Multivalent and Bidirectional Binding of Transcriptional Transactivation Domains to the MED25 Coactivator



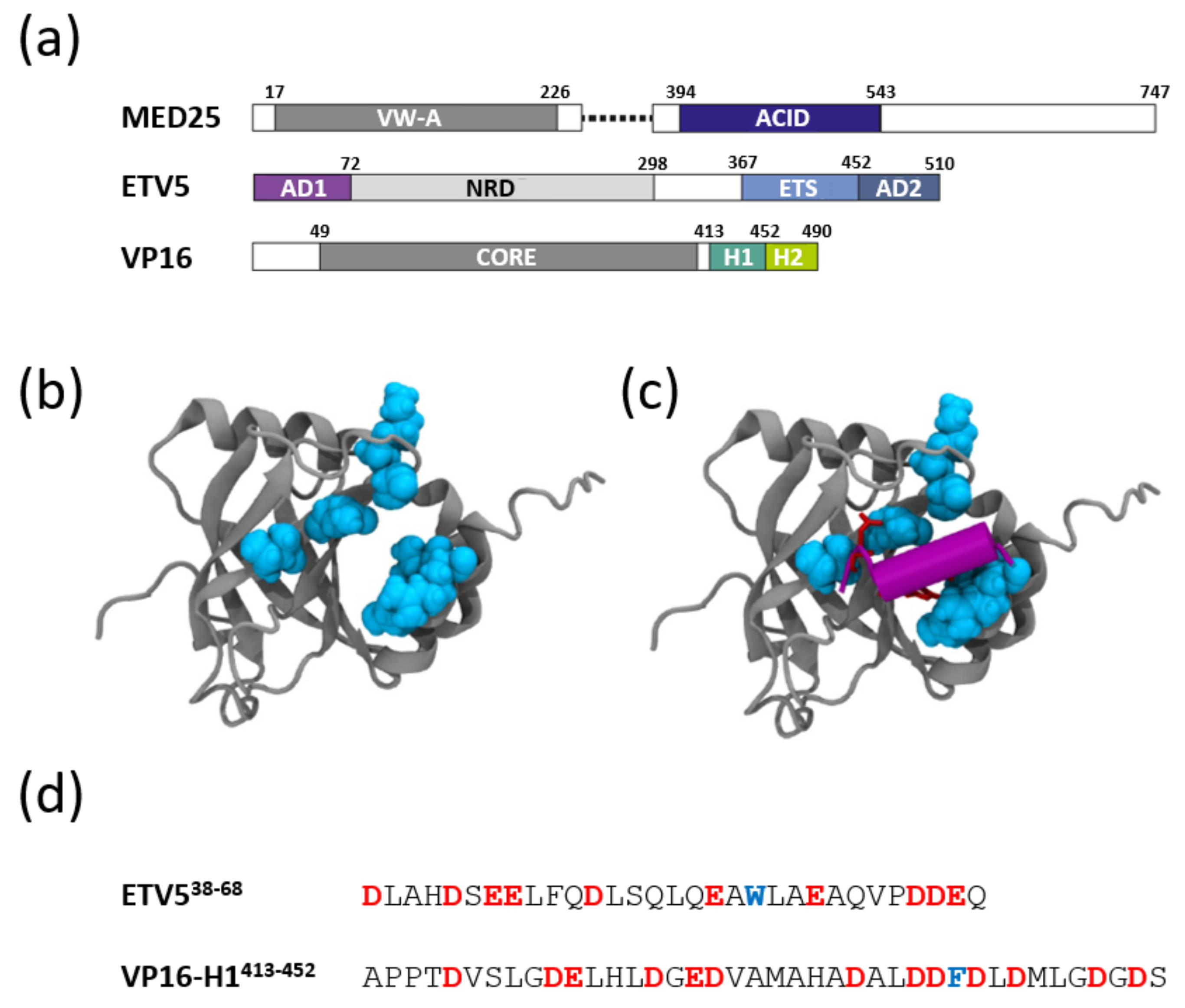{kind=link}
{kind=link}
{kind=link}
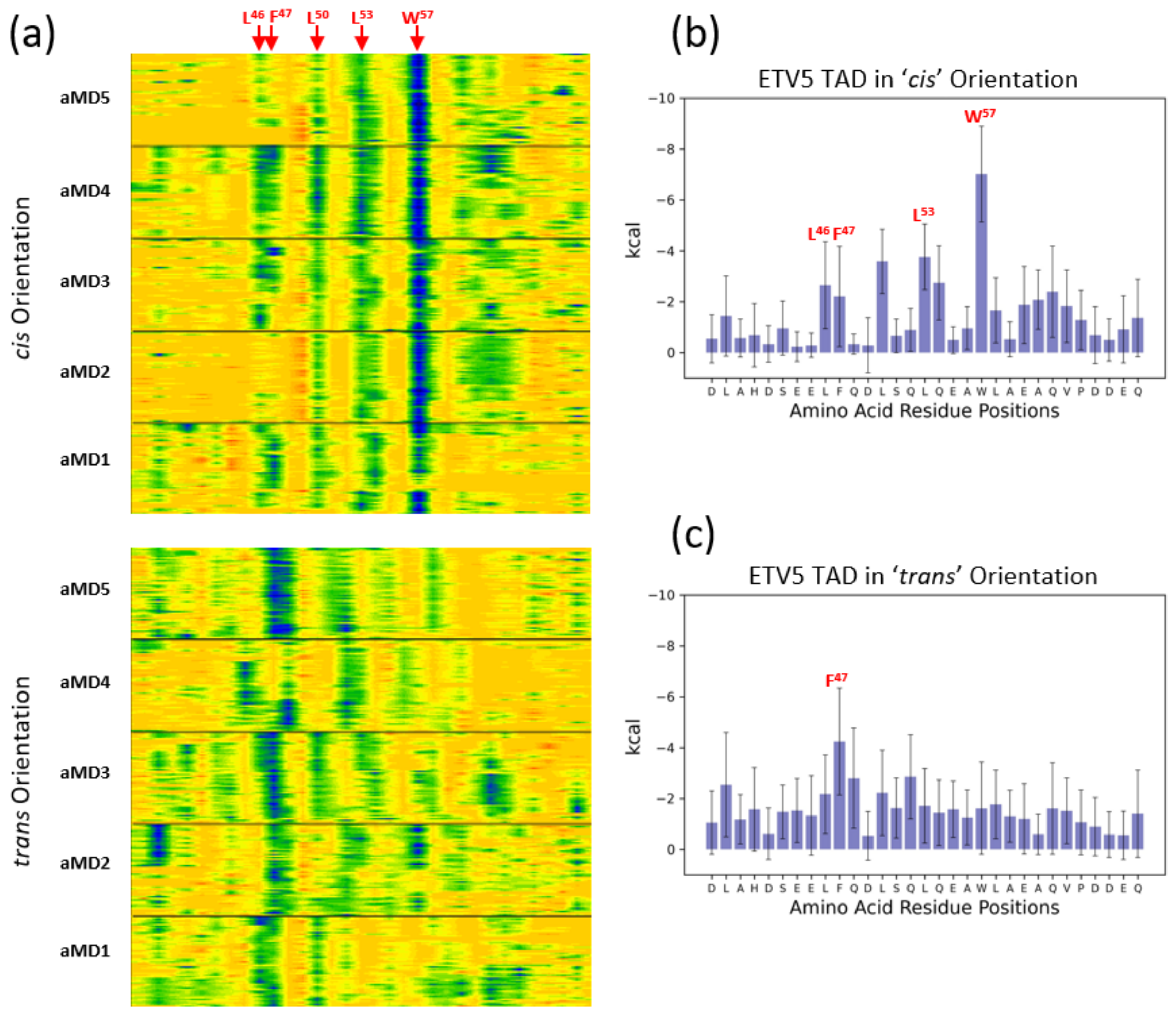{kind=link}
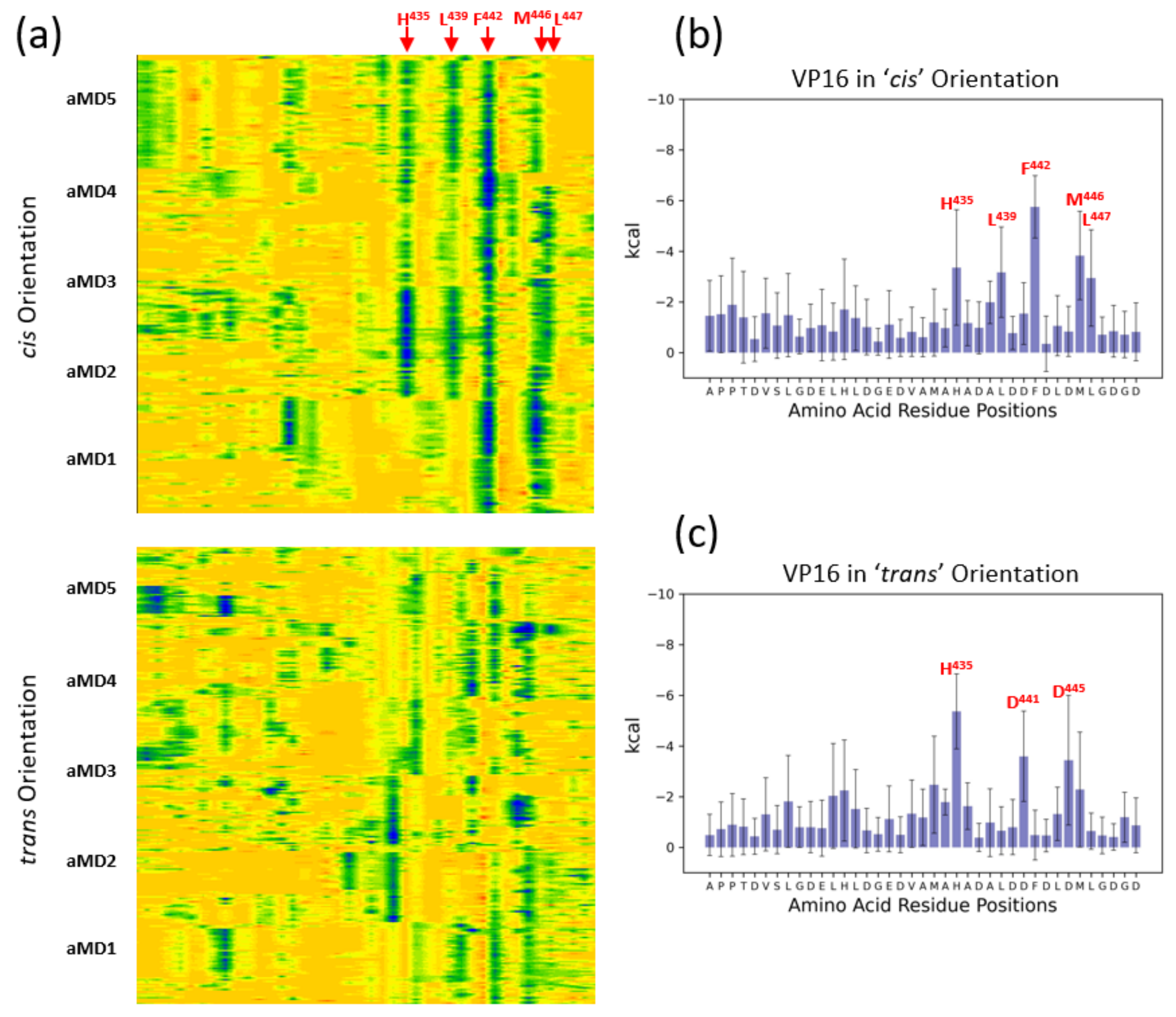{kind=link}
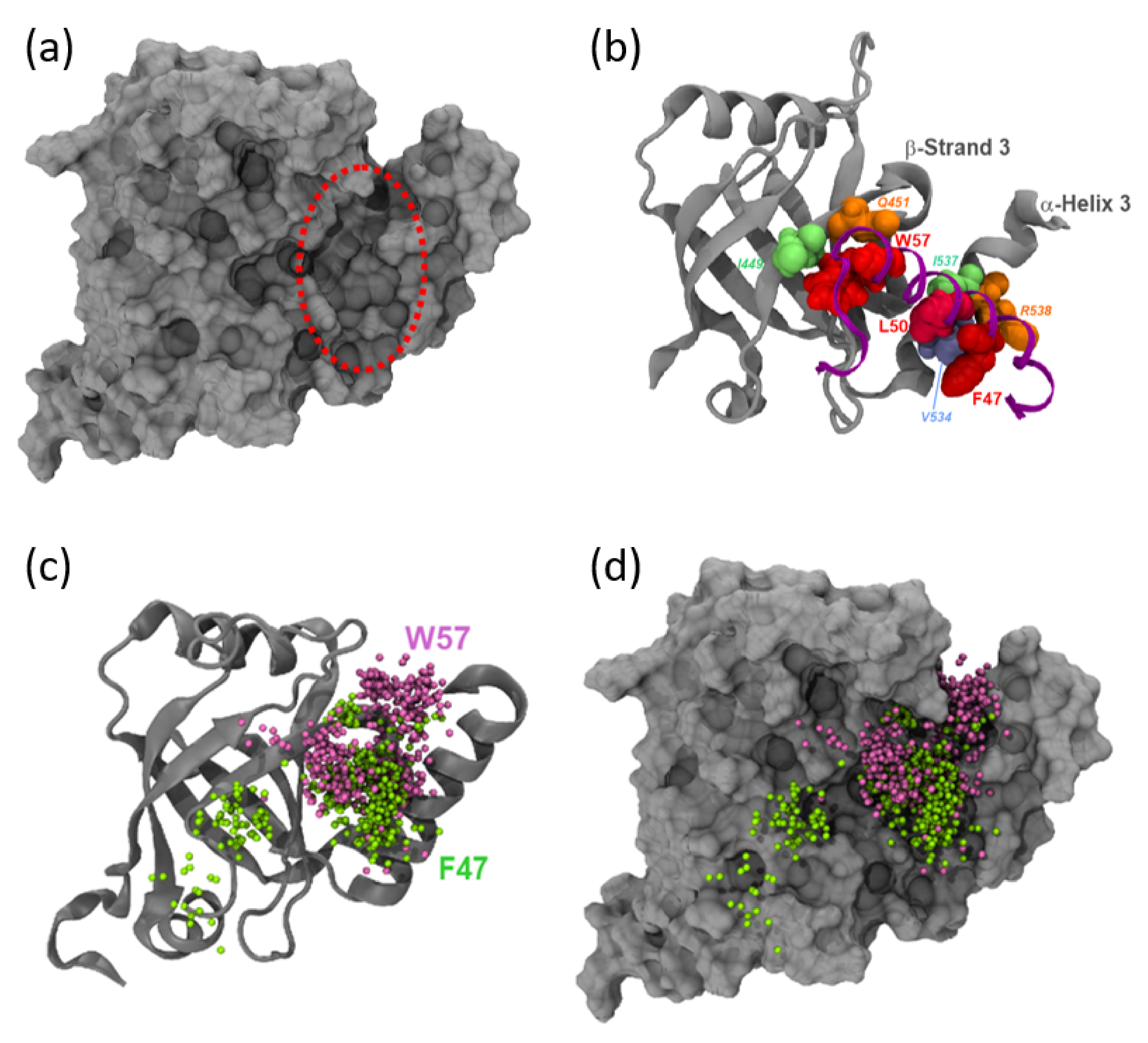{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Modelling, Parameterization and MD Simulation
2.2. Markov Chain Monte Carlo (MCMC) Simulations
2.3. Analysis and Visualization
2.4. MM-GBSA Analysis
3. Results
3.1. Structural Aspects of ETV538–68 and VP16-H1413–452 TADs Prior to Binding to Coactivators
3.2. The ETV538–68 and VP16-H1413–452 TADs Interact with MED25 in an Orientation-Specific Manner
3.3. Energetic Aspects of Bidirectional Interactions of ETV538–68 and VP16-H1413–452—TADs with MED25
3.4. MED25 Coactivator Surfaces Interacting with ETV5- and VP16-H1 TADs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Danino, Y.M.; Even, D.; Ideses, D.; Juven-Gershon, T. The core promoter: At the heart of gene expression. Biochim. Et Biophys. Acta. 2015, 1849, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Piskacek, M.; Havelka, M.; Rezacova, M.; Knight, A. The 9aaTAD Transactivation Domains: From Gal4 to p53. PLoS ONE 2016, 11, e0162842. [Google Scholar] [CrossRef] [PubMed]
