Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection


 , , ,
, , ,  and
and 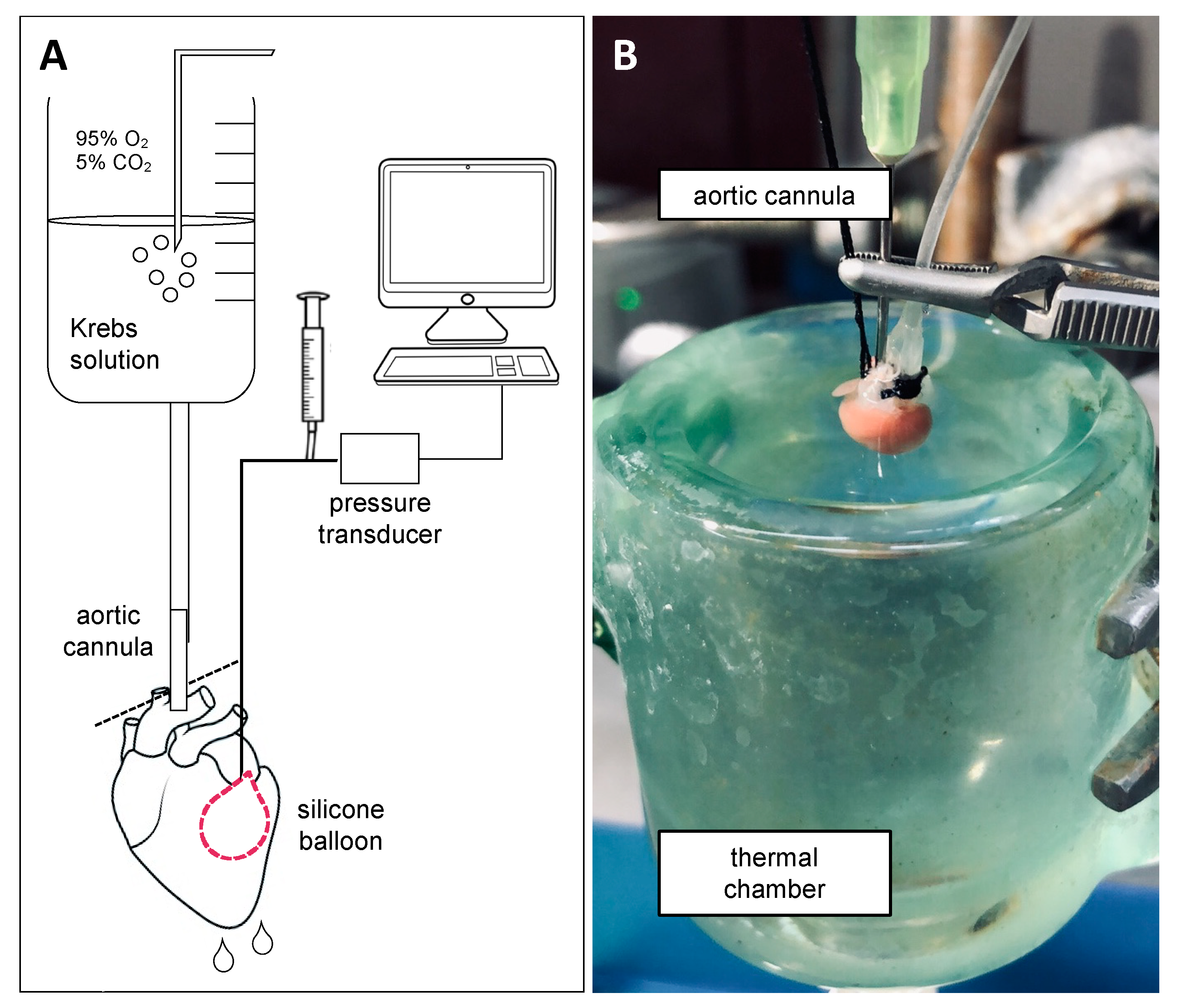{kind=link}
{kind=link}
{kind=link}
{kind=link}
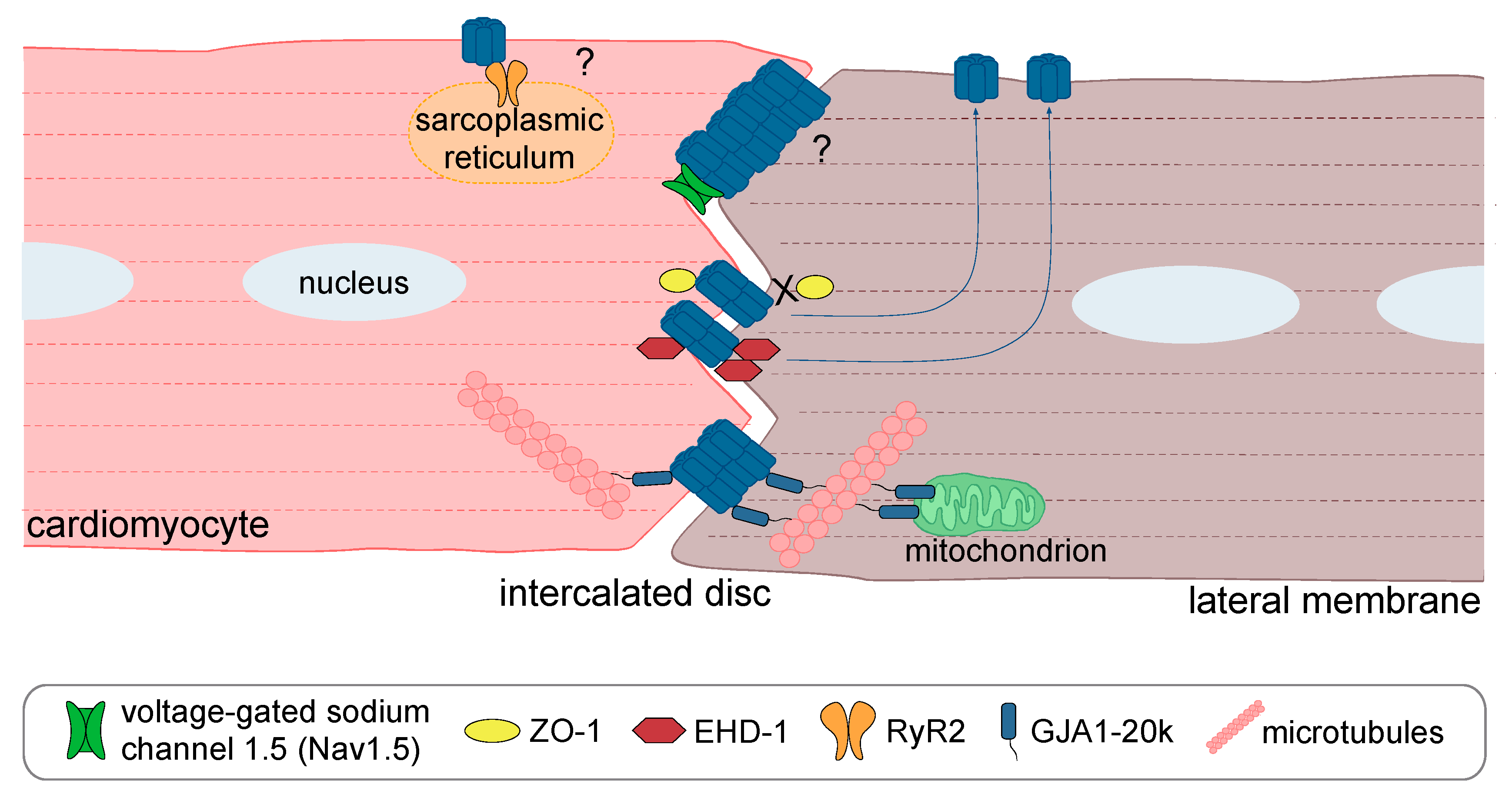{kind=link}
Abstract
1. Introduction
2. Pathophysiology of Cardiac I/R Injury
3. Methods Used to Study Cardiac I/R Injury
3.1. Ex Vivo Perfused Heart as a Model for I/R Injury
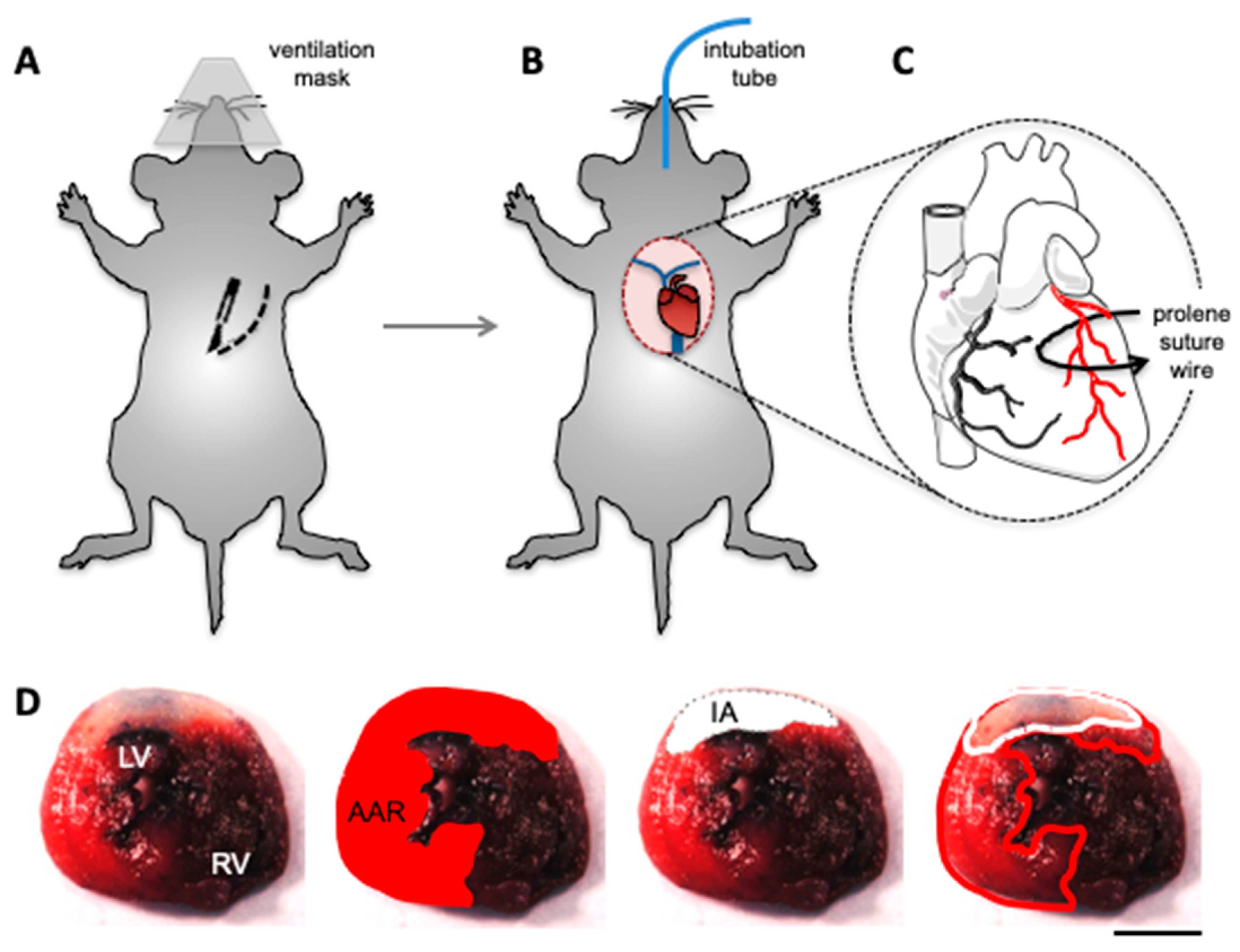
3.2. In Vivo Models of Cardiac I/R Injury
4. Canonical Role of Connexin43 in I/R Injury and Cardioprotection
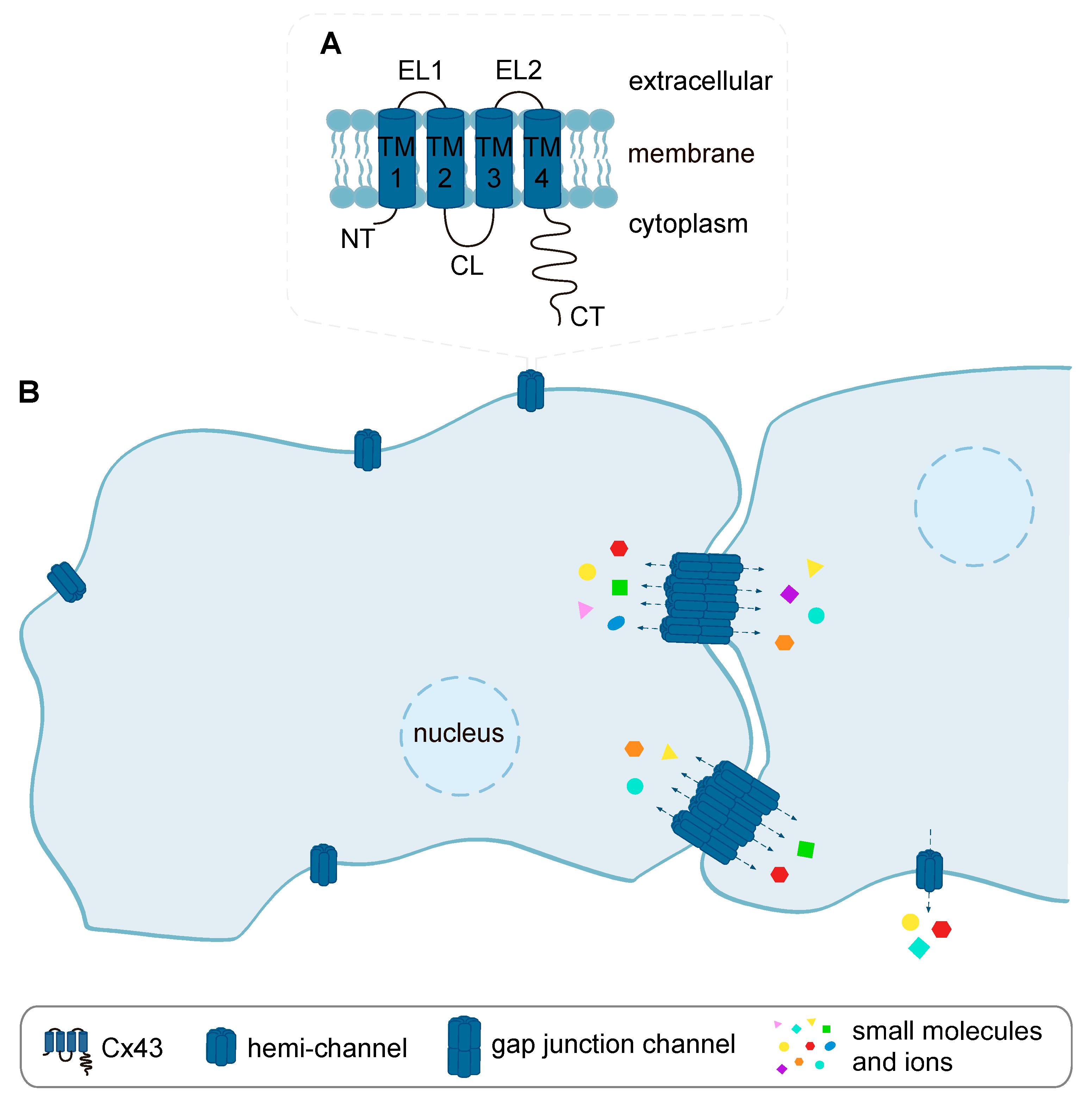
4.1. Cx43 Gap Junction- and Hemi-Channels and Cardiac I/R Injury
4.2. Potential Role of Cx43 in Non-Cardiac Cells of the Heart in I/R Injury
5. Non-Canonical Role of Connexin43 in I/R Injury and Cardioprotection
5.1. Mitochondrial Cx43 and Cardiac I/R Injury
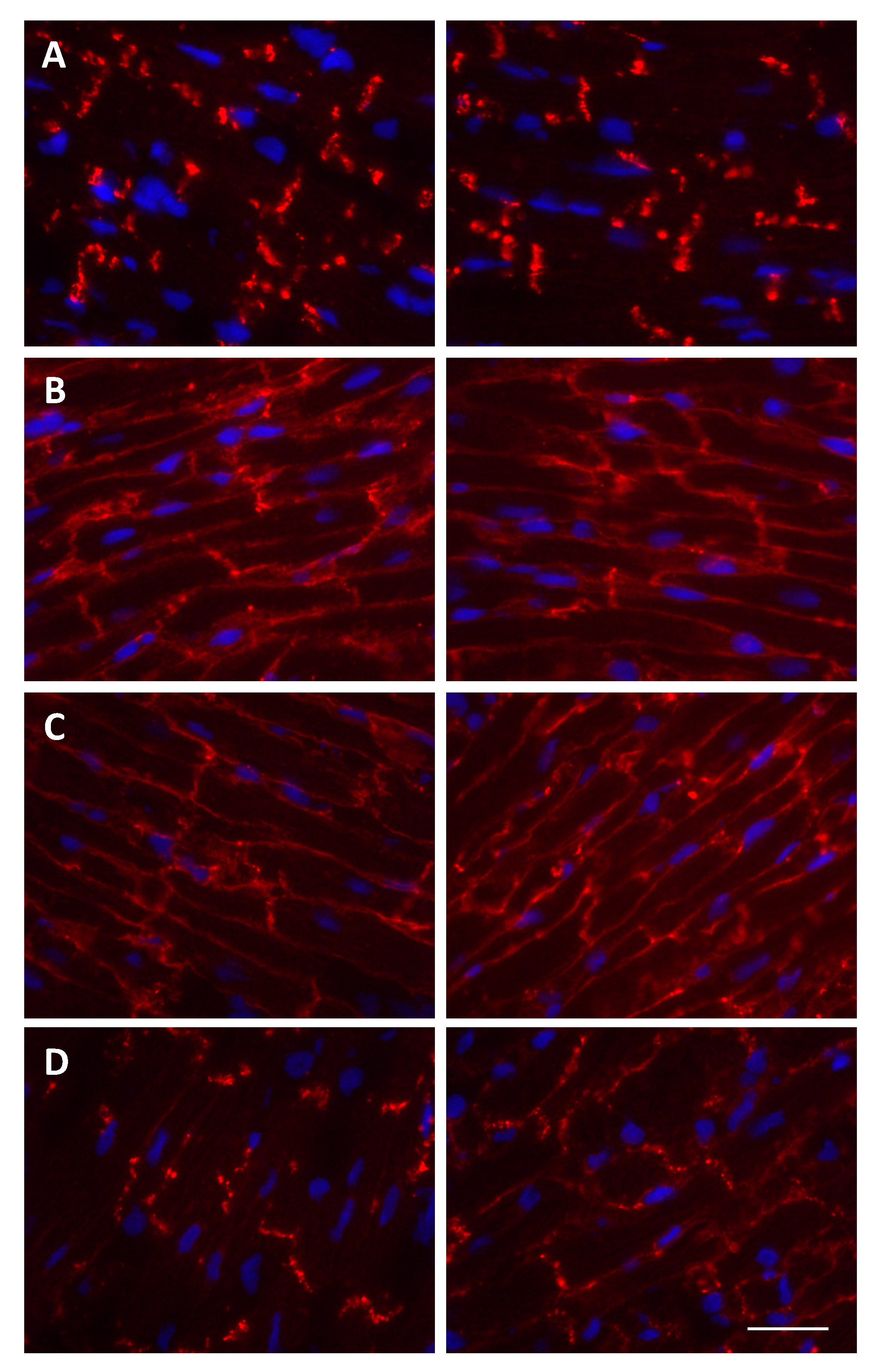
5.2. Cx43 Protein Partners and Their Role in Cardiac I/R Injury-Mediated Gap Junction Remodeling
6. Concluding Remarks: On the Way Towards Cx43-Targeted Strategies for Cardioprotection
Funding
Conflicts of Interest
References
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the global burden of cardiovascular disease, part 1: The epidemiology and risk factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the mechanisms of acute coronary syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Rossello, X.; Yellon, D.M. Cardioprotection: The disconnect between bench and bedside. Circulation 2016, 134, 574–575. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Cardioprotection research must leave its comfort zone. Eur. Heart J. 2018, 39, 3393–3395. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Schulz, R.; Girao, H.; Kwak, B.R.; De Stefani, D.; Rizzuto, R.; Bernardi, P.; Di Lisa, F. Mitochondrial ion channels as targets for cardioprotection. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Montecucco, F.; Carbone, F.; Schindler, T.H. Pathophysiology of ST-segment elevation myocardial infarction: Novel mechanisms and treatments. Eur. Heart J. 2016, 37, 1268–1283. [Google Scholar] [CrossRef]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kasala, E.R.; Bodduluru, L.N.; Dahiya, V.; Sharma, D.; Kumar, V.; Lahkar, M. Animal models of myocardial infarction: Mainstay in clinical translation. Regul. Toxicol. Pharmacol. 2016, 76, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ludman, A.J.; Yellon, D.M.; Hausenloy, D.J. Cardiac preconditioning for ischaemia: Lost in translation. Dis. Models Mech. 2010, 3, 35–38. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McCafferty, K.; Forbes, S.; Thiemermann, C.; Yaqoob, M.M. The challenge of translating ischemic conditioning from animal models to humans: The role of comorbidities. Dis. Models Mech. 2014, 7, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Bolli, R.; Canty, J.M., Jr.; Du, X.J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Podesser, B.K.; Lim, C.C. The continuing evolution of the Langendorff and ejecting murine heart: New advances in cardiac phenotyping. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H156–H167. [Google Scholar] [CrossRef]
- Sutherland, F.J.; Hearse, D.J. The isolated blood and perfusion fluid perfused heart. Pharmacol. Res. 2000, 41, 613–627. [Google Scholar] [CrossRef]
- Langendorff, O. Untersuchungen am überlebenden Säugethierherzen. Arch. Gesamte Physiol. Menschen Tiere 1895, 61, 291–332. [Google Scholar] [CrossRef]
- Sutherland, F.J.; Shattock, M.J.; Baker, K.E.; Hearse, D.J. Mouse isolated perfused heart: Characteristics and cautions. Clin. Exp. Pharmacol. Physiol. 2003, 30, 867–878. [Google Scholar] [CrossRef]
- Bell, R.M.; Mocanu, M.M.; Yellon, D.M. Retrograde heart perfusion: The Langendorff technique of isolated heart perfusion. J. Mol. Cell. Cardiol. 2011, 50, 940–950. [Google Scholar] [CrossRef]
