The Interaction of Temozolomide with Blood Components Suggests the Potential Use of Human Serum Albumin as a Biomimetic Carrier for the Drug

 , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. LUVs Formation
2.3. Labeling of LUVs and Fluorescence Measurements
2.4. Absorption and Fluorescence Experiments
2.5. Fluorescence Anisotropy Measurements
2.6. Quenching Experiments
2.7. Energy Transfer Experiments
2.8. Circular Dichroism Experiments
2.9. Molecular Docking Simulations
2.10. Molecular Dynamics Simulations
3. Results and Discussion
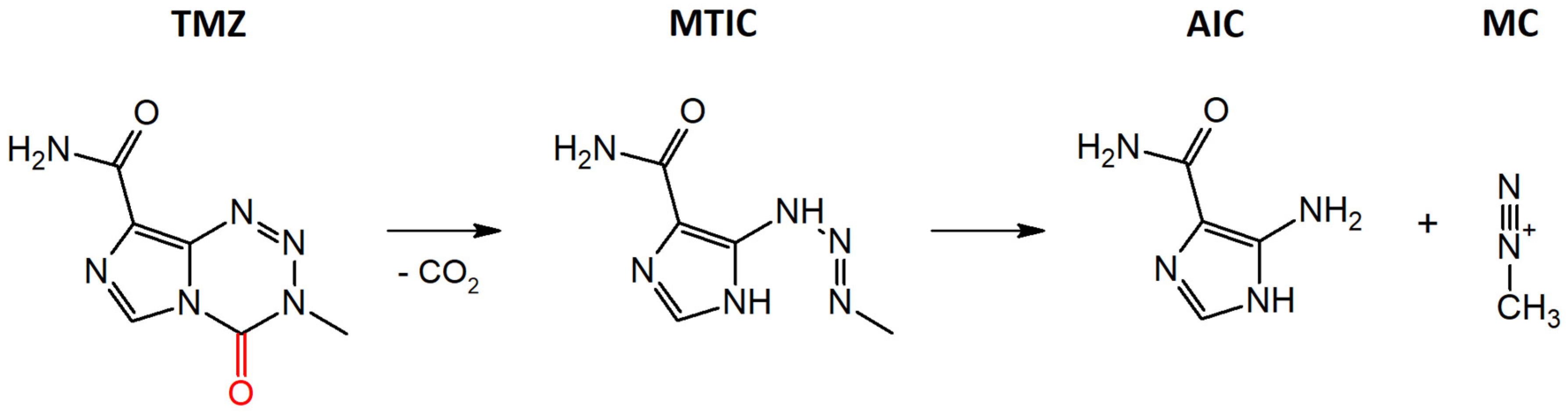
3.1. Stability of TMZ in Aqueous Media
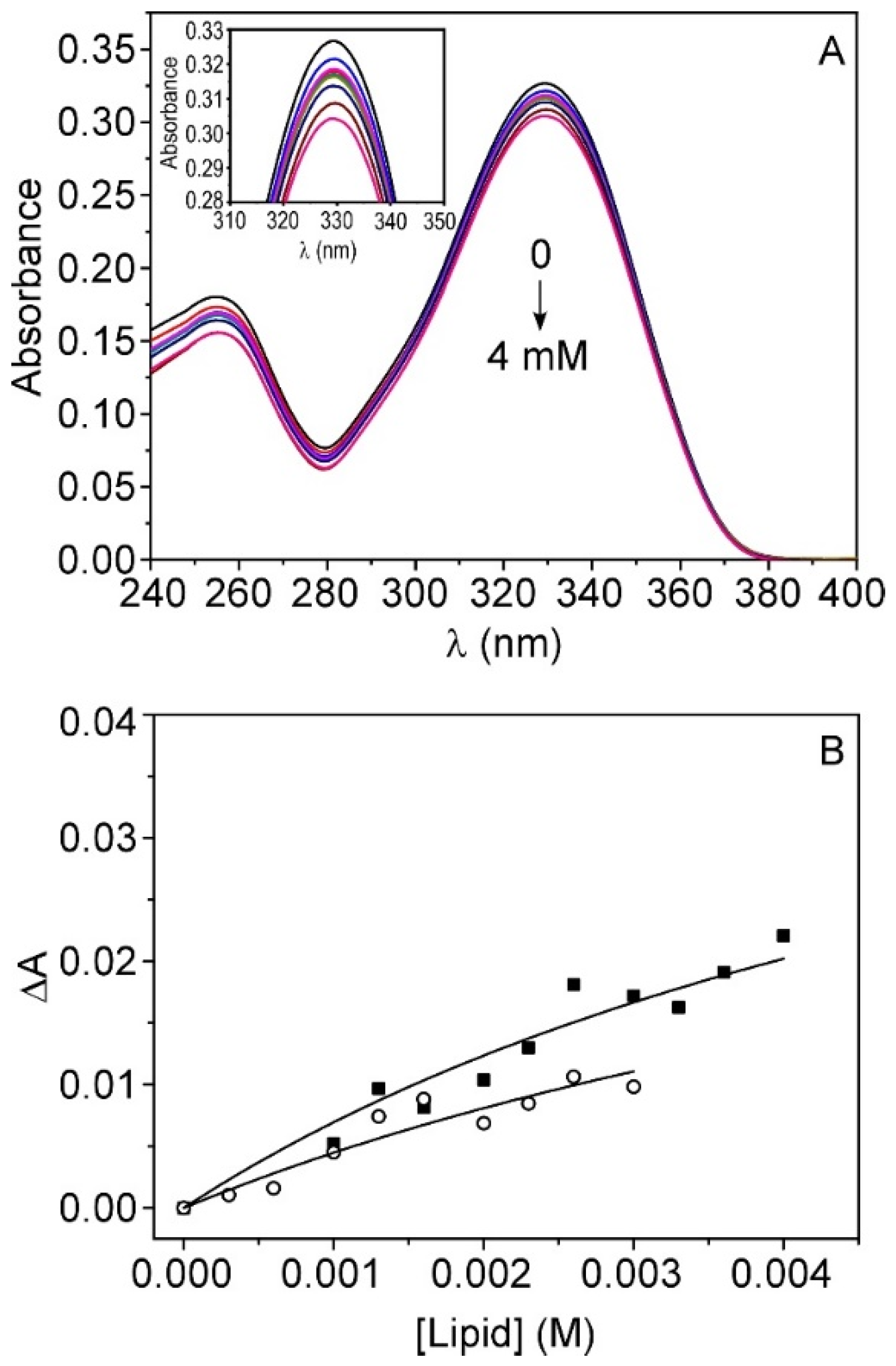
3.2. Interaction of TMZ with Model Membranes
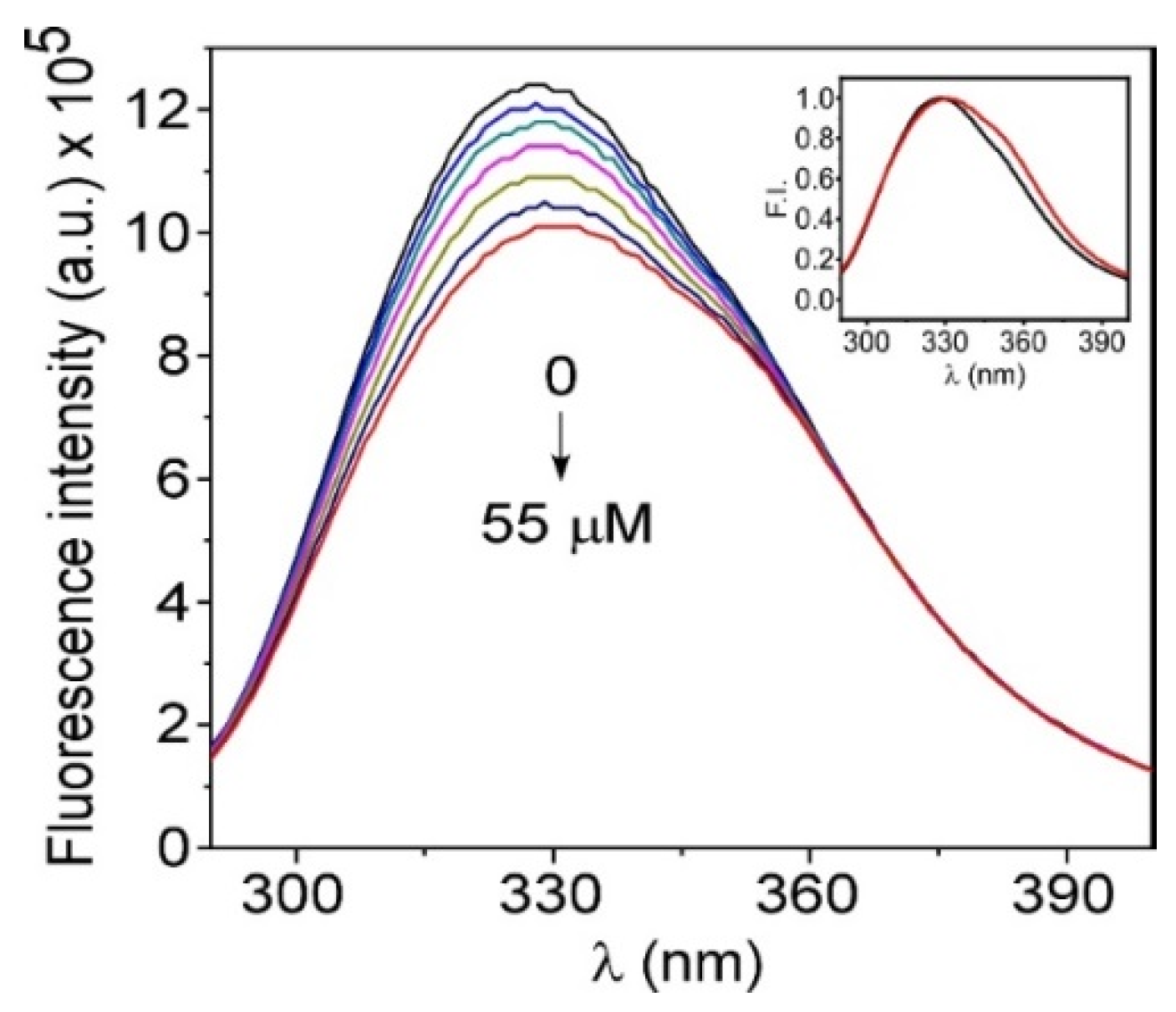
3.3. Interaction of TMZ with HSA
3.4. Interaction of TMZ with AGP
3.5. Stability of TMZ-HSA Complex
3.6. CD Measurements on the TMZ-HSA Complex
3.7. Molecular Docking and Dynamics Simulations of TMZ-HSA Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ward, S.M.; Skinner, M.; Saha, B.; Emrick, T. Polymer-temozolomide conjugates as therapeutics for treating glioblastoma. Mol. Pharm. 2018, 15, 5263–5276. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. HHS public access. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, M.J.; Coelho, M.A.N.; Pereira, M.C. Nanocarriers for the delivery of temozolomide in the treatment of glioblastoma. In Design and Development of New Nanocarriers; Elsevier: Amsterdam, The Netherlands, 2018; pp. 687–722. ISBN 9780128136270. [Google Scholar]
- Quan, R.; Zhang, H.; Li, Z.; Li, X. Survival analysis of patients with glioblastoma treated by long-term administration of temozolomide. Medicine 2020, 99, e18591. [Google Scholar] [CrossRef] [PubMed]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol. Med. Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Lopes, I.C.; De Oliveira, S.C.B.; Oliveira-Brett, A.M. Temozolomide chemical degradation to 5-aminoimidazole-4-carboxamide—Electrochemical study. J. Electroanal. Chem. 2013, 704, 183–189. [Google Scholar] [CrossRef]
- Wei, J.H.; Zhou, R.H.; Peng, Y.; Liu, Y.C. Studies on the Binding Properties of Temozolomide with DNA. Asian, J. Chem. 2013, 25, 2597–2600. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Temozolomide: Therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev. Neurother. 2010, 10, 1537–1544. [Google Scholar] [CrossRef]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Marchesi, F.; Turriziani, M.; Tortorelli, G.; Avvisati, G.; Torino, F.; De Vecchis, L. Triazene compounds: Mechanism of action and related DNA repair systems. Pharmacol. Res. 2007, 56, 275–287. [Google Scholar] [CrossRef]
