The GDP-Bound State of Mitochondrial Mfn1 Induces Membrane Adhesion of Apposing Lipid Vesicles through a Cooperative Binding Mechanism

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Lipids
2.3. Cloning of Full-Length Homo Sapiens Mitofusin 1 (hsMfn1)
2.4. Heterologous Production of hsMfn1 in Escherichia coli
2.5. Isolation of E. coli Inner Membrane Containing hsMfn1
2.6. Preparation of Large Unilamellar Vesicles (LUVs) and Dynamic Light Scattering (DLS) Measurements
2.7. Total Lipid-Mixing Fluorescence Assay
2.8. Electroformation of Giant Unilamellar Vesicles (GUVs).
2.9. Confocal Fluorescence Microscopy
2.10. Quantification of the Adhesion Energy
2.11. Cooperative Binding Adhesion
2.12. Statistical Analysis
3. Results
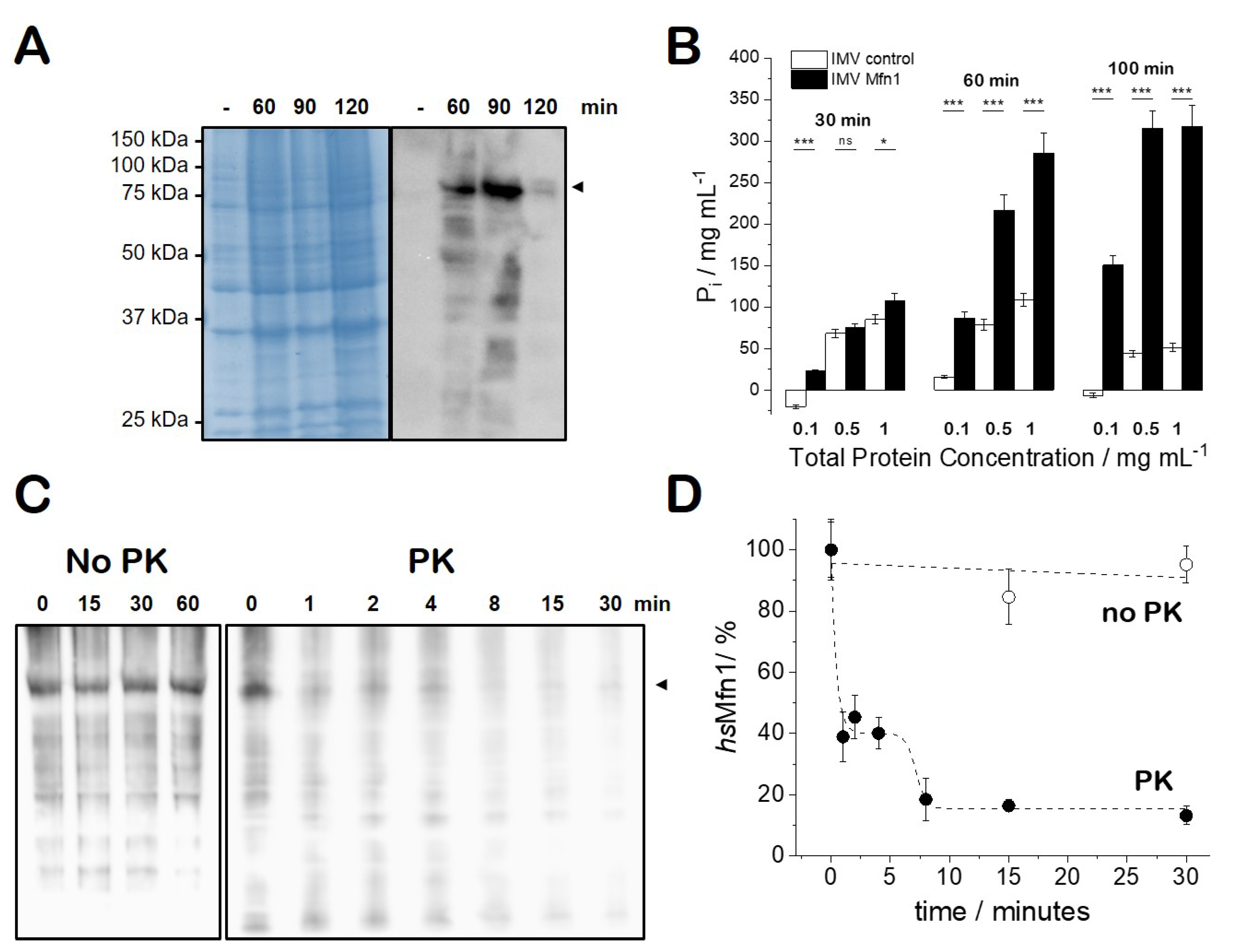
3.1. The Heterologous Production of Human Mfn1 Protein in Escherichia coli
3.2. GTP-Hydrolysis of E. coli IMVs Carrying hsMfn1
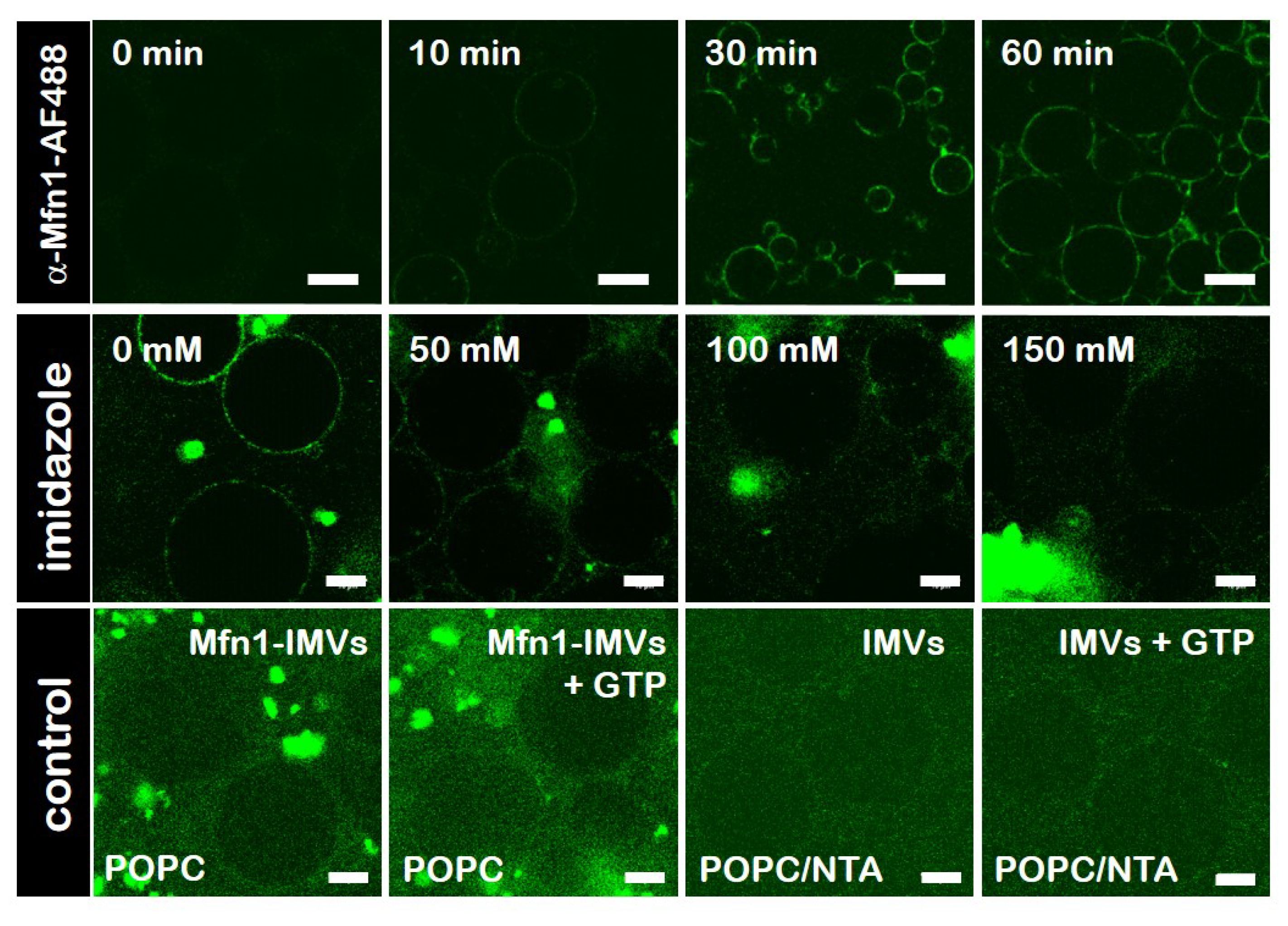
3.3. Characterisation of E. coli IMVs Carrying hsMfn1
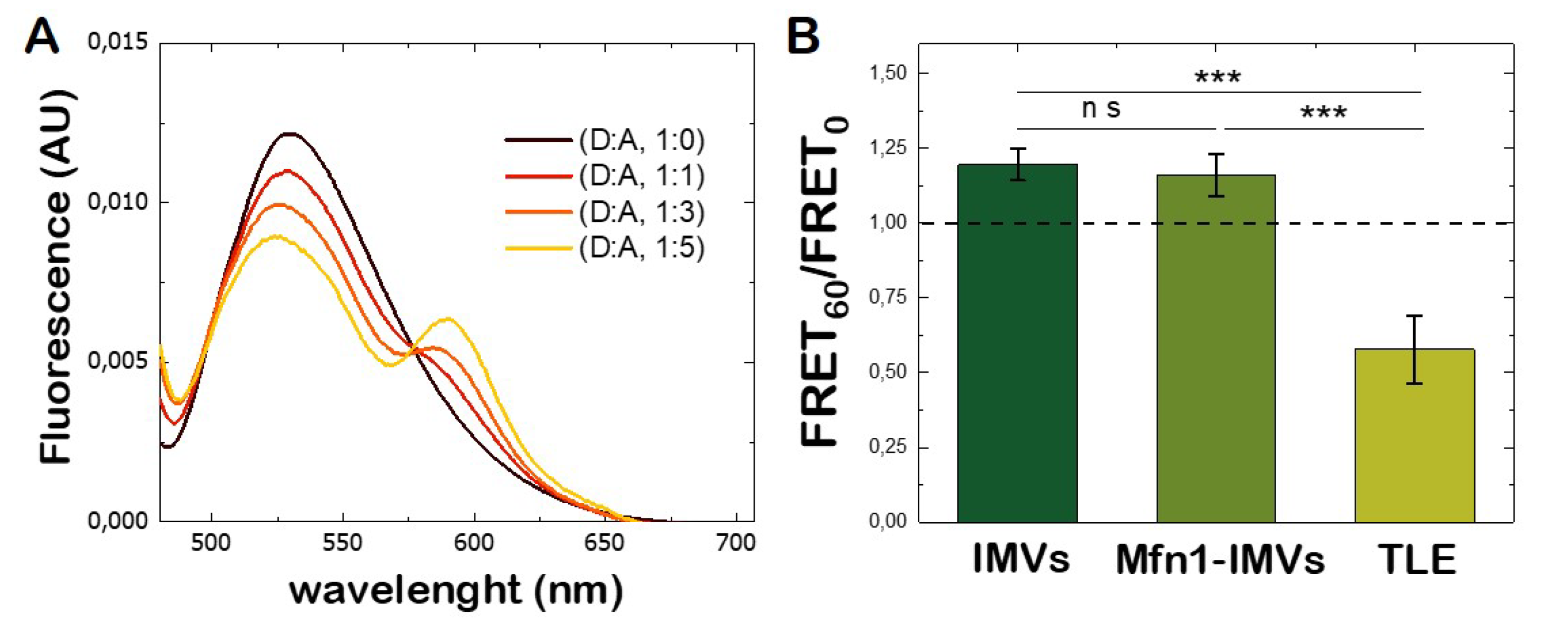
3.4. Lipid Mixing Assay of Mfn1-IMVs
3.5. Specific Binding of hsMfn1 Protein to the GUVs
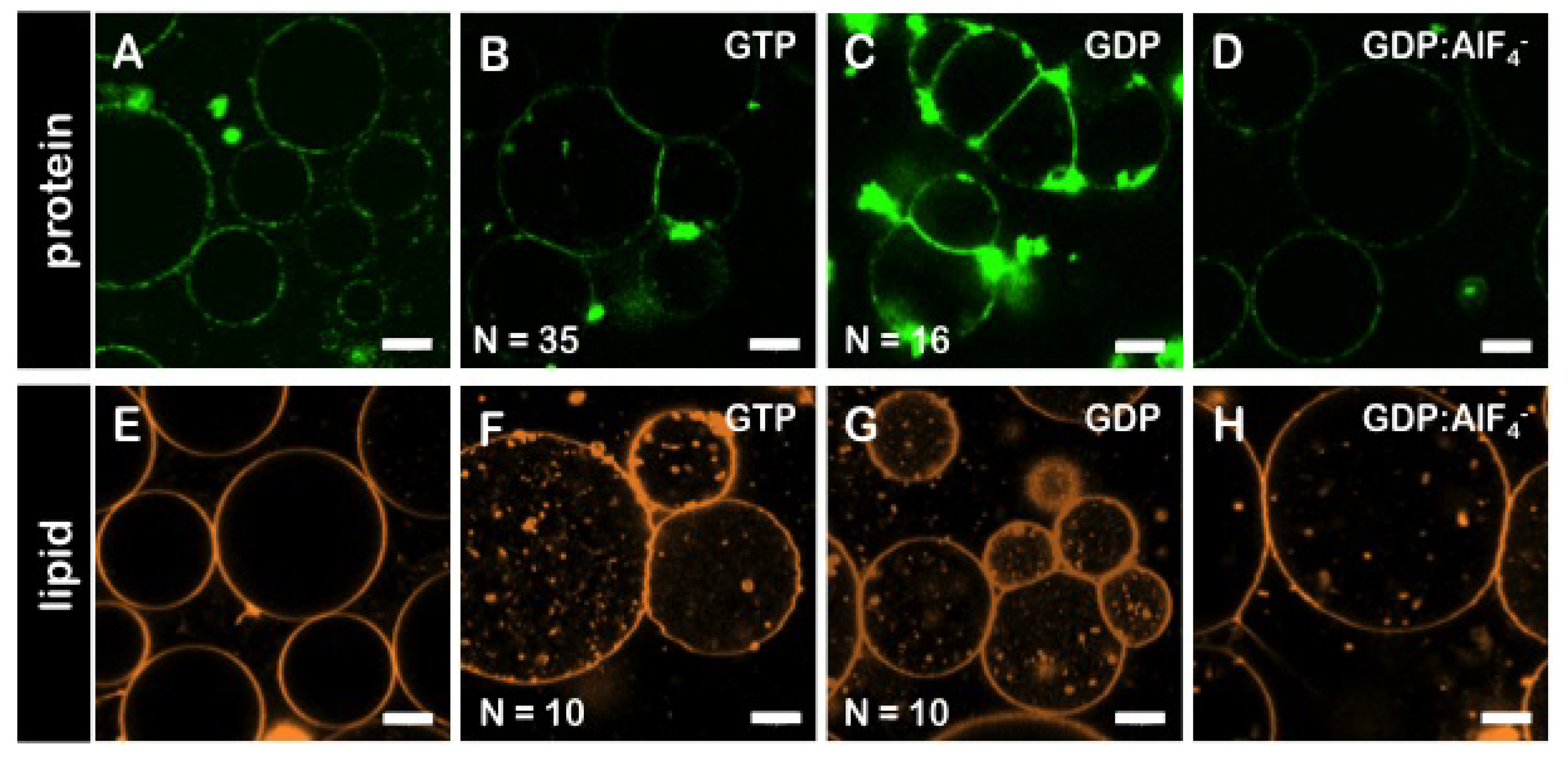
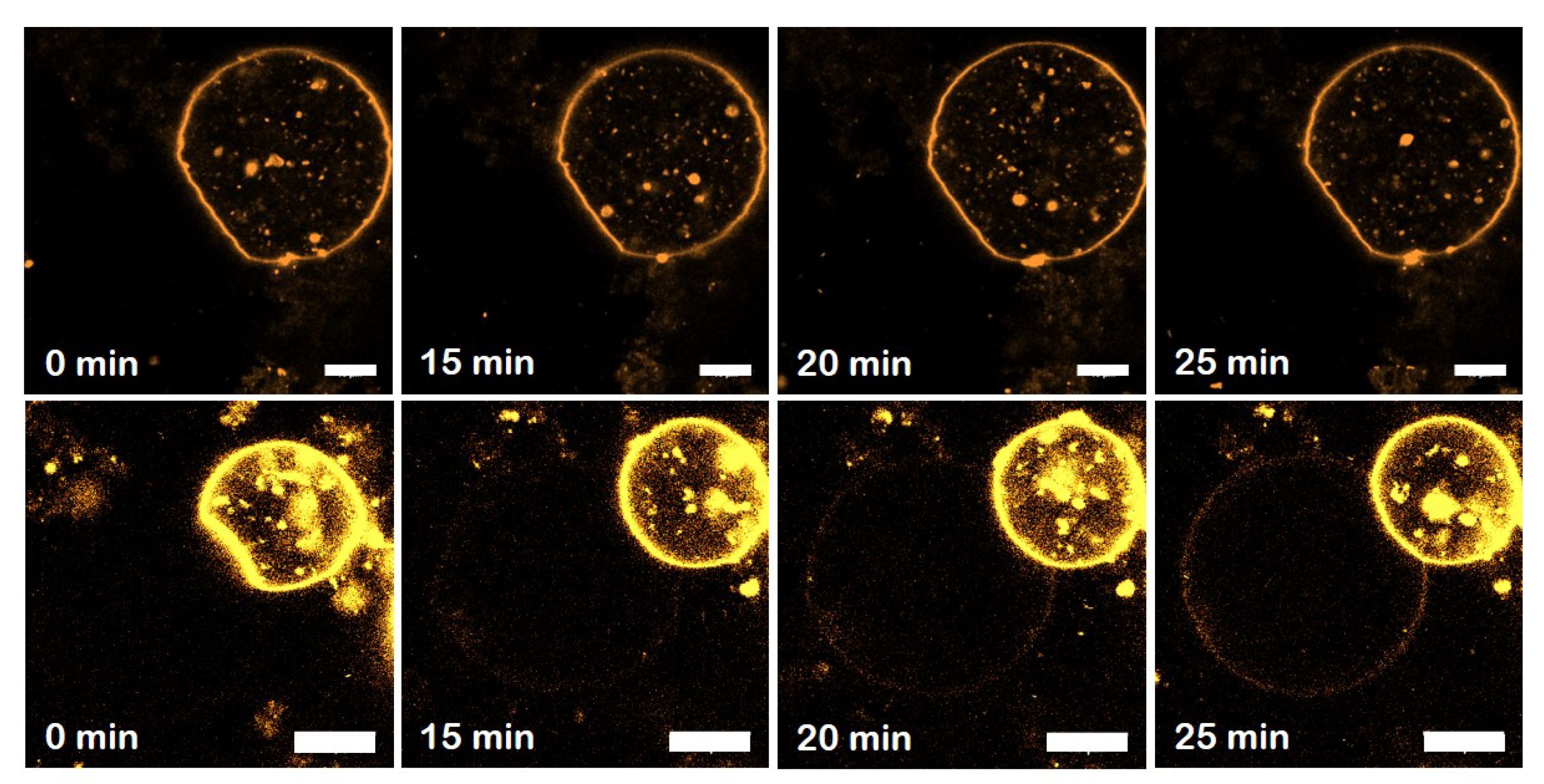
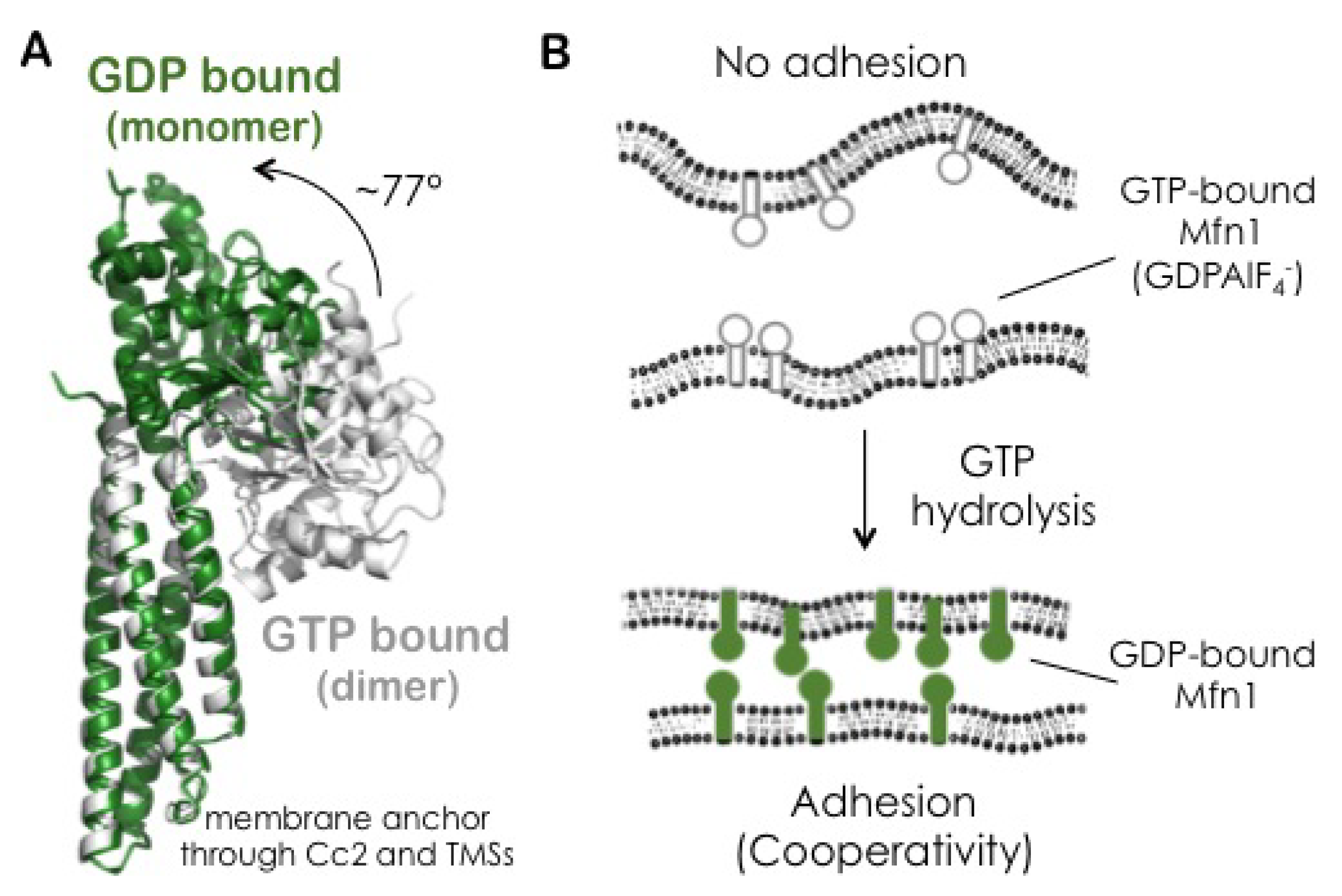
3.6. The GDP-Bound State of Mfn1 Promotes the Adhesion of Lipid Vesicles
3.7. GTP-Dependent Change of the Average Diameters Mfn1-LUVs
3.8. The Adhesion Strength Mediated by GDP-Mfn1 Is Comparable to GTP-Mfn1
3.9. Binding Cooperativity of Mfn1-Mediated Membrane Adhesion
3.10. Lipid Mixing Assay of GUVs Bearing Adhesion
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martens, S.; McMahon, H.T. Mechanisms of membrane fusion: Disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008, 9, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, R.; Schmid, S. The dynamin superfamily. Curr. Biol. 2018, 28, R411–R416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonny, B.; Burd, C.; De Camilli, P.; Chen, E.; Daumke, O.; Faelber, K.; Ford, M.; Frolov, V.A.; Frost, A.; Hinshaw, J.E.; et al. Membrane fission by dynamin: What we know and what we need to know. EMBO J. 2016, 35, 2270–2284. [Google Scholar] [CrossRef] [PubMed]