- Curtis, M.J.; Hancox, J.C.; Farkas, A.; Wainwright, C.L.; Stables, C.L.; Saint, D.A.; Clements-Jewery, H.; Lambiase, P.D.; Billman, G.E.; Janse, M.J.; et al. The Lambeth Conventions (II): Guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol. Ther. 2013, 139, 213–248. [Google Scholar] [CrossRef] [PubMed]
- Mersmann, J.; Latsch, K.; Habeck, K.; Zacharowski, K. Measure for measure-determination of infarct size in murine models of myocardial ischemia and reperfusion: A systematic review. Shock 2011, 35, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, R.; Benhabbouche, S.; Bopassa, J.C.; Li, B.; Ovize, M. One hour reperfusion is enough to assess function and infarct size with TTC staining in Langendorff rat model. Cardiovasc. Drugs Ther. 2009, 23, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Headrick, J.P.; Peart, J.; Hack, B.; Flood, A.; Matherne, G.P. Functional properties and responses to ischaemia-reperfusion in Langendorff perfused mouse heart. Exp. Physiol. 2001, 86, 703–716. [Google Scholar] [CrossRef]
- Fishbein, M.C.; Meerbaum, S.; Rit, J.; Lando, U.; Kanmatsuse, K.; Mercier, J.C.; Corday, E.; Ganz, W. Early phase acute myocardial infarct size quantification: Validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am. Heart J. 1981, 101, 593–600. [Google Scholar] [CrossRef]
- Redfors, B.; Shao, Y.; Omerovic, E. Myocardial infarct size and area at risk assessment in mice. Exp. Clin. Cardiol. 2012, 17, 268–272. [Google Scholar]
- Pitoulis, F.G.; Watson, S.A.; Perbellini, F.; Terracciano, C.M. Myocardial slices come to age: An intermediate complexity in vitro cardiac model for translational research. Cardiovasc. Res. 2020, 116, 1275–1287. [Google Scholar] [CrossRef]
- Kolk, M.V.; Meyberg, D.; Deuse, T.; Tang-Quan, K.R.; Robbins, R.C.; Reichenspurner, H.; Schrepfer, S. LAD-ligation: A murine model of myocardial infarction. J. Vis. Exp. 2009. [Google Scholar] [CrossRef]
- Michael, L.H.; Entman, M.L.; Hartley, C.J.; Youker, K.A.; Zhu, J.; Hall, S.R.; Hawkins, H.K.; Berens, K.; Ballantyne, C.M. Myocardial ischemia and reperfusion: A murine model. Am. J. Physiol. 1995, 269, H2147–H2154. [Google Scholar] [CrossRef]
- Verdouw, P.D.; van den Doel, M.A.; de Zeeuw, S.; Duncker, D.J. Animal models in the study of myocardial ischaemia and ischaemic syndromes. Cardiovasc. Res. 1998, 39, 121–135. [Google Scholar] [CrossRef]
- Xu, Z.; McElhanon, K.E.; Beck, E.X.; Weisleder, N. A murine model of myocardial ischemia-reperfusion injury. Methods Mol. Biol. 2018, 1717, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Johns, T.N.; Olson, B.J. Experimental myocardial infarction. I. A method of coronary occlusion in small animals. Ann. Surg. 1954, 140, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Tarnavski, O.; McMullen, J.R.; Schinke, M.; Nie, Q.; Kong, S.; Izumo, S. Mouse cardiac surgery: Comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol. Genom. 2004, 16, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Gao, E.; Lei, Y.H.; Shang, X.; Huang, Z.M.; Zuo, L.; Boucher, M.; Fan, Q.; Chuprun, J.K.; Ma, X.L.; Koch, W.J. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 2010, 107, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Braunersreuther, V.; Chanson, M.; Bouis, D.; Rochemont, V.; Foglia, B.; Pelli, G.; Sutter, E.; Pinsky, D.J.; Mach, F.; et al. Endothelial Cx40 limits myocardial ischaemia/reperfusion injury in mice. Cardiovasc. Res. 2014, 102, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Alloush, J.; Beck, E.; Weisleder, N. A murine model of myocardial ischemia-reperfusion injury through ligation of the left anterior descending artery. J. Vis. Exp. 2014. [Google Scholar] [CrossRef]
- Hartley, C.J.; Michael, L.H.; Entman, M.L. Noninvasive measurement of ascending aortic blood velocity in mice. Am. J. Physiol. 1995, 268, H499–H505. [Google Scholar] [CrossRef]