- Perazzoli, G.; Prados, J.; Ortiz, R.; Caba, O.; Cabeza, L.; Berdasco, M.; Gónzalez, B.; Melguizo, C. Temozolomide resistance in glioblastoma cell lines: Implication of MGMT, MMR, P-glycoprotein and CD133 expression. PLoS ONE 2015, 10, e0140131. [Google Scholar] [CrossRef]
- Kumari, S.; Ahsan, S.M.; Kumar, J.M.; Kondapi, A.K.; Rao, N.M. Overcoming blood brain barrier with a dual purpose Temozolomide loaded Lactoferrin nanoparticles for combating glioma (SERP-17-12433). Sci. Rep. 2017, 7, 6602. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, B.I.; Jeon, J.H.; Kim, D.K.; Kang, S.G.; Shim, J.K.; Kim, S.Y.; Kang, S.W.; Jang, H. Gossypol suppresses growth of temozolomide-resistant glioblastoma tumor spheres. Biomolecules 2019, 9, 595. [Google Scholar] [CrossRef]
- Groothuis, D.R. The blood-brain and blood-tumor barriers: A review of strategies for increasing drug delivery. Neuro. Oncol. 2000, 2, 45–59. [Google Scholar] [CrossRef]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef]
- Angeli, E.; Nguyen, T.T.; Janin, A.; Bousquet, G. How to make anticancer drugs cross the blood-brain barrier to treat brain metastases. Int. J. Mol. Sci. 2020, 21, 22. [Google Scholar] [CrossRef]
- Sun, Q.; Ojha, T.; Kiessling, F.; Lammers, T.; Shi, Y. Enhancing tumor penetration of nanomedicines. Biomacromolecules 2017, 18, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, M.J.; Andrade, S.; Coelho, M.Á.N.; Loureiro, J.A.; Pereira, M.C. Biophysical interaction of temozolomide and its active metabolite with biomembrane models: The relevance of drug-membrane interaction for Glioblastoma Multiforme therapy. Eur. J. Pharm. Biopharm. 2019, 136, 156–163. [Google Scholar] [CrossRef]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin-drug interaction and its clinical implication. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Huang, Z.; Ung, T. Effect of alpha-1-acid glycoprotein binding on pharmacokinetics and pharmacodynamics. Curr. Drug Metab. 2013, 14, 226–238. [Google Scholar]
- Chaves, O.A.; Ana, A.P.; Castro, L.H.E.; Sant’anna, C.M.R.; De Oliveira, M.C.C.; Cesarin-Sobrinho, D.; Netto-Ferreira, J.C.; Ferreira, A.B.B. Fluorescence and docking studies of the interaction between human serum albumin and pheophytin. Molecules 2015, 20, 19526–19539. [Google Scholar] [CrossRef]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999, 5, 309–317. [Google Scholar] [PubMed]