- Soutourina, J. Mammalian Mediator as a Functional Link between Enhancers and Promoters. Cell 2019, 178, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361, eaar3555. [Google Scholar] [CrossRef]
- Currie, S.L.; Doane, J.J.; Evans, K.S.; Bhachech, N.; Madison, B.J.; Lau, D.K.W. ETV4 and AP1 Transcription Factors Form Multivalent Interactions with three Sites on the MED25 Activator-Interacting Domain. J. Mol. Biol. 2017, 429, 2975–2995. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Heikaus, C.C.; Kisselev, L.; Vernon, R.; Herbig, E.; Pacheco, D. The Acidic Transcription Activator Gcn4 Binds the Mediator Subunit Gal11/Med15 Using a Simple Protein Interface Forming a Fuzzy Complex. Mol. Cell. 2011, 44, 942–953. [Google Scholar] [CrossRef]
- Henderson, A.R.; Henley, M.J.; Foster, N.J.; Peiffer, A.L.; Beyersdorf, M.S.; Stanford, K.D.; Sturlis, S.M.; Linhares, B.M.; Hill, Z.B.; Wells, J.A.; et al. Conservation of coactivator engagement mechanism enables small-molecule allosteric modulators. Proc. Natl. Acad. Sci. USA 2018, 115, 8960–8965. [Google Scholar] [CrossRef]