- Morel, S.; Christoffersen, C.; Axelsen, L.N.; Montecucco, F.; Rochemont, V.; Frias, M.A.; Mach, F.; James, R.W.; Naus, C.C.; Chanson, M.; et al. Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc. Res. 2016, 109, 385–396. [Google Scholar] [CrossRef]
- Stopa, B.; Rybarska, J.; Drozd, A.; Konieczny, L.; Krol, M.; Lisowski, M.; Piekarska, B.; Roterman, I.; Spolnik, P.; Zemanek, G. Albumin binds self-assembling dyes as specific polymolecular ligands. Int. J. Biol. Macromol. 2006, 40, 1–8. [Google Scholar] [CrossRef]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in cardiovascular and neurovascular health and disease: Pharmacological implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef]
- Hoagland, D.T.; Santos, W.; Poelzing, S.; Gourdie, R.G. The role of the gap junction perinexus in cardiac conduction: Potential as a novel anti-arrhythmic drug target. Prog. Biophys. Mol. Biol. 2019, 144, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Gourdie, R.G.; Poelzing, S. Mechanisms of cardiac conduction: A history of revisions. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H619–H627. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Hoeker, G.S.; Alvarez-Laviada, A.; Hoagland, D.; Wan, X.; King, D.R.; Sanchez-Alonso, J.; Chen, C.; Jourdan, J.; Isom, L.L.; et al. The adhesion function of the sodium channel beta subunit (beta1) contributes to cardiac action potential propagation. eLife 2018, 7. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; Batista-Almeida, D.; Aasen, T.; Kwak, B.R.; Girao, H. Biological functions of Connexin43 beyond intercellular communication. Trends Cell Biol. 2019, 29, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Van Campenhout, R.; Cooreman, A.; Leroy, K.; Rusiecka, O.M.; Van Brantegem, P.; Annaert, P.; Muyldermans, S.; Devoogdt, N.; Cogliati, B.; Kwak, B.R.; et al. Non-canonical roles of connexins. Prog. Biophys. Mol. Biol. 2020, 153, 35–41. [Google Scholar] [CrossRef]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meana, M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc. Res. 2004, 61, 386–401. [Google Scholar] [CrossRef]
- Schulz, R.; Gorge, P.M.; Gorbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef]
- Schulz, R.; Gres, P.; Skyschally, A.; Duschin, A.; Belosjorow, S.; Konietzka, I.; Heusch, G. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J. 2003, 17, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, L.N.; Stahlhut, M.; Mohammed, S.; Larsen, B.D.; Nielsen, M.S.; Holstein-Rathlou, N.H.; Andersen, S.; Jensen, O.N.; Hennan, J.K.; Kjolbye, A.L. Identification of ischemia-regulated phosphorylation sites in connexin43: A possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123). J. Mol. Cell. Cardiol. 2006, 40, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin 43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflug. Arch. 2010, 460, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Frias, M.A.; Rosker, C.; James, R.W.; Rohr, S.; Kwak, B.R. The natural cardioprotective particle HDL modulates connexin43 gap junction channels. Cardiovasc. Res. 2012, 93, 41–49. [Google Scholar] [CrossRef]
- Surinkaew, S.; Kumphune, S.; Chattipakorn, S.; Chattipakorn, N. Inhibition of p38 MAPK during ischemia, but not reperfusion, effectively attenuates fatal arrhythmia in ischemia/reperfusion heart. J. Cardiovasc. Pharmacol. 2013, 61, 133–141. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, J.; Hu, W.; Yu, D.; Bai, X. Effects of pinocembrin pretreatment on connexin 43 (Cx43) protein expression after rat myocardial ischemia-reperfusion and cardiac arrhythmia. Med. Sci. Monit. 2018, 24, 5008–5014. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kleber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef]
- Matsuda, T.; Fujio, Y.; Nariai, T.; Ito, T.; Yamane, M.; Takatani, T.; Takahashi, K.; Azuma, J. N-cadherin signals through Rac1 determine the localization of connexin 43 in cardiac myocytes. J. Mol. Cell. Cardiol. 2006, 40, 495–502. [Google Scholar] [CrossRef]