- Dang, M.; Saunders, L.; Niu, X.; Fan, Y.; Ma, P.X. Biomimetic delivery of signals for bone tissue engineering. Bone Res. 2018, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhao, P.; Jiang, Y.; Tang, Y.; Jin, H.; Pan, Z.; He, H.; Yang, V.C.; Huang, Y. Blood-brain-barrier-penetrating albumin nanoparticles for biomimetic drug delivery via albumin-binding protein pathways for antiglioma therapy. ACS Nano 2016, 10, 9999–10012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, J.; Jiang, M.; Zhao, L.; Li, S.; Sun, H.; Yang, F.; Liang, H. Human serum albumin-based dual-agent delivery systems for combination therapy: Acting against cancer cells and inhibiting neovascularization in the tumor microenvironment. Mol. Pharm. 2020, 17, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Bourgeaux, V.; Lanao, J.M.; Bax, B.E.; Godfrin, Y. Drug-loaded erythrocytes: On the road toward marketing approval. Drug Des. Dev. Ther. 2016, 2016, 665–676. [Google Scholar] [CrossRef]
- Muzykantov, V.R. Drug delivery by red blood cells: Vascular carriers designed by mother nature. Expert Opin. Drug Deliv. 2010, 7, 403–427. [Google Scholar] [CrossRef]
- Filip, Z.; Jan, K.; Vendula, S.; Jana, K.Z.; Kamil, M.; Kamil, K. Albumin and α1-acid glycoprotein: Old acquaintances. Expert Opin. Drug Metab. Toxicol. 2013, 9, 943–954. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Rabbani, G.; Lee, E.J.; Ahmad, K.; Baig, M.H.; Choi, I. Binding of tolperisone hydrochloride with human serum albumin: Effects on the conformation, thermodynamics, and activity of HSA. Mol. Pharm. 2018, 15, 1445–1456. [Google Scholar] [CrossRef]
- Mohos, V.; Fliszár-Nyúl, E.; Lemli, B.; Zsidó, B.Z.; Hetényi, C.; Mladěnka, P.; Horký, P.; Pour, M.; Poór, M. Testing the pharmacokinetic interactions of 24 colonic flavonoid metabolites with human serum albumin and cytochrome P450 enzymes. Biomolecules 2020, 10, 409. [Google Scholar] [CrossRef]
- Bern, M.; Sand, K.M.K.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control. Release 2015, 211, 144–162. [Google Scholar] [CrossRef]
- Wang, Z.; Ho, J.X.; Ruble, J.R.; Rose, J.; Rüker, F.; Ellenburg, M.; Murphy, R.; Click, J.; Soistman, E.; Wilkerson, L.; et al. Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 5356–5374. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Y.; Liang, H. Interactive association of drugs binding to human serum albumin. Int. J. Mol. Sci. 2014, 15, 3580–3595. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Li, Z.; Yin, S.; Wang, Q.; Guo, F.; Zhang, Y.; Yu, R.; Liu, Y.; Su, Z. Extending half life of H-ferritin nanoparticle by fusing albumin binding domain for doxorubicin encapsulation. Biomacromolecules 2018, 19, 773–781. [Google Scholar] [CrossRef]