- Scholes, N.S.; Weinzierl, R.O. Molecular Dynamics of “Fuzzy” Transcriptional Activator-Coactivator Interactions. PloS Comput. Biol. 2016, 12, e1004935. [Google Scholar] [CrossRef]
- Borggrefe, T.; Yue, X. Interactions between subunits of the Mediator complex with gene-specific transcription factors. Semin. Cell Dev. Biol. 2011, 22, 759–768. [Google Scholar] [CrossRef]
- Taatjes, D.J. Transcription Factor-Mediator Interfaces: Multiple and Multi-Valent. Journal of molecular biology. 2017, 429, 2996–2998. [Google Scholar] [CrossRef] [PubMed]
- Vojnic, E.; Mourao, A.; Seizl, M.; Simon, B.; Wenzeck, L.; Lariviere, L. Structure and VP16 binding of the Mediator Med25 activator interaction domain. Nat. Struct. Mol. Biol. 2011, 18, 404–U29. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, A.G.; Kulkarni, M.; Yi, T.; Takeuchi, K.; Sun, Z.Y.; Luna, R.E. Structure of the VP16 transactivator target in the Mediator. Nat. Struct. Mol. Biol. 2011, 18, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Lim, K.; Lee, M.K.; Chi, S.W. Structural Basis for the Interaction between p53 Transactivation Domain and the Mediator Subunit MED25. Molecules 2018, 23, 2726. [Google Scholar] [CrossRef]