- Tansey, E.E.; Kwaku, K.F.; Hammer, P.E.; Cowan, D.B.; Federman, M.; Levitsky, S.; McCully, J.D. Reduction and redistribution of gap and adherens junction proteins after ischemia and reperfusion. Ann. Thorac. Surg. 2006, 82, 1472–1479. [Google Scholar] [CrossRef]
- Bodendiek, S.B.; Raman, G. Connexin modulators and their potential targets under the magnifying glass. Curr. Med. Chem. 2010, 17, 4191–4230. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Sanchez, J.A.; Gonzalez-Loyola, A.; Barba, I.; Morente, M.; Aguilar, R.; Agullo, E.; Miro-Casas, E.; Esquerda, N.; Ruiz-Meana, M.; et al. Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischaemia and preconditioning protection. J. Physiol. 2010, 588, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Cooper, C.D.; King, T.J.; Burt, J.M. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 2006, 119, 3435–3442. [Google Scholar] [CrossRef]
- Turner, M.S.; Haywood, G.A.; Andreka, P.; You, L.; Martin, P.E.; Evans, W.H.; Webster, K.A.; Bishopric, N.H. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ. Res. 2004, 95, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Perrelli, M.G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Inserte, J.; Ruiz-Meana, M.; Gonzalez, M.A.; Solares, J.; Julia, M.; Barrabes, J.A.; Soler-Soler, J. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation 1997, 96, 3579–3586. [Google Scholar] [CrossRef]
- Srisakuldee, W.; Jeyaraman, M.M.; Nickel, B.E.; Tanguy, S.; Jiang, Z.S.; Kardami, E. Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc. Res. 2009, 83, 672–681. [Google Scholar] [CrossRef]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef]
- Jiang, J.; Hoagland, D.; Palatinus, J.A.; He, H.; Iyyathurai, J.; Jourdan, L.J.; Bultynck, G.; Wang, Z.; Zhang, Z.; Schey, K.; et al. Interaction of alpha carboxyl terminus 1 peptide with the connexin 43 carboxyl terminus preserves left ventricular function after ischemia-reperfusion injury. J. Am. Heart Assoc. 2019, 8, e012385. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Shiki, K.; Hearse, D.J. Preconditioning of ischemic myocardium: Reperfusion-induced arrhythmias. Am. J. Physiol. 1987, 253, H1470–H1476. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Alkhulaifi, A.M.; Pugsley, W.B. Preconditioning the human myocardium. Lancet 1993, 342, 276–277. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef] [PubMed]
- Schwanke, U.; Konietzka, I.; Duschin, A.; Li, X.; Schulz, R.; Heusch, G. No ischemic preconditioning in heterozygous connexin43-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1740–H1742. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Rodriguez-Sinovas, A.; Barba, I.; Miro-Casas, E.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Alburquerque-Bejar, J.J.; Garcia-Dorado, D. Activation of RISK and SAFE pathways is not involved in the effects of Cx43 deficiency on tolerance to ischemia-reperfusion injury and preconditioning protection. Basic Res. Cardiol. 2013, 108, 351. [Google Scholar] [CrossRef]
- Li, G.; Whittaker, P.; Yao, M.; Kloner, R.A.; Przyklenk, K. The gap junction uncoupler heptanol abrogates infarct size reduction with preconditioning in mouse hearts. Cardiovasc. Pathol. 2002, 11, 158–165. [Google Scholar] [CrossRef]
- Hatanaka, K.; Kawata, H.; Toyofuku, T.; Yoshida, K. Down-regulation of connexin43 in early myocardial ischemia and protective effect by ischemic preconditioning in rat hearts in vivo. Jpn. Heart J. 2004, 45, 1007–1019. [Google Scholar] [CrossRef][Green Version]
- Jain, S.K.; Schuessler, R.B.; Saffitz, J.E. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ. Res. 2003, 92, 1138–1144. [Google Scholar] [CrossRef]