- Chaves, O.A.; Tavares, M.T.; Cunha, M.R.; Parise-Filho, R.; Sant’anna, C.M.R.; Carlos Netto-Ferreira, J. Multi-spectroscopic and theoretical analysis on the interaction between human serum albumin and a Capsaicin derivative—RPF101. Biomolecules 2018, 8, 78. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, Y.; Wu, A.; Rao, Y.; Huang, Y. Roles of albumin-binding proteins in cancer progression and biomimetic targeted drug delivery. ChemBioChem 2018, 19, 1796–1805. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin-more than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef]
- Parodi, A.; Miao, J.; Soond, S.M.; Rudzińska, M.; Zamyatnin, A.A. Albumin nanovectors in cancer therapy and imaging. Biomolecules 2019, 9, 218. [Google Scholar] [CrossRef]
- Kremer, J.M.; Wilting, J.; Janssen, L.H. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol. Rev. 1988, 40, 1–47. [Google Scholar]
- Zsila, F.; Iwao, Y. The drug binding site of human α1-acid glycoprotein: Insight from induced circular dichroism and electronic absorption spectra. Biochim. Biophys. Acta-Gen. Subj. 2007, 1770, 797–809. [Google Scholar] [CrossRef]
- Peetla, C.; Stine, A.; Labhasetwar, V. Biophysical interactions with model lipid membranes: Applications in drug discovery and drug delivery. Mol. Pharm. 2009, 6, 1264–1276. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial peptides: Interaction with model and biological membranes and synergism with chemical antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy Principles of Fluorescence Spectroscopy; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Eftink, M.R. Fluorescence Techniques for Studying Protein Structure. Methods Biochem. Anal. 1991, 35, 127–205. [Google Scholar] [PubMed]
- Abou-Zied, O.K.; Al-Shihi, O.I.K. Characterization of subdomain HA binding site of human serum albumin in its native, unfolded, and refolded states using small molecular probes. J. Am. Chem. Soc. 2008, 130, 10793–10801. [Google Scholar] [CrossRef] [PubMed]
- Van De Weert, M.; Stella, L. Fluorescence quenching and ligand binding: A critical discussion of a popular methodology. J. Mol. Struct. 2011, 998, 144–150. [Google Scholar] [CrossRef]