- Sousounis, K.; Haney, C.E.; Cao, J.; Sunchu, B.; Tsonis, P.A. Conservation of the three-dimensional structure in non-homologous or unrelated proteins. Hum. Genom. 2012, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramkamp, M. Structure and function of bacterial dynamin-like proteins. Biol. Chem. 2012, 393, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- McNew, J.A.; Sondermann, H.; Lee, T.; Stern, M.; Brandizzi, F. GTP-dependent membrane fusion. Annu. Rev. Cell Dev. Biol. 2013, 29, 529–550. [Google Scholar] [CrossRef]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef] [Green Version]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Rojo, M.; Legros, F.; Chateau, D.; Lombes, A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [Google Scholar]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Daumke, O.; Roux, A. Mitochondrial Homeostasis: How Do Dimers of Mitofusins Mediate Mitochondrial Fusion? Curr. Biol. 2017, 27, R353–R356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.M.; Qi, Y.B.; Huang, X.F.; Yu, C.T.; Lan, L.; Guo, X.Y.; Rao, Z.H.; Hu, J.J.; Lou, Z.Y. Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef]
- Low, H.H.; Sachse, C.; Amos, L.A.; Lowe, J. Structure of a bacterial dynamin-like protein lipid tube provides a mechanism for assembly and membrane curving. Cell 2009, 139, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, H.H.; Löwe, J. A bacterial dynamin-like protein. Nature 2006, 444, 766. [Google Scholar] [CrossRef] [PubMed]
- Betancourt-Solis, M.A.; Desai, T.; McNew, J.A. The atlastin membrane anchor forms an intramembrane hairpin that does not span the phospholipid bilayer. J. Biol. Chem. 2018, 293, 18514–18524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeusen, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial fusion intermediates revealed in vitro. Science 2004, 305, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Krobath, H.; Różycki, B.; Lipowsky, R.; Weikl, T.R. Binding cooperativity of membrane adhesion receptors. Soft Matter 2009, 5, 3354–3361. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning. A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Baneyx, F.; Georgiou, G. In vivo degradation of secreted fusion proteins by the Escherichia coli outer membrane protease OmpT. J. Bacteriol. 1990, 172, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Does, C.; de Keyzer, J.; van der Laan, M.; Driessen, A.J. Reconstitution of purified bacterial preprotein translocase in liposomes. Methods Enzym. 2003, 372, 86–98. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [Green Version]
- Lanzetta, P.A.; Alvarez, L.J.; Reinach, P.S.; Candia, O.A. An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 1979, 100, 95. [Google Scholar] [CrossRef]
- Bangham, A.D. Properties and uses of lipid vesicles: An overview. Ann. N. Y. Acad. Sci. 1978, 308, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Kluwer Academic/Plenum Publishers: Dordrecht, The Netherlands, 1999. [Google Scholar]
- Almendro Vedia, V.G.; Natale, P.; Chen, S.; Monroy, F.; Rosilio, V.; López-Montero, I. iGUVs: Preparing Giant Unilamellar Vesicles with a Smartphone and Lipids Easily Extracted from Chicken Eggs. J. Chem. Educ. 2017, 94, 644–649. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Servuss, R.; Helfrich, W. Mutual adhesion of lecithin membranes at ultralow tensions. J. Phys. 1989, 50, 809–827. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, R.; Arriaga, L.R.; Mell, M.; Moleiro, L.H.; Lopez-Montero, I.; Monroy, F. Bimodal spectrum for the curvature fluctuations of bilayer vesicles: Pure bending plus hybrid curvature-dilation modes. Phys. Rev. Lett. 2009, 102, 128101. [Google Scholar] [CrossRef]
- Dembo, M.; Torney, D.C.; Saxman, K.; Hammer, D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B Biol. Sci. 1988, 234, 55–83. [Google Scholar] [PubMed] [Green Version]