- Totzeck, A.; Boengler, K.; van de Sand, A.; Konietzka, I.; Gres, P.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. No impact of protein phosphatases on connexin 43 phosphorylation in ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2106–H2112. [Google Scholar] [CrossRef]
- Hund, T.J.; Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; Saffitz, J.E. Protein kinase Cepsilon mediates salutary effects on electrical coupling induced by ischemic preconditioning. Heart Rhythm 2007, 4, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Katare, R.G.; Kakinuma, Y.; Zhang, D.; Yamasaki, F.; Muramoto, K.; Sato, T. Efferent vagal nerve stimulation protects heart against ischemia-induced arrhythmias by preserving connexin43 protein. Circulation 2005, 112, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.R.; Luo, Y.; Li, X.; Boengler, K.; Buechert, A.; Garcia-Dorado, D.; Di Lisa, F.; Schulz, R.; Heusch, G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ. Res. 2005, 97, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Buchert, A.; Feldhaus, S.; Schulz, R. No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res. Cardiol. 2006, 101, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Shinlapawittayatorn, K.; Chinda, K.; Palee, S.; Surinkaew, S.; Kumfu, S.; Kumphune, S.; Chattipakorn, S.; KenKnight, B.H.; Chattipakorn, N. Vagus nerve stimulation initiated late during ischemia, but not reperfusion, exerts cardioprotection via amelioration of cardiac mitochondrial dysfunction. Heart Rhythm 2014, 11, 2278–2287. [Google Scholar] [CrossRef]
- Yue, P.; Zhang, Y.; Du, Z.; Xiao, J.; Pan, Z.; Wang, N.; Yu, H.; Ma, W.; Qin, H.; Wang, W.H.; et al. Ischemia impairs the association between connexin 43 and M3 subtype of acetylcholine muscarinic receptor (M3-mAChR) in ventricular myocytes. Cell. Physiol. Biochem. 2006, 17, 129–136. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Garcia-Dorado, D.; Botker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position paper of the European society of cardiology working group on cellular biology of the heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Kuper, N.; Karcher, C.; Weissmuller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 2006, 99, 1100–1108. [Google Scholar] [CrossRef]
- Calder, B.W.; Matthew Rhett, J.; Bainbridge, H.; Fann, S.A.; Gourdie, R.G.; Yost, M.J. Inhibition of connexin 43 hemichannel-mediated ATP release attenuates early inflammation during the foreign body response. Tissue Eng. Part A 2015, 21, 1752–1762. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Dolmatova, E.; Spagnol, G.; Boassa, D.; Baum, J.R.; Keith, K.; Ambrosi, C.; Kontaridis, M.I.; Sorgen, P.L.; Sosinsky, G.E.; Duffy, H.S. Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1208–H1218. [Google Scholar] [CrossRef] [PubMed]
- Rook, M.B.; Jongsma, H.J.; de Jonge, B. Single channel currents of homo- and heterologous gap junctions between cardiac fibroblasts and myocytes. Pflug. Arch. 1989, 414, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Gaudesius, G.; Rohr, S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ. Res. 2006, 98, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Ferko, M.; Andelova, N.; Szeiffova Bacova, B.; Jasova, M. Myocardial adaptation in pseudohypoxia: Signaling and regulation of mPTP via mitochondrial connexin 43 and cardiolipin. Cells 2019, 8, 1449. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, H.; Hoppel, C.L. Mitochondria in the human heart. J. Bioenerg. Biomembr. 2009, 41, 99–106. [Google Scholar] [CrossRef]
- Halestrap, A.P. Mitochondria and preconditioning: A connexin connection? Circ. Res. 2006, 99, 10–12. [Google Scholar] [CrossRef]
- Opie, L.H.; Sack, M.N. Metabolic plasticity and the promotion of cardiac protection in ischemia and ischemic preconditioning. J. Mol. Cell. Cardiol. 2002, 34, 1077–1089. [Google Scholar] [CrossRef]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef]