- Förster, T. 10th Spiers Memorial Lecture. Transfer mechanics of Electronic Excitation. Discuss. Faraday Soc. 1959, 27, 7–17. [Google Scholar]
- Encinar, J.A.; Fernández-Ballester, G.; Galiano-Ibarra, V.; Micol, V. In silico approach for the discovery of new PPARγ modulators among plant-derived polyphenols. Drug Des. Dev. Ther. 2015, 9, 5877–5895. [Google Scholar] [CrossRef]
- Galiano, V.; Garcia-Valtanen, P.; Micol, V.; Encinar, J.A. Looking for inhibitors of the dengue virus NS5 RNA-dependent RNA-polymerase using a molecular docking approach. Drug Des. Dev. Ther. 2016, 10, 3163–3181. [Google Scholar] [CrossRef]
- Ruiz-Torres, V.; Losada-Echeberría, M.; Herranz-López, M.; Barrajón-Catalán, E.; Galiano, V.; Micol, V.; Encinar, J.A. New mammalian target of rapamycin (mTOR) modulators derived from natural product databases and marine extracts by using molecular docking techniques. Mar. Drugs 2018, 16, 385. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Revisiting silibinin as a novobiocin-like Hsp90 C-terminal inhibitor: Computational modeling and experimental validation. Food Chem. Toxicol. 2019, 132, 110645. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Lozano-Sánchez, J.; Viciano, I.; Llorach-Parés, L.; Nonell-Canals, A.; Bosch-Barrera, J.; Brunet, J.; Segura-Carretero, A.; Sanchez-Martinez, M.; et al. The extra virgin olive oil phenolic oleacein is a dual substrate-inhibitor of catechol-O-methyltransferase. Food Chem. Toxicol. 2019, 128, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging (Albany NY) 2020, 12, 8–34. [Google Scholar] [CrossRef]
- Payliss, B.J.; Hassanin, M.; Prenner, E.J. The structural and functional effects of Hg(II) and Cd(II) on lipid model systems and human erythrocytes: A review. Chem. Phys. Lipids 2015, 193, 36–51. [Google Scholar] [CrossRef]
- Doucet, K.G.; Glister, J.F.; Pye, C.C. An ab initio study of model triazene-based anticancer agents. Can. J. Chem. 2010, 88, 709–715. [Google Scholar] [CrossRef]
- Khalilian, M.H.; Mirzaei, S.; Taherpour, A.A. The simulation of UV spectroscopy and electronic analysis of temozolomide and dacarbazine chemical decomposition to their metabolites. J. Mol. Model. 2016, 22, 270. [Google Scholar] [CrossRef]
- Santos, N.C.; Prieto, M.; Castanho, M.A.R.B. Quantifying molecular partition into model systems of biomembranes: An emphasis on optical spectroscopic methods. Biochim. Biophys. Acta-Biomembr. 2003, 1612, 123–135. [Google Scholar] [CrossRef]