- Kazan, K. The Multitalented MEDIATOR25. Front. Plant Sci. 2017, 8, 999. [Google Scholar] [CrossRef]
- Landrieu, I.; Verger, A.; Baert, J.L.; Rucktooa, P.; Cantrelle, F.X.; Dewitte, F. Characterization of ERM transactivation domain binding to the ACID/PTOV domain of the Mediator subunit MED25. Nucleic Acids Res. 2015, 43, 7110–7121. [Google Scholar] [CrossRef]
- Bontems, F.; Verger, A.; Dewitte, F.; Lens, Z.; Baert, J.L.; Ferreira, E. NMR structure of the human Mediator MED25 ACID domain. J. Struct. Biol. 2011, 174, 245–251. [Google Scholar] [CrossRef]
- Nicholas, T.R.; Strittmatter, B.G.; Hollenhorst, P.C. Oncogenic ETS Factors in Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 409–436. [Google Scholar]
- Defossez, P.A.; Baert, J.L.; Monnot, M.; de Launoit, Y. The ETS family member ERM contains an alpha-helical acidic activation domain that contacts TAFII60. Nucleic Acids Res. 1997, 25, 4455–4463. [Google Scholar] [CrossRef]
- Verger, A.; Baert, J.L.; Verreman, K.; Dewitte, F.; Ferreira, E.; Lens, Z. The Mediator complex subunit MED25 is targeted by the N-terminal transactivation domain of the PEA3 group members. Nucleic Acids Res. 2013, 41, 4847–4859. [Google Scholar] [CrossRef]
- Regier, J.L.; Shen, F.; Triezenberg, S.J. Pattern of aromatic and hydrophobic amino acids critical for one of two subdomains of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. USA 1993, 90, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Shin, S.; Janknecht, R. ETV1, 4 and 5: An oncogenic subfamily of ETS transcription factors. Biochim. Et Biophys. Acta. 2012, 1826, 1–12. [Google Scholar] [CrossRef]
- Oh, S.; Shin, S.; Song, H.; Grande, J.P.; Janknecht, R. Relationship between ETS Transcription Factor ETV1 and TGF-beta-regulated SMAD Proteins in Prostate Cancer. Sci. Rep. 2019, 9, 8186. [Google Scholar] [CrossRef]
- Roizman, B.; Zhou, G. The 3 facets of regulation of herpes simplex virus gene expression: A critical inquiry. Virology 2015, 479, 562–567. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.A.; Case, K.B.; Ben-Shalom, S.R.; Brozell, D.S.; Cerutti, T.E.; Cheatham, V.W.D. Amber 2020; University of California: San Francisco, CA, USA, 2020; Available online: https://ambermd.org/ (accessed on 17 August 2020).