- Boengler, K.; Schulz, R. Connexin 43 and mitochondria in cardiovascular health and disease. Adv. Exp. Med. Biol. 2017, 982, 227–246. [Google Scholar] [CrossRef]
- Kavazis, A.N.; Alvarez, S.; Talbert, E.; Lee, Y.; Powers, S.K. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H144–H152. [Google Scholar] [CrossRef] [PubMed]
- Kavazis, A.N.; McClung, J.M.; Hood, D.A.; Powers, S.K. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H928–H935. [Google Scholar] [CrossRef] [PubMed]
- Holmuhamedov, E.L.; Oberlin, A.; Short, K.; Terzic, A.; Jahangir, A. Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PLoS ONE 2012, 7, e44667. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Stahlhofen, S.; van de Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; Garcia-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta Biomembr. 2018, 1860, 174–181. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miro, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Cabestrero, A.; Boengler, K.; Heusch, G.; Garcia-Dorado, D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2008, 77, 325–333. [Google Scholar] [CrossRef]
- Miro-Casas, E.; Ruiz-Meana, M.; Agullo, E.; Stahlhofen, S.; Rodriguez-Sinovas, A.; Cabestrero, A.; Jorge, I.; Torre, I.; Vazquez, J.; Boengler, K.; et al. Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc. Res. 2009, 83, 747–756. [Google Scholar] [CrossRef]
- Soetkamp, D.; Nguyen, T.T.; Menazza, S.; Hirschhauser, C.; Hendgen-Cotta, U.B.; Rassaf, T.; Schluter, K.D.; Boengler, K.; Murphy, E.; Schulz, R. S-nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res. Cardiol. 2014, 109, 433. [Google Scholar] [CrossRef]
- Trudeau, K.; Muto, T.; Roy, S. Downregulation of mitochondrial connexin 43 by high glucose triggers mitochondrial shape change and cytochrome C release in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6675–6681. [Google Scholar] [CrossRef]
- Waza, A.A.; Andrabi, K.; Hussain, M.U. Protein kinase C (PKC) mediated interaction between conexin43 (Cx43) and K(+)(ATP) channel subunit (Kir6.1) in cardiomyocyte mitochondria: Implications in cytoprotection against hypoxia induced cell apoptosis. Cell Signal. 2014, 26, 1909–1917. [Google Scholar] [CrossRef]
- Penna, C.; Perrelli, M.G.; Raimondo, S.; Tullio, F.; Merlino, A.; Moro, F.; Geuna, S.; Mancardi, D.; Pagliaro, P. Postconditioning induces an anti-apoptotic effect and preserves mitochondrial integrity in isolated rat hearts. Biochim. Biophys. Acta 2009, 1787, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Tu, R.H.; Li, Q.J.; Huang, Z.; He, Y.; Meng, J.J.; Zheng, H.L.; Zeng, Z.Y.; Zhong, G.Q. Novel functional role of heat shock protein 90 in mitochondrial connexin 43-mediated hypoxic postconditioning. Cell. Physiol. Biochem. 2017, 44, 982–997. [Google Scholar] [CrossRef] [PubMed]
- Martins-Marques, T.; Anjo, S.I.; Pereira, P.; Manadas, B.; Girao, H. Interacting network of the Gap Junction (GJ) Protein Connexin43 (Cx43) is modulated by ischemia and reperfusion in the heart. Mol. Cell. Proteom. 2015, 14, 3040–3055. [Google Scholar] [CrossRef] [PubMed]
- Agullo-Pascual, E.; Lin, X.; Leo-Macias, A.; Zhang, M.; Liang, F.X.; Li, Z.; Pfenniger, A.; Lubkemeier, I.; Keegan, S.; Fenyo, D.; et al. Super-resolution imaging reveals that loss of the C-terminus of connexin43 limits microtubule plus-end capture and NaV1.5 localization at the intercalated disc. Cardiovasc. Res. 2014, 104, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Kieken, F.; Mutsaers, N.; Dolmatova, E.; Virgil, K.; Wit, A.L.; Kellezi, A.; Hirst-Jensen, B.J.; Duffy, H.S.; Sorgen, P.L. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ. Res. 2009, 104, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.W.; Zhang, S.S.; Sanchez, J.M.; Lamouille, S.; Vogan, J.M.; Hesketh, G.G.; Hong, T.; Tomaselli, G.F.; Shaw, R.M. A 14-3-3 mode-1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic 2014, 15, 684–699. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, L.; Xuan, L.; Pan, Z.; Hu, X.; Liu, H.; Bai, Y.; Jiao, L.; Li, Z.; Cui, L.; et al. Long non-coding RNA CCRR controls cardiac conduction via regulating intercellular coupling. Nat. Commun. 2018, 9, 4176. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Catarino, S.; Goncalves, A.; Miranda-Silva, D.; Goncalves, L.; Antunes, P.; Coutinho, G.; Leite Moreira, A.; Falcao Pires, I.; Girao, H. EHD1 modulates Cx43 gap junction remodeling associated with cardiac diseases. Circ. Res. 2020, 126, e97–e113. [Google Scholar] [CrossRef]