- Kahveci, Z.; Martínez-Tomé, M.J.; Esquembre, R.; Mallavia, R.; Mateo, C.R. Selective interaction of a cationic polyfluorene with model lipid membranes: Anionic versus zwitterionic lipids. Materials 2014, 7, 2120–2140. [Google Scholar] [CrossRef]
- Davenport, L.; Dale, R.E.; Bisby, R.H.; Cundall, R.B. Transverse location of the fluorescent probe l,6-Diphenyl-l,3,5-hexatriene in model lipid bilayer membrane systems by resonance excitation energy transfer. Biochemistry 1985, 24, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Imayoshi, N.; Goto, T.; Shiro, H.; Mano, T.; Nakai, Y. Second derivative spectrophotometric determination of partition coefficients of chlorpromazine and promazine between lecithin bilayer vesicles and water. Anal. Chim. Acta 1995, 304, 101–106. [Google Scholar] [CrossRef]
- Parasassi, T.; Di Stefano, M.; Loiero, M.; Ravagnan, G.; Gratton, E. Influence of cholesterol on phospholipid bilayers phase domains as detected by Laurdan fluorescence. Biophys. J. 1994, 66, 120–132. [Google Scholar] [CrossRef]
- Sýkora, J.; Kapusta, P.; Fidler, V.; Hof, M. On what time scale does solvent relaxation in phospholipid bilayers happen? Langmuir 2002, 18, 571–574. [Google Scholar] [CrossRef]
- Esquembre, R.; Ferrer, M.L.; Gutiérrez, M.C.; Mallavia, R.; Mateo, C.R. Fluorescence study of the fluidity and cooperativity of the phase transitions of zwitterionic and anionic liposomes confined in sol-gel glasses. J. Phys. Chem. B 2007, 111, 3665–3673. [Google Scholar] [CrossRef]
- Jurkiewicz, P.; Olzyńska, A.; Langner, M.; Hof, M. Headgroup hydration and mobility of DOTAP/DOPC bilayers: A fluorescence solvent relaxation study. Langmuir 2006, 22, 8741–8749. [Google Scholar] [CrossRef] [PubMed]
- Kasende, O.E.; Matondo, A.; Muzomwe, M.; Muya, J.T.; Scheiner, S. Interaction between temozolomide and water: Preferred binding sites. Comput. Theor. Chem. 2014, 1034, 26–31. [Google Scholar] [CrossRef]
- Kasende, O.E.; Muya, J.T.; de Paul, N.; Nziko, V.; Scheiner, S. Hydrogen bonded and stacked geometries of the temozolomide dimer. J. Mol. Model. 2016, 22, 77. [Google Scholar] [CrossRef]
- Martínez-Tomé, M.J.; Esquembre, R.; Mallavia, R.; Mateo, C.R. Formation of complexes between the conjugated polyelectrolyte Poly{[9,9-bis(6′-N,N,N-trimethylammonium)hexyl]fluorene-phenylene} bromide (HTMA-PFP) and human serum albumin. Biomacromolecules 2010, 11, 1494–1501. [Google Scholar] [CrossRef]