- Nye, J.A.; Groves, J.T. Kinetic Control of Histidine-Tagged Protein Surface Density on Supported Lipid Bilayers. Langmuir 2008, 24, 4145–4149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumon-Seignovert, L.; Cariot, G.; Vuillard, L. The toxicity of recombinant proteins in Escherichia coli: A comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr. Purif. 2004, 37, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Knol, J.; Veenhoff, L.; Liang, W.J.; Henderson, P.F.; Leblanc, G.; Poolman, B. Unidirectional reconstitution into detergent-destabilized liposomes of the purified lactose transport system of Streptococcus thermophilus. J. Biol. Chem. 1996, 271, 15358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattjus, P.; Molotkovsky, J.G.; Smaby, J.M.; Brown, R.E. A fluorescence resonance energy transfer approach for monitoring protein-mediated glycolipid transfer between vesicle membranes. Anal. Biochem. 1999, 268, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science 1997, 277, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Daste, F.; Sauvanet, C.; Bavdek, A.; Baye, J.; Pierre, F.; Le Borgne, R.; David, C.; Rojo, M.; Fuchs, P.; Tareste, D. The heptad repeat domain 1 of Mitofusin has membrane destabilization function in mitochondrial fusion. EMBO Rep. 2018, 19, E43637. [Google Scholar] [CrossRef]
- Seifert, U.; Lipowsky, R. Adhesion of Vesicles. Phys. Rev. A 1990, 42, 4768–4771. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lee, C.C.; Huang, H.W. Adhesion and merging of lipid bilayers: A method for measuring the free energy of adhesion and hemifusion. Biophys. J. 2011, 100, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinkühler, J.; Agudo-Canalejo, J.; Lipowsky, R.; Dimova, R. Modulating Vesicle Adhesion by Electric Fields. Biophys. J. 2016, 111, 1454–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulbitch, A.; Guttenberg, Z.; Sackmann, E. Kinetics of membrane adhesion mediated by ligand-receptor interaction studied with a biomimetic system. Biophys. J. 2001, 81, 2743–2751. [Google Scholar] [CrossRef] [Green Version]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiokas, R.; Klenkar, G.; Tinazli, A.; Reichel, A.; Tampé, R.; Piehler, J.; Liedberg, B. Self-Assembled Monolayers Containing Terminal Mono-, Bis-, and Tris-nitrilotriacetic Acid Groups: Characterization and Application. Langmuir 2008, 4959–4967. [Google Scholar] [CrossRef]