- Lissoni, A.; Hulpiau, P.; Martins-Marques, T.; Wang, N.; Bultynck, G.; Schulz, R.; Witschas, K.; Girao, H.; De Smet, M.; Leybaert, L. RyR2 regulates Cx43 hemichannel intracellular Ca2+-dependent activation in cardiomyocytes. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Basheer, W.A.; Xiao, S.; Epifantseva, I.; Fu, Y.; Kleber, A.G.; Hong, T.; Shaw, R.M. GJA1-20k arranges actin to guide Cx43 delivery to cardiac intercalated discs. Circ. Res. 2017, 121, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Shimura, D.; Baum, R.; Hernandez, D.M.; Agvanian, S.; Nagaoka, Y.; Katsumata, M.; Lampe, P.D.; Kleber, A.G.; Hong, T.; et al. Auxiliary trafficking subunit GJA1-20k protects Connexin43 from degradation and limits ventricular arrhythmias. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Martins-Marques, T.; Catarino, S.; Marques, C.; Matafome, P.; Ribeiro-Rodrigues, T.; Baptista, R.; Pereira, P.; Girao, H. Heart ischemia results in connexin43 ubiquitination localized at the intercalated discs. Biochimie 2015, 112, 196–201. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Catarino, S.; Zuzarte, M.; Marques, C.; Matafome, P.; Pereira, P.; Girao, H. Ischaemia-induced autophagy leads to degradation of gap junction protein connexin43 in cardiomyocytes. Biochem. J. 2015, 467, 231–245. [Google Scholar] [CrossRef]
- Bejarano, E.; Yuste, A.; Patel, B.; Stout, R.F., Jr.; Spray, D.C.; Cuervo, A.M. Connexins modulate autophagosome biogenesis. Nat. Cell Biol. 2014, 16, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Andreadou, I.; Barile, L.; Birnbaum, Y.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Downey, J.M.; Girao, H.; Pagliaro, P.; Penna, C.; et al. Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc. Res. 2019, 115, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Batista-Almeida, D.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Girao, H. The role of proteostasis in the regulation of cardiac intercellular communication. Adv. Exp. Med. Biol. 2020, 1233, 279–302. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Pinho, M.J.; Zuzarte, M.; Oliveira, C.; Pereira, P.; Sluijter, J.P.; Gomes, C.; Girao, H. Presence of Cx43 in extracellular vesicles reduces the cardiotoxicity of the anti-tumour therapeutic approach with doxorubicin. J. Extracell. Vesicles 2016, 5, 32538. [Google Scholar] [CrossRef]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Isabel Anjo, S.; Manadas, B.; Sluijter, J.P.G.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef]
- Almeida Paiva, R.; Martins-Marques, T.; Jesus, K.; Ribeiro-Rodrigues, T.; Zuzarte, M.; Silva, A.; Reis, L.; da Silva, M.; Pereira, P.; Vader, P.; et al. Ischaemia alters the effects of cardiomyocyte-derived extracellular vesicles on macrophage activation. J. Cell. Mol. Med. 2019, 23, 1137–1151. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Laundos, T.L.; Pereira-Carvalho, R.; Batista-Almeida, D.; Pereira, R.; Coelho-Santos, V.; Silva, A.P.; Fernandes, R.; Zuzarte, M.; Enguita, F.J.; et al. Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis. Cardiovasc. Res. 2017, 113, 1338–1350. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W.; Puranam, K.L.; Revel, J.P. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 1991, 273 Pt 1, 67–72. [Google Scholar] [CrossRef]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-based therapeutics for dermal wound healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef]