- Banerjee, P.; Pramanik, S.; Sarkar, A.; Bhattacharya, S.C. Deciphering the fluorescence resonance energy transfer signature of 3-pyrazolyl 2-pyrazoline in transport proteinous environment. J. Phys. Chem. B 2009, 113, 11429–11436. [Google Scholar] [CrossRef]
- Yang, X.; Ye, Z.; Yuan, Y.; Zheng, Z.; Shi, J.; Ying, Y.; Huang, P. Insights into the binding of paclitaxel to human serum albumin: Multispectroscopic studies. Luminescence 2013, 28, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Qu, X.; Li, Y.; Kong, Y.; Jia, B.; Yao, X.; Jiang, B. Binding of citreoviridin to human serum albumin: Multispectroscopic and molecular docking. Biomed. Res. Int. 2015, 2015, 162391. [Google Scholar] [CrossRef] [PubMed]
- Jachimska, B.; Wasilewska, M.; Adamczyk, Z. Characterization of globular protein solutions by dynamic light scattering, electrophoretic mobility, and viscosity measurements. Langmuir 2008, 24, 6866–6872. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Appel, E.A.; Rowland, M.J.; Loh, X.J.; Heywood, R.M.; Watts, C.; Scherman, O.A. Enhanced stability and activity of temozolomide in primary glioblastoma multiforme cells with cucurbit[n]uril. Chem. Commun. 2012, 48, 9843–9845. [Google Scholar] [CrossRef]
- Di Martino, A.; Kucharczyk, P.; Capakova, Z.; Humpolicek, P.; Sedlarik, V. Enhancement of temozolomide stability by loading in chitosan-carboxylated polylactide-based nanoparticles. J. Nanopart. Res. 2017, 19, 71. [Google Scholar] [CrossRef]
- Wang, C.; Nguyen, P.H.; Pham, K.; Huynh, D.; Le, T.B.N.; Wang, H.; Ren, P.; Luo, R. Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 2016, 37, 2436–2446. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Li, X.; Shao, F.; Sun, J.; Du, K.; Sun, Y.; Feng, F. Enhanced copper-temozolomide interactions by protein for chemotherapy against glioblastoma multiforme. ACS Appl. Mater. Interfaces 2019, 11, 41935–41945. [Google Scholar] [CrossRef]
- Khosa, A.; Krishna, K.V.; Dubey, S.K.; Saha, R.N. Lipid Nanocarriers for Enhanced Delivery of Temozolomide to the Brain. Methods Mol. Biol. 2020, 2059, 285–298. [Google Scholar]
- Afzalipour, R.; Khoei, S.; Khoee, S.; Shirvalilou, S.; Jamali Raoufi, N.; Motevalian, M.; Karimi, M.R. Dual-targeting temozolomide loaded in folate-conjugated magnetic triblock copolymer nanoparticles to improve the therapeutic efficiency of rat brain gliomas. ACS Biomater. Sci. Eng. 2019, 5, 6000–6011. [Google Scholar] [CrossRef]