- Kuo, S.C.; Lauffenburger, D.A. Relationship between receptor/ligand binding affinity and adhesion strength. Biophys. J. 1993, 65, 2191–2200. [Google Scholar] [CrossRef] [Green Version]
- Steinkühler, J.; Różycki, B.; Alvey, C.; Lipowsky, R.; Weikl, T.R.; Dimova, R.; Discher, D.E. Membrane fluctuations and acidosis regulate cooperative binding of ’marker of self’ protein CD47 with the macrophage checkpoint receptor SIRPα. J. Cell Sci. 2018, 132, Jcs216770. [Google Scholar] [CrossRef] [Green Version]
- Heuvingh, J.; Pincet, F.; Cribier, S. Hemifusion and fusion of giant vesicles induced by reduction of inter-membrane distance. Eur. Phys. J. E Soft Matter 2004, 14, 269–276. [Google Scholar] [CrossRef]
- Nishizawa, M.; Nishizawa, K. Curvature-driven lipid sorting: Coarse-grained dynamics simulations of a membrane mimicking a hemifusion intermediate. J. Biophys. Chem. 2010, 1, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Rappolt, M.; Hickel, A.; Bringezu, F.; Lohner, K. Mechanism of the Lamellar/Inverse Hexagonal Phase Transition Examined by High Resolution X-Ray Diffraction. Biophys. J. 2003, 84, 3111–3122. [Google Scholar] [CrossRef] [Green Version]
- Lonez, C.; Lensink, M.F.; Kleiren, E.; Vanderwinden, J.M.; Ruysschaert, J.M.; Vandenbranden, M. Fusogenic activity of cationic lipids and lipid shape distribution. Cell Mol. Life Sci. 2010, 67, 483–494. [Google Scholar] [CrossRef]
- Dorn, G.W. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, I.E. Mitochondria, 2nd ed.; Wiley-Liss: Hoboken, NJ, USA, 2008. [Google Scholar]
- Ramachandran, R. Mitochondrial dynamics: The dynamin superfamily and execution by collusion. Semin. Cell Dev. Biol. 2018, 76, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Gasper, R.; Meyer, S.; Gotthardt, K.; Sirajuddin, M.; Wittinghofer, A. It takes two to tango: Regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 