- Willebrords, J.; Crespo Yanguas, S.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Kwak, B.R.; Green, C.R.; Cogliati, B.; Vinken, M. Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.L.; Phillips, A.R.; Duft, B.J.; Kim, Y.; Green, C.R. Translating connexin biology into therapeutics. Semin. Cell Dev. Biol. 2016, 50, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; Manicone, N.; Muller, A.; Gerwin, R.; Ziskoven, U.; Irankhahi, A.; Minke, C.; Klaus, W. A new synthetic antiarrhythmic peptide reduces dispersion of epicardial activation recovery interval and diminishes alterations of epicardial activation patterns induced by regional ischemia. A mapping study. Naunyn Schmiedebergs Arch. Pharmacol. 1994, 350, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.M.; Venkatasubramanian, S.; Vase, H.; Hyldebrandt, J.A.; Contractor, H.; Schmidt, M.R.; Botker, H.E.; Cruden, N.L.; Newby, D.E.; Kharbanda, R.K.; et al. Rotigaptide protects the myocardium and arterial vasculature from ischaemia reperfusion injury. Br. J. Clin. Pharmacol. 2016, 81, 1037–1045. [Google Scholar] [CrossRef]
- Skyschally, A.; Walter, B.; Schultz Hansen, R.; Heusch, G. The antiarrhythmic dipeptide ZP1609 (danegaptide) when given at reperfusion reduces myocardial infarct size in pigs. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 383–391. [Google Scholar] [CrossRef]
- Boengler, K.; Bulic, M.; Schreckenberg, R.; Schluter, K.D.; Schulz, R. The gap junction modifier ZP1609 decreases cardiomyocyte hypercontracture following ischaemia/reperfusion independent from mitochondrial connexin 43. Br. J. Pharmacol. 2017, 174, 2060–2073. [Google Scholar] [CrossRef]
- Engstrom, T.; Nepper-Christensen, L.; Helqvist, S.; Klovgaard, L.; Holmvang, L.; Jorgensen, E.; Pedersen, F.; Saunamaki, K.; Tilsted, H.H.; Steensberg, A.; et al. Danegaptide for primary percutaneous coronary intervention in acute myocardial infarction patients: A phase 2 randomised clinical trial. Heart 2018, 104, 1593–1599. [Google Scholar] [CrossRef]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Prasad, G.M.; Viswanathan, V.; Armstrong, D.G.; Gourdie, R.G.; Ghatnekar, G.S. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen. 2015, 23, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Montgomery, J.; Sharma, M.; Ravi, A.; Rajkumar, J.S.; Moyer, K.E.; Gourdie, R.G.; Ghatnekar, G.S. A multicenter randomized controlled trial evaluating a Cx43-mimetic peptide in cutaneous scarring. J. Investig. Dermatol. 2017, 137, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Brisset, A.C.; Isakson, B.E.; Kwak, B.R. Connexins in vascular physiology and pathology. Antioxid. Redox Signal. 2009, 11, 267–282. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusiecka, O.M.; Montgomery, J.; Morel, S.; Batista-Almeida, D.; Van Campenhout, R.; Vinken, M.; Girao, H.; Kwak, B.R. Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules 2020, 10, 1225. https://doi.org/10.3390/biom10091225
Rusiecka OM, Montgomery J, Morel S, Batista-Almeida D, Van Campenhout R, Vinken M, Girao H, Kwak BR. Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules. 2020; 10(9):1225. https://doi.org/10.3390/biom10091225
Chicago/Turabian StyleRusiecka, Olga M., Jade Montgomery, Sandrine Morel, Daniela Batista-Almeida, Raf Van Campenhout, Mathieu Vinken, Henrique Girao, and Brenda R. Kwak. 2020. "Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection" Biomolecules 10, no. 9: 1225. https://doi.org/10.3390/biom10091225
APA StyleRusiecka, O. M., Montgomery, J., Morel, S., Batista-Almeida, D., Van Campenhout, R., Vinken, M., Girao, H., & Kwak, B. R. (2020). Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules, 10(9), 1225. https://doi.org/10.3390/biom10091225