Methods and Software Tools for Functional Analysis of miRNA Data. Biomolecules, 10(9), 1252. https://doi.org/10.3390/biom10091252