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wires Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Pierce, L.C.; Salomon-Ferrer, R.; Augusto, F.O.C.; McCammon, J.A.; Walker, R.C. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef]
- Boomsma, W.; Frellsen, J.; Harder, T.; Bottaro, S.; Johansson, K.E.; Tian, P.F. PHAISTOS: A framework for Markov chain Monte Carlo simulation and inference of protein structure. J. Comput. Chem. 2013, 34, 1697–1705. [Google Scholar] [CrossRef]
- Sullivan, S.S.; Weinzierl, R.O.J. Optimization of Molecular Dynamics Simulations of c-MYC (1-88)-An Intrinsically Disordered System. Life 2020, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dahl, A.C.; Chavent, M.; Sansom, M.S. Bendix: Intuitive helix geometry analysis and abstraction. Bioinformatics 2012, 28, 2193–2194. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Kim, D.H.; Han, K.H. Transient Secondary Structures as General Target-Binding Motifs in Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2018, 19, 3614. [Google Scholar] [CrossRef]
- Piskacek, S.; Gregor, M.; Nemethova, M.; Grabner, M.; Kovarik, P.; Piskacek, M. Nine-amino-acid transactivation domain: Establishment and prediction utilities. Genom. 2007, 89, 756–768. [Google Scholar] [CrossRef]
- Erijman, A.; Kozlowski, L.; Sohrabi-Jahromi, S.; Fishburn, J.; Warfield, L.; Schreiber, J. A High-Throughput Screen for Transcription Activation Domains Reveals Their Sequence Features and Permits Prediction by Deep Learning. Mol. Cell. 2020, 78, 890–902. [Google Scholar] [CrossRef]
- Cress, W.D.; Triezenberg, S.J. Critical structural elements of the VP16 transcriptional activation domain. Science 1991, 251, 87–90. [Google Scholar] [CrossRef]
- Warfield, L.; Tuttle, L.M.; Pacheco, D.; Klevit, R.E.; Hahn, S. A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. USA 2014, 111, E3506–E3513. [Google Scholar] [CrossRef]
- Meyer, K.D.; Lin, S.C.; Bernecky, C.; Gao, Y.; Taatjes, D.J. p53 activates transcription by directing structural shifts in Mediator. Nat. Struct. Mol. Biol. 2010, 17, 753–760. [Google Scholar] [CrossRef]
- Thakur, J.K.; Arthanari, H.; Yang, F.; Pan, S.J.; Fan, X.; Breger, J. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 2008, 452, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, J.L.; Boeszoermenyi, A.; Vale-Silva, L.A.; Torelli, R.; Posteraro, B.; Sohn, Y.J. Inhibiting fungal multidrug resistance by disrupting an activator-Mediator interaction. Nature 2016, 530, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Eletsky, A.; Szyperski, T.; Hay, J.; Ruyechan, W.T. Analysis of the varicella-zoster virus IE62 N-terminal acidic transactivating domain and its interaction with the human mediator complex. J. Virol. 2009, 83, 6300–6305. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Park, U.H.; Kim, E.J.; Um, S.J. MED25 is distinct from TRAP220/MED1 in cooperating with CBP for retinoid receptor activation. EMBO J. 2007, 26, 3545–3557. [Google Scholar] [CrossRef] [PubMed]
- Mittler, G.; Stuhler, T.; Santolin, L.; Uhlmann, T.; Kremmer, E.; Lottspeich, F. A novel docking site on Mediator is critical for activation by VP16 in mammalian cells. EMBO J. 2003, 22, 6494–6504. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; de Beaumont, R.; Zhou, S.; Naar, A.M. The activator-recruited cofactor/Mediator coactivator subunit ARC92 is a functionally important target of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. USA 2004, 101, 2339–2344. [Google Scholar] [CrossRef]