2009, 10, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: Role of GEF and GAP systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Molt, R.W.J.; Blackburn, G.M. Metal Fluorides: Tools for Structural and Computational Analysis of Phosphoryl Transfer Enzymes. Top. Curr. Chem. 2017, 375, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigay, J.; Deterre, P.; Pfister, C.; Chabre, M. Fluoroaluminates Activate Transducin-Gdp by Mimicking the Gamma-Phosphate of Gtp in Its Binding-Site. FEBS Lett. 1985, 191, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, T.L.; Martin, R.B. Aluminum ion in biological systems. Trends Biochem. Sci. 1988, 13, 15–19. [Google Scholar] [CrossRef]
- Miller, J.L.; Hubbard, C.M.; Litman, B.J.; Macdonald, T.L. Inhibition of transducin activation and guanosine triphosphatase activity by aluminum ion. J. Biol. Chem. 1989, 264, 243–250. [Google Scholar] [PubMed]
- Chappie, J.S.; Acharya, S.; Leonard, M.; Schmid, S.L.; Dyda, F. G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature 2010, 465, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Zorzano, A.; Liesa, M.; Sebastian, D.; Segales, J.; Palacin, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef] [Green Version]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Hoppins, S.; Edlich, F.; Cleland, M.M.; Banerjee, S.; McCaffery, J.M.; Youle, R.J.; Nunnari, J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol. Cell 2011, 41, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Sloat, S.; Whitley, B.; Engelhart, E.; Hoppins, S. Identification of a mitofusin specificity region that confers unique activities to Mfn1 and Mfn2. Mol. Biol. Cell 2019, 30, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.L.; Meng, S.X.; Chen, Y.; Feng, J.X.; Gu, D.D.; Yu, B.; Li, Y.J.; Yang, J.Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, X.; Klemm, R.W.; Liu, T.Y.; Zhang, M.; Sun, S.; Sui, X.; Liu, X.; Rapoport, T.A.; Hu, J. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 3976–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrnes, L.J.; Sondermann, H. Structural basis for the nucleotide-dependent dimerization of the large G protein atlastin-1/SPG3A. Proc. Natl. Acad. Sci. USA 2011, 108, 2216–2221. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Rapoport, T.A. Fusion of the endoplasmic reticulum by membrane-bound GTPases. Semin. Cell Dev. Biol. 2016, 60, 105–111. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. On the role of Mitofusin 2 in endoplasmic reticulum-mitochondria tethering. Proc. Natl. Acad. Sci. USA 2017, 114, E2266–E2267. [Google Scholar] [CrossRef] [Green Version]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [Green Version]
- Cosson, P.; Marchetti, A.; Ravazzola, M.; Orci, L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: An ultrastructural study. PLoS ONE 2012, 7, e46293. [Google Scholar] [CrossRef] [Green Version]
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER–mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 2016, 20, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.T.C.; Garcin, P.O.; Fu, M.; Masoudi, M.; St-Pierre, P.; Panté, N.; Nabi, I.R. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J. Cell Sci. 2015, 128, 2759–2765. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, P.; Song, M.; Chen, Y.; Burelle, Y.; Dorn, G.W. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ. Res. 2014, 114, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francois-Martin, C.; Rothman, J.E.; Pincet, F. Low energy cost for optimal speed and control of membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 1238–1241. [Google Scholar] [CrossRef] [Green Version]
- Daumke, O.; Praefcke, G.J. Invited review: Mechanisms of GTP hydrolysis and conformational transitions in the dynamin superfamily. Biopolymers 2016, 105, 580–593. [Google Scholar] [CrossRef] [Green Version]
- Brandt, T.; Cavellini, L.; Kühlbrandt, W.; Cohen, M.M. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. Elife 2016, 5, E14618. [Google Scholar] [CrossRef]
- Wittinghofer, A. Signaling mechanistics: Aluminum fluoride for molecule of the year. Curr. Biol. 1997, 7, R682–R685. [Google Scholar] [CrossRef] [Green Version]