- Fu, W.; You, C.; Ma, L.; Li, H.; Ju, Y.; Guo, X.; Shi, S.; Zhang, T.; Zhou, R.; Lin, Y. Enhanced efficacy of temozolomide loaded by a tetrahedral framework DNA nanoparticle in the therapy for glioblastoma. ACS Appl. Mater. Interfaces 2019, 11, 39525–39533. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Italiya, K.; Mittal, A.; Chitkara, D. New strategies for cancer management: How can temozolomide carrier modifications improve its delivery? Ther. Deliv. 2017, 8, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Imam, S.S.; Aqil, M.; Ahad, A.; Sultana, Y.; Ali, A.; Khan, K. Brain targeting of temozolomide via the intranasal route using lipid-based nanoparticles: Brain pharmacokinetic and scintigraphic analyses. Mol. Pharm. 2016, 13, 3773–3782. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TMZ (μM) | <r> (15 °C) ± SD | <r> (37 °C) ± SD |
|---|---|---|
| 0 | 0.339 ± 0.001 | 0.092 ± 0.002 |
| 100 | 0.339 ± 0.003 | 0.091 ± 0.002 |
| 150 | 0.340 ± 0.001 | 0.093 ± 0.001 |
| Stern–Volmer Constants | Binding Parameter | Thermodynamic Parameters | ||||
|---|---|---|---|---|---|---|
| Protein | T(K) | KSV (M−1) a | Ka (M−1) a | ΔG (J mol−1) a | ΔH (J mol−1) a | ΔS (J mol−1K−1) a |
| HSA | 288 | 4462 ± 334 | 5442 ± 140 | −2.06 × 104 ± 62 | −4571 ± 2644 | 57 ± 9 |
| 298 | 5232 ± 245 | 5103 ± 58 | −2.11 × 104 ± 28 | |||
| AGP | 298 | 3093 ± 127 | 8184 ± 983 | −2.23 × 104 ± 298 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio-Camacho, M.; Encinar, J.A.; Martínez-Tomé, M.J.; Esquembre, R.; Mateo, C.R. The Interaction of Temozolomide with Blood Components Suggests the Potential Use of Human Serum Albumin as a Biomimetic Carrier for the Drug. Biomolecules 2020, 10, 1015. https://doi.org/10.3390/biom10071015
Rubio-Camacho M, Encinar JA, Martínez-Tomé MJ, Esquembre R, Mateo CR. The Interaction of Temozolomide with Blood Components Suggests the Potential Use of Human Serum Albumin as a Biomimetic Carrier for the Drug. Biomolecules. 2020; 10(7):1015. https://doi.org/10.3390/biom10071015
Chicago/Turabian StyleRubio-Camacho, Marta, José A. Encinar, María José Martínez-Tomé, Rocío Esquembre, and C. Reyes Mateo. 2020. "The Interaction of Temozolomide with Blood Components Suggests the Potential Use of Human Serum Albumin as a Biomimetic Carrier for the Drug" Biomolecules 10, no. 7: 1015. https://doi.org/10.3390/biom10071015
APA StyleRubio-Camacho, M., Encinar, J. A., Martínez-Tomé, M. J., Esquembre, R., & Mateo, C. R. (2020). The Interaction of Temozolomide with Blood Components Suggests the Potential Use of Human Serum Albumin as a Biomimetic Carrier for the Drug. Biomolecules, 10(7), 1015. https://doi.org/10.3390/biom10071015