- Roupelieva, M.; Griffiths, S.J.; Kremmer, E.; Meisterernst, M.; Viejo-Borbolla, A.; Schulz, T. Kaposi’s sarcoma-associated herpesvirus Lana-1 is a major activator of the serum response element and mitogen-activated protein kinase pathways via interactions with the Mediator complex. J. Gen. Virol. 2010, 91, 1138–1149. [Google Scholar] [CrossRef]
- Cumbo, F.; Vergni, D.; Santoni, D. Investigating transcription factor synergism in humans. DNA Res. 2018, 25, 103–112. [Google Scholar] [CrossRef]
- Lens, Z.; Cantrelle, F.X.; Peruzzini, R.; Hanoulle, X.; Dewitte, F.; Ferreira, E. Solution Structure of the N-Terminal Domain of Mediator Subunit MED26 and Molecular Characterization of Its Interaction with EAF1 and TAF7. J. Mol. Biol. 2017, 429, 3043–3055. [Google Scholar] [CrossRef]
- Radhakrishnan, I.; PerezAlvarado, G.C.; Parker, D.; Dyson, H.J.; Montminy, M.R.; Wright, P.E. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator: Coactivator interactions. Cell 1997, 91, 741–752. [Google Scholar] [CrossRef]
- Hua, Q.X.; Jia, W.H.; Bullock, B.P.; Habener, J.F.; Weiss, M.A. Transcriptional activator-coactivator recognition: Nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry 1998, 37, 5858–5866. [Google Scholar] [CrossRef] [PubMed]
- Triezenberg, S.J. Structure and function of transcriptional activation domains. Curr. Opin. Genet. Dev. 1995, 5, 190–196. [Google Scholar] [CrossRef]
- Odoux, A.; Jindal, D.; Tamas, T.C.; Lim, B.W.; Pollard, D.; Xu, W. Experimental and molecular dynamics studies showed that CBP KIX mutation affects the stability of CBP:C-Myb complex. Comput. Biol. Chem. 2016, 62, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Yazar, M.; Ozbek, P. Revisiting allostery in CREB-binding protein (CBP) using residue-based interaction energy. J. Comput. Aided Mol. Des. 2020, 34, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, W.; Guo, Y.; Wang, J.; Fu, W.; Sun, H. Molecular Dynamics Simulations Revealed the Regulation of Ligands to the Interactions between Androgen Receptor and Its Coactivator. J. Chem. Inf. Modeling 2018, 58, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Piskacek, M.; Havelka, M.; Rezacova, M.; Knight, A. The 9aaTAD Is Exclusive Activation Domain in Gal4. PLoS ONE 2017, 12, e0169261. [Google Scholar] [CrossRef]
- Arnold, C.D.; Nemcko, F.; Woodfin, A.R.; Wienerroither, S.; Vlasova, A.; Schleiffer, A. A high-throughput method to identify trans-activation domains within transcription factor sequences. EMBO J. 2018, 37, e98896. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol. Rep. 2013, 5, 1. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeffery, H.M.; Weinzierl, R.O.J. Multivalent and Bidirectional Binding of Transcriptional Transactivation Domains to the MED25 Coactivator. Biomolecules 2020, 10, 1205. https://doi.org/10.3390/biom10091205
Jeffery HM, Weinzierl ROJ. Multivalent and Bidirectional Binding of Transcriptional Transactivation Domains to the MED25 Coactivator. Biomolecules. 2020; 10(9):1205. https://doi.org/10.3390/biom10091205
Chicago/Turabian StyleJeffery, Heather M., and Robert O. J. Weinzierl. 2020. "Multivalent and Bidirectional Binding of Transcriptional Transactivation Domains to the MED25 Coactivator" Biomolecules 10, no. 9: 1205. https://doi.org/10.3390/biom10091205
APA StyleJeffery, H. M., & Weinzierl, R. O. J. (2020). Multivalent and Bidirectional Binding of Transcriptional Transactivation Domains to the MED25 Coactivator. Biomolecules, 10(9), 1205. https://doi.org/10.3390/biom10091205