- Fenz, S.F.; Bihr, T.; Schmidt, D.; Merkel, R.; Seifert, U.; Sengupta, K.; Smith, A.S. Membrane fluctuations mediate lateral interaction between cadherin bonds. Nat. Phys. 2017, 13, 906–913. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhou, X.; Hu, X.; Joshi, A.S.; Guo, X.; Zhu, Y.; Chen, Q.; Prinz, W.A.; Hu, J. Sequences flanking the transmembrane segments facilitate mitochondrial localization and membrane fusion by mitofusin. Proc. Natl. Acad. Sci. USA 2017, 114, E9863–E9872. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.Y.; Bian, X.; Sun, S.; Hu, X.; Klemm, R.W.; Prinz, W.A.; Rapoport, T.A.; Hu, J. Lipid interaction of the C terminus and association of the transmembrane segments facilitate atlastin-mediated homotypic endoplasmic reticulum fusion. Proc. Natl. Acad. Sci. USA 2012, 109, E2146–E2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolosa-Díaz, A.; Almendro-Vedia, V.G.; Natale, P.; López-Montero, I. The GDP-Bound State of Mitochondrial Mfn1 Induces Membrane Adhesion of Apposing Lipid Vesicles through a Cooperative Binding Mechanism. Biomolecules 2020, 10, 1085. https://doi.org/10.3390/biom10071085
Tolosa-Díaz A, Almendro-Vedia VG, Natale P, López-Montero I. The GDP-Bound State of Mitochondrial Mfn1 Induces Membrane Adhesion of Apposing Lipid Vesicles through a Cooperative Binding Mechanism. Biomolecules. 2020; 10(7):1085. https://doi.org/10.3390/biom10071085
Chicago/Turabian StyleTolosa-Díaz, Andrés, Víctor G. Almendro-Vedia, Paolo Natale, and Iván López-Montero. 2020. "The GDP-Bound State of Mitochondrial Mfn1 Induces Membrane Adhesion of Apposing Lipid Vesicles through a Cooperative Binding Mechanism" Biomolecules 10, no. 7: 1085. https://doi.org/10.3390/biom10071085
APA StyleTolosa-Díaz, A., Almendro-Vedia, V. G., Natale, P., & López-Montero, I. (2020). The GDP-Bound State of Mitochondrial Mfn1 Induces Membrane Adhesion of Apposing Lipid Vesicles through a Cooperative Binding Mechanism. Biomolecules, 10(7), 1085. https://doi.org/10.3390/biom10071085